
Abstract
Hypoxia contributes to the initiation and progression of glioblastoma by regulating a cohort of genes called hypoxia-regulated genes (HRGs) which form a complex molecular interacting network (HRG-MINW). Transcription factors (TFs) often play central roles for MINW. The key TFs for hypoxia induced reactions were explored using proteomic analysis to identify a set of hypoxia-regulated proteins (HRPs) in GBM cells. Next, systematic TF analysis identified CEBPD as a top TF that regulates the greatest number of HRPs and HRGs. Clinical sample and public database analysis revealed that CEBPD is significantly up-regulated in GBM, high levels of CEBPD predict poor prognosis. In addition, CEBPD is highly expressed in hypoxic condition both in GBM tissue and cell lines. For molecular mechanisms, HIF1α and HIF2α can activate the CEBPD promotor. In vitro and in vivo experiments demonstrated that CEBPD knockdown impaired the invasion and growth capacity of GBM cells, especially in hypoxia condition. Next, proteomic analysis identified that CEBPD target proteins are mainly involved in the EGFR/PI3K pathway and extracellular matrix (ECM) functions. WB assays revealed that CEBPD significantly positively regulated EGFR/PI3K pathway. Chromatin immunoprecipitation (ChIP) qPCR/Seq analysis and Luciferase reporter assay demonstrated that CEBPD binds and activates the promotor of a key ECM protein FN1 (fibronectin). In addition, the interactions of FN1 and its integrin receptors are necessary for CEBPD-induced EGFR/PI3K activation by promoting EGFR phosphorylation. Furthermore, GBM sample analysis in the database corroborated that CEBPD is positively correlated with the pathway activities of EGFR/PI3K and HIF1α, especially in highly hypoxic samples. At last, HRPs are also enriched in ECM proteins, indicating that ECM activities are important components of hypoxia induced responses in GBM. In conclusion, CEPBD plays important regulatory roles in the GBM HRG-MINW as a key TF, which activates the EGFR/PI3K pathway through ECM, especially FN1, mediated EGFR phosphorylation.
Similar content being viewed by others

Introduction
As one of the most malignant tumors in human, effective therapeutic approach is very limited for glioblastoma (GBM) [1], mainly because of the incomplete understanding of the mechanisms of GBM development. Hypoxia played important roles in the progression of GBM in several aspects, including invasion, angiogenesis, therapeutic resistance, and stem cell maintenance [2, 3]. Hypoxia is related to the progression, as well as the initiation of GBM. Therefore, great efforts have been made to understand the mechanisms in which the hypoxia promotes the progression of GBM, and develop novel therapeutic targets against hypoxia-induced molecular responses. Hypoxia inducible factors 1A (HIF1A or HIF1α), and 2A (EPAS1, or HIF2α) are the key factors that induce gene expression levels under hypoxic condition and are important therapeutic targets [4,5,6]. However, HIF-targeted therapies have not been successful in patients [22], a GBM dataset from Lee et al. (GSE13041) [23], and a GBM data from Gravendeel et al. (GSE16011) [24], which contained 197, 260, 228, 191, and 159 samples, respectively). Then target genes that were identified in at least one dataset were used as the targets for each TF.
Glioma samples
Glioma or normal brain samples used for real-time quantitative (q)PCR and Western blot were provided by the Department of Neurosurgery, **, in which 35.29% and 64.71% CEBPD targets in HRPs are among that in CEBPD targets in HRGs and PANSGs, respectively.
Interestingly, analysis of all the three kinds of HRTs lead to the same conclusion that CEBPD is the top TF. Then, we examined whether this would be a biased results introduced by the algorithm. Accordingly, top TF analysis was performed on random target gene sets containing the same number of corresponding HRTs (Fig. 1D). Based on the examination of the rank position distribution of each TF in repeated random analysis, the probabilities of CEBPD with a rank position smaller than 5 is very low (P < 0.05; Fig. 1E) in all of the three HRTs, while those of CEBPB are higher than that of CEBPD, because CEBPB has more target genes in the entire genome (Fig. 1E). Therefore, it is an intrinsic nature for CEBPD as a top TF for HRTs.
