
Abstract
Background
Ovarian cancer is a leading cause of the death from gynecologic malignancies. Hypoxia is closely related to the malignant growth of cells. However, the molecular mechanism of hypoxia-regulated ovarian cancer cells remains unclear. Thus, this study was conducted to identify the key genes and pathways implicated in the regulation of hypoxia by bioinformatics analysis.
Methods
Using the datasets of GSE53012 downloaded from the Gene Expression Omnibus (GEO), the differentially expressed genes (DEGs) were screened by comparing the RNA expression from cycling hypoxia group, chronic hypoxia group, and control group. Subsequently, cluster analysis was performed followed by the construction of the protein-protein interaction (PPI) network of the overlap** DEGs between the cycling hypoxia and chronic hypoxia using ClusterONE. In addition, gene ontology (GO) functional and pathway enrichment analyses of the DEGs in the most remarkable module were performed using Database for Annotation, Visualization and Integrated Discovery (DAVID) software. Ultimately, the signaling pathways associated with hypoxia were verified by RT-PCR, WB, and MTT assays.
Results
A total of 931 overlap** DEGs were identified. Nine hub genes and seven node genes were screened by analyzing the PPI and pathway integration networks, including ESR1, MMP2, ErbB2, MYC, VIM, CYBB, EDN1, SERPINE1, and PDK. Additionally, 11 key pathways closely associated with hypoxia were identified, including focal adhesion, ErbB signaling, and proteoglycans in cancer, among which the ErbB signaling pathway was verified by RT-PCR, WB, and MTT assays. Furthermore, functional enrichment analysis revealed that these genes were mainly involved in the proliferation of ovarian cancer cells, such as regulation of cell proliferation, cell adhesion, positive regulation of cell migration, focal adhesion, and extracellular matrix binding.
Conclusion
The results show that hypoxia can promote the proliferation of ovarian cancer cells by affecting the invasion and adhesion functions through the dysregulation of ErbB signaling, which may be governed by the HIF-1α-TGFA-EGFR-ErbB2-MYC axis. These findings will contribute to the identification of new targets for the diagnosis and treatment of ovarian cancer.
Similar content being viewed by others


Background
Ovarian cancer, the most lethal gynecological malignancy, is a major cause of cancer-related mortality in women, with an estimated 22,280 new cases and 14,240 deaths predicted for 2016 in the United States [1]. Ovarian cancer is often called the “silent killer” because its signs and symptoms are frequently absent until it has reached advanced stages where outcome is poor [2]. Accumulating evidence suggests that a hypoxic environment in vivo, i.e., the absence of a sufficient oxygen supply, is tightly associated with a poor prognosis and a high mortality in patients with ovarian cancer [3]. However, the detailed mechanisms by which hypoxia regulates the status of ovarian cancer cells leading to a series of physiological changes are still unknown. Therefore, exploring the effect of hypoxia on ovarian cancer cells will likely have important implications and offer opportunities to solve this problem for potential therapeutic purposes.
The oxygen tension of normal tissues is in the region of 1-4%, while hypoxia is < 1% [4]. Generally, rapidly growing tumors outstrip their vascular supply and become hypoxic. Under hypoxic conditions, tumor cells adapt by generating energy in oxygen independent ways and minimize cellular damage by inducing the expression of genes involved in angiogenesis, glycolysis, cell survival, invasion, tumor progression, and pH regulation, which can observably influence cell metabolism by activating the hypoxia inducible factor-1 (HIF-1) signaling pathway [5]. In recent decades, more and more researchers have devoted themselves to exploring the potential mechanisms by which hypoxia regulates the progression of ovarian cancer cells. For instance, it has been demonstrated that the endothelin-1/ endothelin A receptor (ET-1/ETAR) axis in epithelial ovarian cancer (EOC) cells induces vascular-endothelial growth factor (VEGF) expression through HIF-1α nuclear accumulation, resulting in the invasion of cancer cells [6]. Rab25, a small GTPase of the Rab11 subfamily, has been functionally linked to the progression and aggressiveness of ovarian cancer. Enhanced Rab25 expression in ovarian cancer cell lines results in increased cell proliferation, inhibition of apoptosis and anoikis, as well as increased aggressiveness in vivo [7], which has been found to rely on the activation of HIF-1 [8]. In addition, hypoxia is well-known in increasing the resistance to chemotherapy and radiotherapy and result in the decline of cell immunity, which both contribute to the survival and growth of cancer cells [9,10,11]. Moreover, hypoxia was found to induce the expression of HIF-1α and G-protein estrogen receptor (GPER) that were involved in the regulation of VEGF expression in breast cancer cells and carcinoma-associated fibroblasts (CAFs), leading to the release of angiogenic factors and the growth of new blood vessels [12]. Hypoxia can also regulate the frequency of tumor-initiating cells by promoting epithelial-mesenchymal transition (EMT) and metastasis formation [13, 14]. Overall, the effects of hypoxia on cells are regulated in a variety of ways depending on the external environment and cell type. Therefore, it is of great practical significance to explore the specific action modes and pathways of hypoxia on ovarian cancer cells.