The above conclusion was further validated by establishing more accurate TF targets by combining the ARACNe inferred TF targets and the TF target database (ARACNe_hTFtarget_combined, Supplementary Table S3). In the TF target database, the targets of each TF were identified by integrating huge human TF target resources and epigenetic modification information [22], The Cancer Genome Atlas (TCGA) data combing GBM and LGG expression [39], and the Chinese Glioma Genome Atlas (CGGA) databases [40] supports that CEBPD is more highly expressed in GBM than in normal and LGGs (Fig. 2B).

A The mRNA levels of CEBPD, as revealed by qPCR, in a cohort of glioma samples with different grades (**p < 0.001 compared to Normal brain samples; ##p < 0.001 compared to grade 2 astrocytoma (A); !p < 0.05 compared to grade 3 anaplastic astrocytoma (AA). B Expression of CEBPD mRNA levels in different grades of gliomas in TCGA, Rembrandt, and CGGA datasets. (**p < 0.001 compared to Grade 4 GBM; ##p < 0.001 compared to Grade 3 AA; !p < 0.05, !!p < 0.001, compared to Normal brain samples.) C Representative WB results showing protein levels of CEBPD and HIF1α in different grade gliomas. D, E Representative IHC staining and quantitative staining scores of CEBPD in glioma samples (*p < 0.05, **p < 0.001, compared to grade 2 astrocytoma). F Survival analysis and progression-free analysis of GBM patients in different databases (TCGA, CGGA, and Rembrandt data). G WB results showing CEBPD protein levels in core hypoxic and peripheral less hypoxic areas in GBM samples (n = 3 independent experiments). H IHC staining showing CEBPD stain in hypoxic pseudo-palisading areas in GBM tissues; *, T, indicated pseudo-palisading areas and tumor area, respectively. I CEBPD mRNA levels in different areas in GBM tissues, including leading edge (LE), infiltrating tumor (IT), cellular tumor (CT), pseudopalisading cells around necrosis (PAN), and microvascular proliferation (MVP) areas, in the glioblastoma atlas data [26]; **p < 0.001, compared to PAN. J WB results showing CEBPD protein levels in GBM U87 and U251 cells under hypoxia condition. K Luciferase reporter assay showing bind and activation of HIF1α and HIF2α to the CEBPD promotor region (***p < 0.0001 compared to CEBPD-promotor-HIF-NC group). (pGL3: pGL3-Basic vector containing blank control; CEBPD promotor: pGL3-Basic vector containing CEBPD promoter sequence; pRL: pRL-TK vector containing blank control; HIF1A or EPAS1: pRL-TK vector containing HIF1A or EPAS1 TF sequence). L The DNA fragment with the highly reliable HIF1A or EPAS1 binding sites (Hypoxia-responsive Element, HRE) in the CEBPD promoter, as predicted by the JASPAR database [43]. There are 4 highly reliable HREs (bold letters) in the fragment. For each HRE, the sequence, location, and the binding scores (expressed as score/relative score) were labeled; red letters represent positive strand, while yellow shaded letters represent negative strand. Greed shaded letters represent sequences for primers. M Chip-qPCR showing that HIF1A bind to the CEBPD promoter region shown in (L). A grade 2 astrocytoma; O grade 2 oligodendroglioma; AA grade 3 astrocytoma; GBM grade 4 glioblastoma; peri peripheral.
The protein levels of CEBPD in glioma tissues were further investigated. Western blot (WB) analysis in glioma tissues, including 2 grade II A, 2 grade II oligodendroglioma (O), 2 grade III AA, and 2 GBM samples, revealed a trend that CEBPD increased along with tumor grade (Fig. 2C).
The increased expression of CEBPD protein levels in GBMs was confirmed by performing immunohistochemistry (IHC) in a tissue chip, including 5 normal brain samples, 30 grade II A, 20 grade III AA, and 50 GBM samples, and the IHC intensity was semi-quantified (Fig. 2D). No cell staining of CEBPD was observed in normal brain tissue besides background staining (Fig. 2D). Semi-quantitative analysis revealed that CEBPD staining scores increased along with tumor grade and is significantly higher in GBM than in normal brain and LGGs (Fig. 2E).
The clinical significance of high levels of CEBPD in GBM was investigated demonstrating that the high expression of CEBPD predicted the poor prognosis of patients with GBM and LGGs (Fig. 2F; Supplementary Figure S2) in different datasets, and is an independent risk factor for patients with GBM (Supplementary Table S4).
CEBPD is highly expressed in hypoxic condition in GBM samples and cells
As a master TF for hypoxia-induced targets, the influence of hypoxia on CEBPD expression was investigated. First, a significant positive correlation was observed between CEBPD and HIFs (HIF1α and HIF2α) in GBMs (Fig. 1G, H; Supplementary Figure S3). Next, the expression of CEBPD was compared in 6 GBM tissues derived from core necrosis area (CA) or peripheral less-hypoxic area (PA). CEBPD is more highly expressed in CA than in PA in both mRNA and protein levels (Fig. 2G, Supplementary Figure S4). Moreover, the HIF1α protein level was more highly expressed in GBM than in LGGs (Fig. 2C), and could be more highly expressed in CA than in PA (Fig. 2G, Supplementary Figure S4). Furthermore, pseudopalisades areas are more hypoxic in GBM [41, 42], and the IHC staining revealed that CEBPD is more strongly expressed in cells in the pseudopalisades area than in other areas (Fig. 2H). Based on the investigation of GAP data, CEBPD is highly expressed in the hypoxic PAN area than in the other areas (Fig. 2I). Therefore, CEBPD is more highly expressed in hypoxic condition in GBM tissues.
Next, the influence of hypoxia on CEBPD expression was investigated in GBM cells. Accordingly, U87 and U251 glioma cell lines were cultured under different oxygen levels (20%, 5%, 1%, or 0.3% O2 levels) for 24 h. Then, q-PCR and WB assays revealed that CEBPD was up-regulated with the decrease in oxygen levels in both cell lines, in protein and mRNA levels (Fig. 2J, Supplementary Figure S5).
The ability of HIF1α and HIF2α to bind to and activate the promotor of CEBPD was determined. Luciferase reporter assay, including a 2000 bp up-stream promoter region of CEBPD, revealed that both HIF1α and HIF2α significantly activated the promoter region of CEBPD compared with the control (Fig. 2K). In addition, chromatin immunoprecipitation (Chip)-qPCR confirmed that HIF1α could bind to the CEBPD promotor region with a high score predicted with JASPAR database [43] (Fig. 2L, M). Therefore, CEBPD can be transcriptionally regulated by HIF1α or HIF2α directly.
CEBPD is important for the tumorigenic potential of GBM cells in vitro, and contributes to hypoxia enhanced invasion capacity
Then, the use of CEBPD for the tumorigenic potential of GBM cells in vitro was determined. U87 and U251 cells were knocked-down with CEBPD shRNA lentivirus (Fig. 3A). Two shRNA sequences, namely, shCEBPD-KD1 and shCEBPD-KD2, with significant CEBPD knockdown efficiency were developed. The knockdown efficiencies of the lentiviruses were validated in both mRNA (Supplementary Figure S6) and protein levels in both U87 and U251 cells (Fig. 3A). Considering that shCEBPD-KD1 shRNA did not exhibit validated knockdown effect in U251 in normoxic condition (Fig. 3A), it was not used for further experiments in U251 cells.

A WB assays showing proteins levels of CEBPD after shCEBPD lentiviruses infection in U87 (up panel) and U251 (bottom panel) cells, in normoxia (20% O2) or hypoxia (1% O2) conditions. B In vitro CCK8 assays showing the effect of CEBPD knockdown on growth curves for U87 (up panel) and U251 cells (bottom panel). C In vitro colony formation assays of U87 (up panel) and U251 (bottom panel) cells with shCtrl or shCEBPD infection. D, E Transwell invasion assays of U87 (D) and U251 (E) cells infected with shCtrl or shCEBPD lentiviruses in normoxia (20% O2) or hypoxia (1% O2) conditions. **p < 0.001, *p < 0.05, compared with corresponding normoxia (20% O2) group; #p < 0.05 compared with shCtrl group in normoxia (20% O2); !p < 0.05 compared with shCtrl group in hypoxia (1% O2). Right images in (D, E): Fold increase of number of hypoxia induced invasion cells after shCtrl or shCEBPD treatment in U87 (D, right image) and U251 (E, right imag) cells. Repeated data for each group were obtained by calculating the ratio of each hypoxia data to the averaged value of normoxia data. *p < 0.05, compared with shCtrl group. Experiments were repeated in triplicate.
Then, growth curve experiments revealed that compared with the scramble-shRNA control group (shCtrl), the growth of GBM cells was significantly inhibited in shCEBPD-KD groups in both cell lines (Fig. 3B). In addition, shCEBPD-KD inhibited the colony formation of the two GBM cell lines (Fig. 3C).