Although ovarian cancer has been long studied from the perspective of single genes and their specific properties, it has become clear that more integrative, systematic analyses are necessary to better understand how this serious disease develops and how it may respond to hypoxia. Recently, gene expression profile data associated with ovarian cancer have been studied by many researchers. For example, Fu et al. indicated that ovarian cancer was closely associated with dysregulation of the p53 signaling pathway, drug metabolism, tyrosine metabolism, and cell cycle by screening the differentially expressed genes (DEGs) between human ovarian cancer samples and healthy controls based on the microarray data of GSE14407. Further, a series of genes, such as cyclin E1 (CCNE1), cyclin B2 (CCNB2), cytochrome P450 family 3 subfamily A member 5 (CYP3A5), and vascular endothelial growth factor A (VEGFA), have been predicted as target genes for diagnosing ovarian cancer [15]. Additionally, Xue et al. explored the molecular mechanisms of NSC319726, a newly discovered anticancer small-molecule drug, in ovarian cancer by bioinformatics analyses and found that it might play a role against ovarian cancer via targeting genes involved in the oocyte meiosis pathway, such as ribosomal protein S6 kinase A6 (RPS6KA6), B-cell CLL/lymphoma 6 (BCL6), forkhead box O3 (FOXO3), cyclin B1 (CCNB1), and cell division cycle 20 (CDC20) [16, 17]. However, studies have not been yet performed on the relationship between target genes and hypoxia, nor on the main effect of hypoxia on the function and relevant regulatory pathways of ovarian cancer cells. In this context, investigating the regulation of hypoxia on the progression of ovarian cancer by bioinformatics analyses supposes to be necessary.
In the present study, RNA expression in the ovarian cancer cell line SK-OV-3 were compared between untreated and hypoxia treated (including cycling hypoxia and chronic hypoxia) samples to identify the DEGs related to hypoxia effects based on the microarray datasets. Subsequently, the protein-protein interaction (PPI) network of the overlap** DEGs was constructed and the hub genes in the network with wide influence on others were identified. Then, the most remarkable module was screened followed by cluster analysis of the PPI network. Afterwards, the Gene Ontology (GO) functional and Kyoto Encyclopedia of Genes and Genomes (KEGG) (http://www.genome.jp/kegg/) pathway enrichment analyses of the most remarkable module were performed. Finally, an integrated pathway network associated with hypoxia in ovarian cancer was constructed and verified. The findings of this study improve the understanding of the role of hypoxia and initially validate a subset of these markers in ovarian cancer cells. This investigation also provides a resource for building new hypotheses for further follow-up studies.
Methods
Data normalization and identification of DEGs
The microarray expression profile datasets (GEO access number: GSE53012), contributed by Olbryt et al. [18], were downloaded from the National Center of Biotechnology Information (NCBI) Gene Expression Omnibus (GEO) database (http://www.ncbi.nlm.nih.gov/geo/), which was currently the largest fully public gene expression resource [19]. There were a total of nine samples in this analysis, including three untreated samples, three samples treated with cycling hypoxia and three with chronic hypoxia. After background correction and quartile data normalization were performed [20, 21], the DEGs were identified between untreated samples and cycling hypoxia treated samples (DEGs1) and between untreated samples and chronic hypoxia treated samples (DEGs2) [22], with the criteria of |log fold change (FC)| > 1 and p-value < 0.05 followed by multiple-testing correction using the Benjamini-Hochberg procedure to obtain the adjusted p-value [18]. Based on these data, the overlap** DEGs between DEGs1 and DEGs2 were further analyzed.
PPI network construction
Considering that proteins rarely work alone, it is necessary to study the interactions among proteins. Therefore, the protein-protein interaction network (PPI) of the overlap** DEGs was constructed with a confidence score of > 0.4 based on the Search Tool for the Retrieval of Interacting Genes (STRING) database [23] (STRING, version 9.1, http://string91.embl.de/), which was a precomputed global resource to predicted and known interaction information, by the use of Cytoscape [24] (version 3.0; http://cytoscape.org/), which was a general bioinformatics package. In the PPI network, each node stands for a gene and edges represent the interactions between the nodes. In view of the fact that most of the networks were scale-free, the hub genes were then selected with a connectivity degree ≥35 after calculating the degree of each node.
Module analysis of the PPI network
Module analysis of PPI network was performed with the parameters of minimum size > 5 and minimum density < 0.05 using ClusterONE, a Cytoscape plugin that unified different clustering techniques and displayed them in a single interface [25]. Subsequently, to assess the function of the overlap** DEGs at the molecular level, on the basis of the Database for Annotation, Visualization and Integrated Discovery (DAVID) [26], the genes obtained from the most significant module were selected for pathway enrichment analysis using the KEGG database, to classify the correlated gene sets into their respective pathways [27], and for functional enrichment analysis using the Gene Ontology database (GO, http://www.geneontology.org/), to collect functions of genes and gene products from the aspects of biological process (BP), molecular function (MF) and cellular component (CC) [28].
Pathway integration
To further interpret and visualize the molecular changes of ovarian cancer in hypoxia at the pathway level, a comprehensive network that integrated the HIF-1 signaling pathway and other signaling pathways was described by applying the combined and visualized functionality in Cytoscape based on the pathway analysis of the most significant module. Furthermore, to elucidate the regulation network of hypoxia in ovarian cancer in detail, the significant pathways inextricably linked with HIF-1 signaling pathway were screened with a carrying degree larger than 3 for the further analysis.
Gene integration
As above, a network of integrated genes from the critical pathways and the HIF-1 signaling pathway was constructed to obtain better insight into how biological processes were regulated in human ovarian cancer, which might contribute to identifying new markers and drug targets for the diagnosis and treatment of ovarian cancer. Subsequently, in order to verify the reliability of the results of the integrated network constructed by bioinformatics analysis, a signaling pathway (ErbB signaling pathway) was selected for validation through testing the mRNA expression levels of the relevant critical genes, such as transforming growth factor alpha (TGFA), erb-b2 receptor tyrosine kinase 2 (ErbB2), and v-myc avian myelocytomatosis viral oncogene homolog (MYC), through regulating the HIF-1α expression in order to establish the relationship between the HIF-1 and ErbB signaling pathways.
Cell culture and transfection
The human ovarian cancer cell line, Caov3, a kind gift from department of pathology, cancer hospital of Guizhou province, was cultured in a monolayer in Dulbecco’s modified Eagle’s medium (DMEM) (Gibco, Life Technologies, Carlsbad, CA, USA) supplemented with 10% fetal bovine serum (FBS) and 100 mg/mL penicillin/streptomycin (Life Technologies), and was maintained at 37 °C in an atmosphere of 5% CO2, as previously described [29]. Then, the cells were split into 12-well plates at a final concentration of 5 × 105 viable cells/ml culture medium. The medium was replaced with RPMI only after 12 h, then the overexpression vector PMX-Flag-hif-1α and interference vectors pSUPER-TGFA/EGFR were transfected using Lipofectamine 2000. 8 h later, cells were subsequently treated with the complete medium for 24 h, followed by extraction of total RNA.
Quantitative real-time PCR
Total RNA was extracted using a TRIzol reagent kit (TaKaRa) according to the manufacturer’s protocol as described previously [30]. Subsequently, complementary DNA was synthesized from the RNA by reverse transcription using the SuperScript III First-Standard Synthesis System for Reverse Transcription-Polymerase Chain Reaction (RT-PCR) (Invitrogen Co.). Following the protocol provided by the manufacturer, the expression of the selected genes was quantified by the PCR System 7500 (Promega) and SYBR green using the following procedures: initial denaturation and enzyme activation at 95 °C for 30 s, followed by 40 cycles of denaturation at 95 °C for 5 s and annealing at 60 °C for 30 s [1).