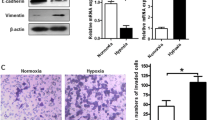
Next, trans-well invasion assays were performed to investigate the influence of CEBPD knockdown on the invasion capacity of GBM cells, especially in hypoxia. First, shCEBPD-KD significantly decreased the invasion capacity of both U87 and U251 cells (Fig. 3D, E) either under normoxic or hypoxic condition. Notably, hypoxia induced an increase in the number of cells invading across the transwell (Fig. 3D, E), which supports previous findings [4]. Further analysis revealed that shCEBPD-KD attenuated hypoxia enhanced invasion, because the fold change of hypoxia-induced invading cell numbers in the shCtrl group are significantly greater than that in shCEBPD-KD groups in both cell lines (Fig. 3D, E). Therefore, CEBPD contributes to hypoxia-promoted invasion of GBM cells.
CEBPD promotes the tumorigenic potential and invasion capacity of GBM cells in vivo
The use of CEBPD in the tumorigenesis of GBM cells in vivo was explored, by establishing in vivo flank xenografts in nude mice by implanting U87 cells transfected with shCEBPD-KD1 or shCtrl lentivirus. The in vivo tumor volume growth curves during the 8 weeks of investigation revealed that the mice in the shCtrl group developed significant larger xenografts in later stages compared with the shCEBPD-KD1 group (Fig. 4A). Consistently, at the last investigation time point, the tumor weights in the shCtrl group were significantly greater than those in the shCEBPD-KD1 group (Fig. 4B).

A, B Flank xenograft experiments showing the in vivo growth curves of U87 cells in shCtrl (n = 5) and shCEBPD groups (n = 5). C Intracranial xenografts tumor showing the tumor volumes in shCtrl (n = 5) and shCEBPD KD (n = 5) groups. The bottom binary images showed the tumor areas. D The invasion capacity of U87 cells in vivo in shCtrl and shCEBPD KD groups was evaluated by examining the number of tumor fingers in per field. The bottom binary images showed the tumor areas, where islands and protruded areas were indicated by arrows. *p < 0.05, compared with shCtrl group.
Next, U87 cells infected with shCtrl or shCEBPD-KD1 lentivirus were injected into the brains of nude mice to establish brain xenografts. At the end point, the shCtrl groups developed larger xenografts than the shCEBPD-KD1 groups (Fig. 4C). The invasive capacity of the xenografts was quantitively examined by estimating the relative invasive fingers microscopically by counting the protruded tumor tissue fingers and disseminated areas [44]. As a result, mice brain tumors in the shCEBPD-KD1 group exhibited significantly less invasive fingers compared with the shCtrl group (Fig. 4D). Together with the in vitro study, these results imply that CEBPD expression is important for the invasive capacity of GBM cells.
CEBPD positively regulated a cohort of proteins involved in the EGFR/PI3K/AKT pathway and ECM function under hypoxic conditions
The underlining molecular mechanisms of the CEBPD function were studied. Protein mass spectrometry analysis identified 121 proteins that were significantly down-regulated by at least 1.2-folds after shCEBPD-KD1 knockdown in U87 cells (Fig. 5A). Then, DAVID analysis was performed to decipher the biological functions of these proteins [45]. As a result, the top Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways that were inhibited after CEBPD knockdown included ECM-receptor interaction, focal adhesion, and PI3K-AKT signaling pathways, containing 11, 11, and 13 proteins, respectively (Fig. 5A, B). Notably, almost all the proteins in the ECM-receptor interaction and focal adhesion pathway also belong to the PI3K-AKT signaling pathway (Fig. 5C).