Schematic venn diagram of differentially expressed genes
PPI network construction and hub gene identification
On the basis of the information obtained from the STRING database, the PPI framework containing a total of 3279 protein pairs and 603 nodes were generated with the threshold of combined score > 0.4 (Fig. 2), in which nodes represented proteins and edges represented interactions between proteins [32]; this is helpful for understanding the regulation of hypoxia in ovarian cancer in the aspect of proteomics. Furthermore, proteins with very high degree may play a central regulatory role in this process, and are commonly called “hubs”. Therefore, the hub genes of the PPI network were screened with the cut off criterion of connectivity degree ≥35. The results of some nodes are shown in the Table 2, including jun proto-oncogene (JUN), FBJ osteosarcoma oncogene (FOS), estrogen receptor 1 (ESR1), matrix metallopeptidase 2 (MMP2), erb-b2 receptor tyrosine kinase 2 (ErbB2), and v-myc avian myelocytomatosis viral oncogene homolog (MYC).

The PPI network of overlap** DEGs under cycling hypoxia and chronic hypoxia in ovarian cancer. Nodes stand for the proteins (genes), and edges stand for the interactions of proteins
Module analysis of the PPI network
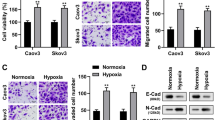
To study and identify the function of the overlap** DEGs in detail, cluster analysis of the PPI network was conducted based on the ClusterONE Cytoscape plugin, an important tool for the analysis of densely connected and possibly overlap** regions within the Cytoscape network, which would contribute to the classification of protein network and relevant analysis. There were a total of 51 functional modules given with the parameters of minimum size > 5 and minimum density < 0.05 and the most significant module (nodes = 223, density = 0.051, quality = 0.802, p value = 0.000, Fig. 3) was selected for the further analysis of functions and pathways to deeply understand the main effect of hypoxia in the course of ovarian cancer progression. Based on DAVID, GO functional annotation of genes obtained from the most significant module was performed (Fig. 4) and found that they mainly participated in cell proliferation, migration, and adhesion. These results suggest hypoxia may promote the proliferation of ovarian cancer cells by affecting cell invasion and adhesion. To further verify the accuracy of this inference, the module genes were submitted into DAVID to perform the KEGG pathway enrichment analysis. The results showed that they were significantly enriched in PI3K-Akt, MAPK, Wnt and ErbB signaling pathways, as well as ECM-receptor interactions, focal adhesion, pathways in cancer. Not surprisingly, the HIF-1 signaling pathway was also enriched (Additional file 1: Figure S1). It is well established that these signaling pathways were closely related to cell proliferation, differentiation, and apoptosis. The proliferation and clone-forming ability of periodontal ligament stem cells (PDLSCs) were markedly enhanced by hypoxia, which may be implicated in the activation of p38/MAPK and ERK/MAPK signaling pathways [33]. In addition, the PI3K-Akt and Wnt signaling pathways have been reported to play an important role in the process of mesenchymal stem cells (MSCs) and MC3T3-E1 cell proliferation induced by hypoxia [34,35,4: Figure S4).
The ErbB pathway involves a family of tyrosine kinases and contributes to resistance to radiation and chemotherapy in many tumor types, including ovarian cancer; its stimulation results in the proliferation of cells [43]. ErbB2, a member of the human epidermal growth factor receptor (HER/EGFR/ERBB) family, has been reported to be closely related to the occurrence and development of breast cancer [44, 45]. Moreover, a previous study indicated that the expression of ErbB2 was positively correlated with the malignant potential of serous ovarian neoplasms [46]. However, there are also studies showing that serum VEGF levels might be used for diagnosis in ovarian cancer patients, while serum ErbB2 levels do not have a clinical significance in terms of ovarian cancer [47]. Interestingly, it has been reported that the level of VEGF in serum may depend on the expression of ErbB2 in patients with ovarian cancer [48]. Therefore, the specific regulation of ErbB2 in ovarian cancer remains to be studied further. MYC, known as a regulatory gene, has been found to encode a multifunctional nuclear phosphoprotein which plays an important role in cell cycle progression, apoptosis, and cellular transformation [49]. The expression of MYC was increased by hypoxia in ovarian cancer cells, which may contributed to the observed resistance to platinum compounds. Moreover, ovarian cancer patients with high MYC mRNA levels tend to have lower disease-free (DFS) and the overall survival (OS) compared with their counterparts; thus, MYC may serve as a potential therapeutic target for ovarian cancers expressing high levels of this oncoprotein [50]. It has also been found that the disordered expression of MYC is common in human cancers and is closely related to the maintenance of aggressive of cancer stem cell populations. Additionally, HIF-1α expression has been significantly correlated with the expression of MYC and survivin in lung cancer [51]. EGFR, as an important receptor of TGFA, has attracted wide attentions from researchers. Previous studies have found that the frequency of oncogenic mutations in the EGFR gene is closely linked to the occurrence of non-small cell lung cancer (NSCLC). Moreover, EGFR has been proposed as a crucial molecular target for cancer therapy, promoting considerable research into the development of pharmacological inhibitors of EGFR [52,53,54]. Furthermore, Du et al. found that the expression of EGFR is closely correlated with progression-free survival (PFS) in post-operative patients with colorectal carcinoma, as patients with high EGFR expression were at a higher risk of tumor progression when comparing with their counterparts [55]. To our knowledge, however, there is no published study that has explored the effect of EGFR expression on ovarian cancer cell lines that have been exposed to hypoxia. Thus, EGFR may be a new therapeutic target for ovarian cancer, and may break through the bottleneck of current ovarian cancer treatment.
Admittedly, there were also some limitations to this study, since only the networks between genes and signaling pathways were explored in hypoxia while relevant transcription factor (TFs) and genes were not. Moreover, our results are mostly based on literature searches or bioinformatics predictions; thus, further validation is required and necessary. In addition, further in vivo studies are needed because of the growth mechanisms of cancer cells are so diverse that they can modify their migration mechanisms in response to different conditions in vitro.
Conclusions
In conclusion, hub genes (such as TGFA, EGFR, ErbB2, and MYC) and key pathways (such as the ErbB signaling pathway) closely related to the proliferation of ovarian cancer cells in hypoxia were identified by bioinformatics analysis. Functional enrichment analysis revealed that these genes were mainly involved in the processes of cells proliferation, invasion, and adhesion. Additionally, the regulation of ErbB signaling by hypoxia was demonstrated by RT-PCR, WB, and MTT assays. These findings indicated that hypoxia regulated the growth of ovarian cancer cells mainly through regulation of the HIF-1α-TGFA-EGFR-ErbB2-MYC axis, which might provide a new drug target and biomarker and lead to improved insights on diagnosis and treatment of ovarian cancer.
Abbreviations
- BCL6:
-
B-cell CLL/lymphoma 6
- BP:
-
Biological process
- CC:
-
Cellular component
- CCNB1:
-
Cyclin B1
- CCNB2:
-
Cyclin B2
- CCNE1:
-
Cyclin E1
- CDC20:
-
Cell division cycle 20
- CoCl2:
-
Cobalt chloride
- CYP3A5:
-
Cytochrome P450 family 3 subfamily A member 5
- DAVID:
-
Database for annotation, Visualization and integrated discovery
- DEGs:
-
Differentially expressed genes
- EOC:
-
Epithelial ovarian cancer
- FDG:
-
Fluorodeoxyglucose
- FOXO3:
-
Forkhead box O3
- GEO:
-
Gene expression omnibus
- GO:
-
Gene ontology
- hif-1:
-
Hypoxia inducible factor-1
- IL-8:
-
Interleukin-8
- KEGG:
-
Kyoto encyclopedia of genes and genomes
- MF:
-
Molecular function
- NCBI:
-
National center of biotechnology information
- PPI:
-
Protein-protein interaction
- RPS6KA6:
-
Ribosomal protein S6 kinase A6
- STRING:
-
The Search Tool for the retrieval of interacting genes database
- VEGF:
-
Vascular-endothelial growth factor
- VEGFA:
-
Vascular endothelial growth factor A
References
Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA Cancer J Clin. 2016;66:7.
Shiner A, Burbos N. Ovarian cysts and ovarian cancer. Innovait. 2012;2:24–36.
Ai Z, Lu Y, Qiu S, Fan Z. Overcoming cisplatin resistance of ovarian cancer cells by targeting HIF-1-regulated cancer metabolism. Cancer Lett. 2016;373:36–44.
Ebbesen P, Eckardt K-U, Ciampor F, Pettersen EO. Linking measured intercellular oxygen concentration to human cell functions. Acta Oncol. 2004;43:598–600.
Dang CV, Semenza GL. Oncogenic alterations of metabolism. Trends Biochem Sci. 1999;24:68.
Cianfrocca R, Tocci P, Rosanò L, Caprara V, Sestito R, Castro VD, et al. Nuclear β-arrestin1 is a critical cofactor of hypoxia-inducible factor-1α signaling in endothelin-1-induced ovarian tumor progression. Oncotarget. 2016;7:17790–804.