A TMT quantitative proteomic data, showing heatmap of protein levels in shCEBPD KD and shCtrl U87 cells, in both normoxia (20% O2) and hypoxia (1% O2) conditions. Proteins that are significantly down-regulated at least 1.2-folds after shCEBPD KD were chosen as the CEBPD-regulated proteins. The proteins were sorted in descending order from top to bottom according to their folds changes. B KEGG pathway analysis and Gene Ontology (GO) analysis of CEBPD positively regulated proteins in both normoxia and hypoxia conditions in U87 cells. Analyzed GO Categories include Biological process (BP), Cellular component (CM), and Molecular function (MF). The colored bars represent the −log10(p value) (left panel) and count of protein (right panel) for each enriched items for CEBPD-regulated protein in normoxia. The black bars, which are covered by the colored bars, represent the corresponding values for CEBPD-regulated proteins in hypoxia. The black parts that are not covered by the colored ones indicated that, for the specific item, the value of the item has a greater value in hypoxia condition than in normoxia condition. The black and red integers in the right panel indicate counts of proteins regulated by CEBPD in normoxia and hypoxia conditions, respectively. C Detailed proteins that are belong to the key KEGG pathways affected by CEBPD knockdown in both normoxia and hypoxia conditions. Proteins labeled with red are those which are also belong to the PI3K/Akt signal pathway. D Quantitative analysis of hypoxia impact on the protein levels that are belong to the key KEGG pathways, in shCtrl (up panel) or shCEBPD KD (bottom panel) conditions. *p < 0.05, compared with normoxia (20% O2) group. E Fold changes of hypoxia induced protein levels of the CEBPD-regulated genes in shCtrl and shCEBPD KD groups. *p < 0.05 compared with the shCtrl group. Repeated data for each group were obtained by calculating the ratio of each hypoxia data to the averaged value of normoxia data.
CEBPD-regulated proteins in hypoxia were also analyzed, and 132 proteins were down-regulated by at least 1.2-folds after CEBPD knockdown (Fig. 5A). In total, 99 proteins overlapped in the CEBPD-regulated proteins under normoxic and hypoxic conditions. The top KEGG pathways involving these proteins include ECM-receptor interaction, PI3K-Akt, and focal adhesion pathways (Fig. 5B, C). Interestingly, the value of (−log p value) and the number of CEBPD-regulated proteins belong to the PI3K-Akt pathway in hypoxia, are higher than that in normoxic condition (Fig. 5B), indicating the important role of CEBPD for the PI3K-AKT pathway in hypoxia.
In detail, a considerable amount of the CEBPD target proteins were belong to the EGFR/PI3K pathway (Fig. 5C). Interestingly, most of these proteins are ECM molecules and contribute to the invasiveness of glioma cells, including FN1 [46], TNC [47], LAMC1 [48], COL1A1 [19, 48], EGFR [49], CDK6 [50], and COL6A2 [51]. Gene Ontology (GO) analysis also revealed that, the target proteins regulated by CEBPD are significantly enriched in genes for ECM constituent and functions either in normoxic or hypoxic condition (Fig. 5B). Several CEBPD target proteins, whose roles in GBM or invasive capacity have not been clearly studied, were identified, and these proteins include HSP90B1, GNG10, and GNG5.
The effect of hypoxia on the CEBPD target proteins involved in the EGFR/PI3K pathways was analyzed. Strikingly, most of these proteins are significantly up-regulated in hypoxia (15 out of 17 up-regulated, with 12 of them exhibited p < 0.05; Fig. 5D), either in shCtrl or shCEBPD condition. Interestingly, most of the 15 up-regulated proteins in hypoxia exhibited a reduced fold increase in hypoxia after CEBPD knockdown (9 out of 15, with 3 of them exhibited p < 0.05; Fig. 5E), including FN1 and TNC (p = 0.033, 0.038, respectively, Fig. 5E). In addition, 8 and 10 HRPs (22.22% and 27.78% HRPs) were CEBPD-responsive proteins under normoxic and hypoxic conditions, respectively. Importantly, HIF1A is also a CEBPD-responsive protein based on WB analysis (Fig. 6A). Collectively, these results support that CEBPD played important roles for hypoxia-induced responses, especially for the EGFR/PI3K pathway, ECM function, and invasion capacity.