Cheng KW, Lahad JP, Kuo W, Lapuk A, Yamada K, Auersperg N, et al. The RAB25 small GTPase determines aggressiveness of ovarian and breast cancers. Nat Med. 2004;10:1251–6.
Natividad GR, Mohan SN, Fiona MG, Chalmers AJ, Jim C, Jane P. Hypoxia-inducible factor 1 alpha is required for the tumourigenic and aggressive phenotype associated with Rab25 expression in ovarian cancer. Oncotarget. 2016;7:22650–64.
Lee CT, Mace T, Repasky EA. Hypoxia-driven immunosuppression: a new reason to use thermal therapy in the treatment of cancer? Int J Hyperth. 2010;26:232–46.
Semenza GL. Hypoxia-inducible factors: mediators of cancer progression and targets for cancer therapy. Trends Pharmacol Sci. 2012;33:207.
Salem Chouaib YM. Hypoxia promotes tumor growth in linking angiogenesis to immune escape. Front Immunol. 2012;3:21.
Francesco EMD, Lappano R, Santolla MF, Marsico S, Caruso A, Maggiolini M. HIF-1α/GPER signaling mediates the expression of VEGF induced by hypoxia in breast cancer associated fibroblasts (CAFs). Breast Cancer Res. 2013;15:R64.
Weilbaecher KN, Guise TA, Mccauley LK. Cancer to bone: a fatal attraction. Nat Rev Cancer. 2011;11:411.
Kizaka-Kondoh S, Inoue M, Harada H, Hiraoka M. Tumor hypoxia: a target for selective cancer therapy. Cancer Sci. 2003;94:1021–8.
Fu LJ, Wang B. RETRACTED ARTICLE: investigation of the hub genes and related mechanism in ovarian cancer via bioinformatics analysis. J Ovarian Res. 2013;6:218.
Mori H, Yao Y, Learman BS, Kurozumi K, Ishida J, Ramakrishnan SK, et al. Induction of WNT11 by hypoxia and hypoxia-inducible factor-1α regulates cell proliferation, migration and invasion. Sci Rep. 2016;6:21520.
Xue J, Yang G, Ding H, Wang P, Wang C. Role of NSC319726 in ovarian cancer based on the bioinformatics analyses. Oncotargets Ther. 2015;8:3757.
Olbryt M, Habryka A, Student S, Jarząb M, Tyszkiewicz T, Lisowska KM. Global gene expression profiling in three tumor cell lines subjected to experimental cycling and chronic hypoxia. PLoS One. 2014;9:e105104.
Barrett T, Wilhite SE, Ledoux P, Evangelista C, Kim IF, Tomashevsky M, et al. NCBI GEO: archive for functional genomics data sets—update. Nucleic Acids Res. 2013;41:991–5.
Gautier L, Cope L, Bolstad BM, Irizarry RA. Affy--analysis of Affymetrix GeneChip data at the probe level. Bioinformatics. 2004;20:307–15.
Irizarry RA, Hobbs B, Collin F, Beazer-Barclay YD, Antonellis KJ, Scherf U, et al. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics. 2003;4:249–64.
Romano P, Giugno R, Pulvirenti A. Tools and collaborative environments for bioinformatics research. Brief Bioinform. 2011;12:549–61.
Franceschini A, Szklarczyk D, Frankild S, Kuhn M, Simonovic M, Roth A, et al. STRING v9.1: protein-protein interaction networks, with increased coverage and integration. Nucleic Acids Res. 2013;41:D808.
Carter GW, Thorsson V, Galitski T. Integrated network modeling of molecular and genetic interactions. Sourcebook of Models for Biomedical Research. Humana Press; 2008;67–74.
Maraziotis IA, Konstantina D, Anastasios B. An in silico method for detecting overlap** functional modules from composite biological networks. BMC Syst Biol. 2008;2:1–14.
Huang DW, Sherman BT, Tan Q, Collins JR, Alvord WG, Roayaei J, et al. The DAVID gene functional classification tool: a novel biological module-centric algorithm to functionally analyze large gene lists. Genome Biol. 2007;8:R183.
Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, et al. Gene ontology: tool for the unification of biology. Nat Genet. 2000;25:25–9.
Michael MD, Kilgore MW, Morohashi K, Simpson ER. Ad4BP/SF-1 regulates cyclic AMP-induced transcription from the proximal promoter (PII) of the human aromatase P450 (CYP19) gene in the ovary. J Biol Chem. 1995;270:13561.
Masson N, Willam C, Maxwell PH, Pugh CW, Ratcliffe PJ. Independent function of two destruction domains in hypoxia-inducible factor-α chains activated by prolyl hydroxylation. EMBO J. 2001;20:5197.
Hilscherova K, Jones PD, Gracia T, Newsted JL, Zhang X, Sanderson JT, et al. Assessment of the effects of chemicals on the expression of ten steroidogenic genes in the H295R cell line using real-time PCR. Toxicol Sci Off J Soc Toxicol. 2004;81:78.
**ang W, Zhang B, Lv F, Ma Y, Chen H, Chen L, et al. The inhibitory effects of RFamide-related peptide 3 on luteinizing hormone release involves an estradiol-dependent manner in Prepubertal but not in adult female Mice1. Biol Reprod. 2015;93:30.
** B, Wang W, Du G, Huang GZ, Han LT, Tang ZY, et al. Identifying hub genes and dysregulated pathways in hepatocellular carcinoma. Eur Rev Med Pharmacol Sci. 2015;19:592.
He Y, Jian CX, Zhang HY, Zhou Y, Wu X, Zhang G, et al. Hypoxia enhances periodontal ligament stem cell proliferation via the MAPK signaling pathway. Genet Mol Res. 2016;15(4). https://doi.org/10.4238/gmr15048965.
Zhang Y, Lv J, Guo H, Wei X, Li W, Xu Z. Hypoxia-induced proliferation in mesenchymal stem cells and angiotensin II-mediated PI3K/AKT pathway. Cell Biochem Funct. 2015;33:51–8.
Wang L, Wu B, Zhang Y, Tian Z. Hypoxia promotes the proliferation of MC3T3-E1 cells via the hypoxia-inducible factor-1α signaling pathway. Mol Med Rep. 2015;12:5267–73.
Chen-Tian L, Jian-**u L, Bo Y, Rui L, Chao D, Song-Jian L. Notch signaling represses hypoxia-inducible factor-1α-induced activation of Wnt/β-catenin signaling in osteoblasts under cobalt-mimicked hypoxia: Mol. Med Rep. 2016;14:689–96.
Brader S, Eccles SA. Phosphoinositide 3-kinase signalling pathways in tumor progression, invasion and angiogenesis. Tumori. 2004;90:2–8.
Lee SH, Lee YJ, Han HJ. Role of hypoxia-induced fibronectin-integrin β1 expression in embryonic stem cell proliferation and migration: involvement of PI3K/Akt and FAK. J Cell Physiol. 2011;226:484.
Yang SH, Hu MH, Lo WY, Sun YH, Wu CC, Yang KC. The influence of oxygen concentration on the extracellular matrix production of human nucleus pulposus cells during isolation-expansion process. J Biomed Mater Res A. 2017;105:1575.
Whelan KA, Schwab LP, Karakashev SV, Franchetti L, Johannes GJ, Seagroves TN, et al. The oncogene HER2/neu (ERBB2) requires the hypoxia-inducible factor HIF-1 for mammary tumor growth and Anoikis resistance. J Biol Chem. 2013;288:15865.
Clarke-Pearson DL. Screening for ovarian cancer. BJOG Int J Obstet Gynaecol. 2000;107:170–7.
Laughner E, Taghavi P, Chiles K, Mahon PC, Semenza GL. HER2 (neu) signaling increases the rate of hypoxia-inducible factor 1 (HIF-1 ) synthesis: novel mechanism for HIF-1-mediated vascular endothelial growth factor expression. Mol Cell Biol. 2001;21:3995–4004.
Karakashev SV, Reginato MJ. Hypoxia/HIF1α induces lapatinib resistance in ERBB2-positive breast cancer cells via regulation of DUSP2. Oncotarget. 2015;6:1967–80.
Marano L, Roviello F. The distinctive nature of HER2-positive gastric cancers. Eur J Surg Oncol. 2015;41:271–3.
Mitri Z. The HER2 receptor in breast cancer: pathophysiology, clinical use, and new advances in therapy. Chemother Res Pract. 2012;2012:743193.
Turan G, Usta CS, Usta A, Kanter M, Tavli L, Karacan M, et al. The expression of HER-2/neu (c-erbB2), survivin and cycline D1 in serous ovarian neoplasms: their correlation with clinicopathological variables. J Mol Histol. 2014;45:679–87.