A WB results showing the protein levels of CEBPD and key transducers of the EGFR/PI3K pathway, especially phosphorylated EGFR, ERK1/2, AKT, mTOR, STAT3, and essential genes for the invasion capacity, after CEBPD knockdown in normoxia and hypoxia conditions. B, C Correlation coefficients and corresponding p values (labels above each bar) between HIF1α (B) or EPAS1 (C) and genes in EGFR/PI3K/Akt signal pathways, which are regulated by CEBPD and contribute to tumor invasion capacity. Pearson correlation was performed between HIF1α or EPAS1and indicated genes labeled in the X axis, in three GBM databases (TCGA, Rembrandt, and CGGA databases). D Chip-seq analysis of CEBPD binding sites in CEBPD-regulated proteins belong to the EGFR/PI3K pathway. Bars represented number of CEBPD binding sites identified by Chip-seq in the promoters of the indicated proteins. E The DNA fragment of CEBPD binding peaks in the FN1 promoter identified by Chip-seq, which also contained highly reliable CEBPD binding sites as predicted by the JASPAR database [43]. There are 4 highly reliable CEBPD binding consensus sequences (red letters, site 1–4) in the fragment, and the site (1) and (2) had the greatest scores. For each binding site, the sequence, location, and the binding scores (expressed as score/relative score) were labeled; red letters represent positive strand, and yellow shaded letters represent negative strand. Greed shaded letters represent sequences for primers. F mRNA levels of FN1 after shCEBPD treatment in U87 cells in normoxia condition. G Chip-qPCR showing that CEBPD bind to the FN1 promoter region shown in E in normoxic condition. H Luciferase reporter assay showing bind and activation of CEBPD on the promotor of FN1 in normoxia condition. The promoter region of FN1 is shown in (E). For mutation analysis, the putative binding sites (site 1 and 2) with the greatest scores were mutated, where the sequence of “TGTTGCTAAATGA” (as shown in E) were replaced by “CTGCCGCGGCGGG”. (pGL3: pGL3-Basic vector containing blank control; FN1 prom: pGL3-Basic vector containing FN1 promoter sequence; FN1 promMut: pGL3-Basic vector containing FN1 promoter with mutation; pRL: pRL-TK vector containing blank control; CEBPD: pRL-TK vector containing CEBPD sequence; CEBPD_KD: pRL-TK vector containing CEBPD shRNA sequence.) *p < 0.05, ***p < 0.0001 compared to the FN1 promoter + pRL group; ###p < 0.0001 compared to the FN1 promoter + CEBPD group. All of the last 6 groups had p < 0.0001 compared to the first 2 groups. I WB assays of rescue experiments after CEBPD KD in U87 cells. Protein levels of key transducers of the EGFR/PI3K pathway, including p-EGFR, were examined in U87 cells, U87 cells treated with shCtrl (U87 shCtrl) and shCEBPD lentivirus (U87 shKD1, U87 shKD2), or U87-shCEBPD KD cells which are recused with the α5β1 integrin agonist Pyrintegrin (U87 shKD1+Pyr, U87 shKD2+Pyr) or a recombinated FN1 protein (U87 shKD1 + rFN1, U87 shKD2 + rFN1). The α5β1 integrin is the receptor for FN1. J Transwell invasion assays of rescue experiments after CEBPD KD for U87 cells. The cell groups were the same as described in (I), and experiments were repeated in triplicate.
CEBPD activates the EGFR/PI3K/AKT pathway through FN1-mediated phosphorylation of EGFR
EGFR/PI3K/AKT pathway is among the core pathways altered in GBM [52] and is important for the invasive capacity of GBM [53]. In addition, EGFR, an important therapeutic target for GBM [16, 17, 49], is among the altered proteins that belong to the PI3K/AKT pathway (Fig. 5D). Therefore, the regulation of EGFR/PI3K activity by CEBPD was further studied.
First, WB assays revealed that CEBPD knockdown reduced the protein levels of phosphorylated/activated form of EGFR, AKT, mTOR, and STAT3, namely p-EGFR, p-AKT, p-mTOR, and p-STAT3 (Fig. 6A, Supplementary Figure S7), either in normoxic or hypoxic condition. Notably, the PI3K/AKT pathway is more activated under hypoxic condition considering that the levels of p-EGFR, p-AKT, p-mTOR, and p-STAT3 were all up-regulated in hypoxia either under shCtrl or shCEBPD-KD condition. Correlation analysis demonstrated that HIF1α and HIF2α were positively correlated with the above genes involved in the EGFR/PI3K/AKT pathway (Fig. 6B, C), confirming that the EGFR/PI3K/AKT pathway is more activated under hypoxic condition. These results strengthened our above conclusion that CEBPD is responsible for the activation of the PI3K/AKT pathway, especially under hypoxic condition.
Second, the genes that contribute to the invasion capacity of GBM cells in the EGFR/PI3K/AKT pathway, including TNC, FN1, and COL1A1, were also down-regulated after CEBPD knockdown in both normoxic and hypoxic conditions (Fig. 6A, Supplementary Figure S7). These genes were also up-regulated under hypoxic condition (Fig. 6A).
The mechanisms of CEBPD-augmented EGFR/PI3K activity were then examined. Firstly, EGFR is a CEBPD target protein in the proteomic results (Fig. 5A), and CEBPD can bind to and activate the EGFR promoter based on Chip-qPCR and luciferase experiments (Supplementary Figure S8). However, WB assays showed that the total EGFR protein level was only marginally reduced after CEBPD knockdown, while the p-EGFR level was significantly reduced (Fig. 6A, Supplementary Figure S7). Therefore, CEBPD promoted the EGFR/PI3K pathway mainly by regulating the phosphorylation but not the transcriptional expression of EGFR. Accordingly, we further deciphered other mechanisms underlining the EGFR/PI3K pathway activation promoted by CEBPD. Considering that ECM-receptor pathway is one of the most apparent KEGG pathways influenced by CEBPD, the associations between them were examined, because interactions between ECMs and their integrin receptors are important for the activation of EGFR/PI3K pathway [54,55,56].
Chip-seq analysis revealed that the peek binding sites of CEBPD were identified in the promotor region of most of the above proteins, in which the FN1 promotor contained the highest number of CEBPD binding sites (Fig. 6D). Considering that FN1 is among the top proteins that are regulated by CEBPD (Fig. 6A), we then further analyzed whether CEBPD transcriptionally regulated FN1 expression. By comparing the results of predicted TF binding sites from the JASPAR database [43], a peak fragment was identified from the Chip-seq data in the FN1 promotor region, which contained putative CEBPD binding sites with high scores (Fig. 6E). Notably, the mRNA level of FN1 was significantly reduced after CEBPD KD (Fig. 6F), and Chip-qPCR confirmed the binding of CEBPD to this FN1 promoter region (Fig. 6G). Luciferase reporter assay results demonstrated that CEBPD can bind and activate this sequence (Fig. 6H). In addition, the mutation of the putative binding sites 1 and 2 with the greatest scores confirmed the specificity of binding and activation (Fig. 6H).
Interestingly, α5β1 integrin (ITGA5 and ITGB1), the receptor of FN1 (fibronectin) and is a key component in ECM, can activate EGFR phosphorylation and downstream PI3K signals [54]. FN1 and its receptor α5β1 integrin also contribute to the tumorigenesis of glioma [20]. Considering that FN1 is a direct target of CEBPD (Fig. 5A, C; Fig. 6A, D–H), we next investigated whether CEBPD promotes EGFR phosphorylation through the FN1-activated α5β1 integrin. To this, we used α5β1 integrin agonist Pyrintegrin or a recombinant FN1 protein (rFN1) to rescue the α5β1 integrin activity in U87 cells after CEBPD knockdown. As a result, CEBPD knockdown reduced the protein level of p-EGFR but not that of total EGFR, while both Pyrintegrin and rFN1 can rescue the p-EGFR level and PI3K/Akt pathway activity, as indicated by the elevated levels of p-EGFR, p-Akt and p-Erk, which were reduced by CEBPD knockdown (Fig. 6I, Supplementary Figure S9). Transwell invasion assay further demonstrated that both Pyrintegrin and rFN1 can rescue the invasion capacity of U87 cells inhibited by CEBPD knockdown (Fig. 6J).
Overall, CEBPD activated the EGFR/PI3K/AKT pathway and invasion capacity through the ECM (especially FN1)-mediated phosphorylation of EGFR.
CEBPD remarkably affected the EGFR/PI3K pathway activity under hypoxic condition in GBM patient samples
Whether the regulatory effects of CEBPD on EGFR/PI3K pathway would be hold in GBM patients need to be considered. Correlation analysis revealed that CEBPD is positively correlated with the expression level of CEBPD target genes that are involved in the EGFR/PI3K/AKT pathway (Fig. 7A), even in each GBM subtype (Proneural, Neural, Classical, and Mesenchymal; Fig. 7A, right panel). In addition, combined with pathway activity analysis [38], we found that both the expression level and pathway activity of CEBPD are positively correlated with the pathway activities of EGFR/PI3K/AKT and HIF1A (Fig. 7B, C). CEBPD also exhibited strong correlation with the pathway activity of itself (Supplementary Fig. S10), thus supporting the reliability of the pathway activity analysis.