Akcay T, Yasar O, Kuseyri MA, Dincer Y, Aydinli K, Benian A, et al. Significance of serum c-erbB-2 oncoprotein, insulin-like growth factor-1 and vascular endothelial growth factor levels in ovarian cancer. Bratisl Lekárske Listy. 2016;117:156.
Wang J, Li G, Wang Y, Tang S, **n S, Feng X, et al. Suppression of tumor angiogenesis by metformin treatment via a mechanism linked to targeting of HER2/HIF-1α/VEGF secretion axis. Oncotarget. 2015;6:44579–92.
Galardi S, Savino M, Scagnoli F, Pellegatta S, Pisati F, Zambelli F, et al. Resetting cancer stem cell regulatory nodes upon MYC inhibition. EMBO Rep. 2016;17:1872–89.
Reyesgonzález JM, Armaizpeña GN, Mangala LS, Valiyeva F, Ivan C, Pradeep S, et al. Targeting c-MYC in platinum-resistant ovarian cancer. Mol Cancer Ther. 2015;14:2260.
Chihiro T, Masashi K, Hironori I, Takumi A, Kenichi O. Expression of hypoxia-inducible factor-1α affects tumor proliferation and antiapoptosis in surgically resected lung cancer: Mol. Clin Oncol. 2016;5:295–300.
Goss GD, Spaans JN. Epidermal growth factor receptor inhibition in the Management of Squamous Cell Carcinoma of the lung. Oncologist. 2016;21:205–13.
Brait M, Rosenbaum E. Genetic and epigenetic analysis of erbB signaling pathway genes in lung cancer. J Thorac Oncol. 2010;5:1887–93.
Wu TH, Hsiue EH, Lee JH, Lin CC, Yang JC. New data on clinical decisions in NSCLC patients with uncommon EGFR mutations. Expert Rev Respir Med. 2017;11:51–5.
Du P, Xu B, Zhang D, Shao Y, Zheng X, Li X, et al. Hierarchical investigating the predictive value of p53, COX2, EGFR, nm23 in the post-operative patients with colorectal carcinoma. Oncotarget. 2017;8:954–66.
Acknowledgements
Not applicable.
Funding
This work was supported by the National Natural Science Foundation of China (Grant no. 31372287), Ministry of Agriculture Transgenic Major Projects of China (2014ZX0800952B), the Agricultural Science and Technology Innovation Program of China (ASTIP-IAS13), and National Biological Breeding Capacity Building and Industrialization Projects (2014-2573).
Availability of data and materials
The datasets used and/or analyzed during the current study available from the corresponding author on reasonable request.
Author information
Authors and Affiliations
Contributions
KZ, BYZ, XJK, GDF, WX, LC, FY and CYC analyzed and interpreted data, drafted or revised the manuscript, read and approved the final manuscript, and agreed to be accountable for all aspects of the work. MXC and PQW made substantial contributions to the conception and design.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interest.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional files
Additional file 1:
Figure S1. The KEGG pathway analysis of the most significant module. The number of DEGs in each signaling pathway was presented in different gradually changing color from red to blue. (PNG 563 kb)
Additional file 2:
Figure S2. Integration of all signaling pathways. Gene products were visualized as blue ellipses. The HIF-1 signaling pathway was marked by a purple ellipse, and signaling pathways marked as orange ellipses represented pathways closely related to the HIF-1 signaling pathway, and other signaling pathways were marked by red ellipses. (PNG 952 kb)
Additional file 3:
Figure S3. Integration of genes of signaling pathways. Genes in the network were indicated as blue ellipses and pathways as red ellipses. In addition, HIF-1 signaling pathway, ErbB2, TGFA, MYC were tagged with purple ellipses as they would be validated in the following tests. (PNG 900 kb)
Additional file 4:
Figure S4. ErbB signaling pathway. This map of ErbB signaling pathway was obtained based on KEGG. The red boxes represented overlap** DEGs between cycling hypoxia and chronic hypoxia which were verified by previous analysis. (PNG 799 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Cite this article
Zhang, K., Kong, X., Feng, G. et al. Investigation of hypoxia networks in ovarian cancer via bioinformatics analysis. J Ovarian Res 11, 16 (2018). https://doi.org/10.1186/s13048-018-0388-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13048-018-0388-x