A Correlations between expression levels of CEBPD and genes in EGFR/PI3K pathways (left image) in different databases (TCGA, Rembrandt, and CGGA). *P < 0.05, **(vertical) P < 10−5, ***(vertical) P < 10−10. In addition, correlations of CEBPD and these genes in each GBM subtype (Proneural, Neural, Classical, and Mesenchymal) were further performed in the TCGA database to test the impact of subtype on the correlations. B Correlations between CEBPD expression level and pathway activities of EGFR/PI3K and HIF1A. C Correlations between pathway activities of CEBPD and EGFR/PI3K related pathways, HIF1A pathways. D Expression levels of HIFs (HIF1A and EPAS1), CEBPD and genes in EGFR/PI3K pathways in high hypoxic samples (HHSs) and low hypoxic samples (LHSs) in the TCGA database. E Coefficients between CEBPD level and EGFR, HIF1A pathway activities in HHSs and LHSs. Digital labels for each bar indicate the coefficients (R, left label) and p values (right label). F KEGG pathway analysis and Gene Ontology (GO) analysis of HRPs in U87 cells. Analyzed GO Categories include Biological process (BP), Cellular component (CM), and Molecular function (MF). The colored bars represent the −log10(p Value) (left panel) and count of protein (right panel) for each enriched items. G Top TF analysis of ECM proteins belong to the HRPs (HRP-ECM), as described in Fig. 1. H Chip-seq analysis of CEBPD binding sites in HRP-ECM proteins. Bars represented number of CEBPD binding sites identified by Chip-seq in the promoters of the indicated proteins.
The effect of hypoxia on the functions of CEBPD in patients was studied by develo** a hypoxia-score for each GBM samples in TCGA datasets. The hypoxia-score for all the TCGA GBM samples were defined by their combined rank index of each gene in the HRGs. Therefore, samples with high hypoxia-scores are more hypoxic than those with lower scores. Then, the GBM samples were split into high-hypoxia samples (HHSs, top 45% samples with hypoxia-score) and low-hypoxia samples (LHS, bottom 45% samples with hypoxia-score). The gene expression levels and pathway activities were analyzed in both HHSs and LHSs.
HIF1A, EPAS1, CEBPD and EGFR were more highly expressed in HHSs than in LHSs (Fig. 7D), indicating the high and low hypoxic features of HHSs and LHSs, respectively. In addition, most of the above genes involved in the EGFR/PI3K/Akt pathway were more highly expressed in HHSs than in LHSs (Fig. 7D), thus confirming that this pathway is more activated under hypoxic condition.
Next, the coefficients between CEBPD and EGFR pathway activities in HHSs and LHSs were calculated. Interestingly, CEBPD exhibited stronger positive correlations with pathway activities of EGFR, ERK1-2, MAPK1, and HIF1A in HHSs than in LHSs (Fig. 7E), indicating that CEBPD remarkably affected these pathways under hypoxic condition.
ECM-related activities are important components of hypoxia induced responses
Considering that ECM activities are important mediators for CEBPD-induced EGFR/PI3K signal activation, its relationships with hypoxia-induced responses were studied. Interestingly, KEGG and GO analysis revealed that HRPs were enriched in proteins that are involved in ECM-receptor interaction and ECM components (HRP-ECM proteins; Fig. 7F), including essential ECM proteins FN1, collagen family (COL1A2, COL2A1, and COL5A1) and their receptor ITGA5. Interestingly, CEBPD is the top TF that regulates greatest number of HRP-ECM proteins as revealed by the top TF analysis (Fig. 7G). In addition, the promotors of most HRP-ECM proteins contained multiple CEBPD binding peaks based on Chip-seq analysis (Fig. 7H). Therefore, ECM-related activities are important components of hypoxia-induced responses, and CEBPD is a key TF that regulates these activities.
Discussion
Hypoxia is important therapeutic target for cancer, including GBM [8, 9, 57]. However, considering that hypoxia induces many molecular responses, which form a complex MINW [2], limited is known about the function and structure organization of the HRGs-MINW despite of the availability of a high volume of data about the roles of many single genes, which are actually hub or key genes for the HRGs-MINW. Accordingly, the present study explored and uncovered certain key features of the HRGs-MINW in GBM. In summary, by starting with proteomic technologies, we uncovered that ECM and its activated EGFR/PI3K pathway are important responses induced by hypoxia, and CEBPD is a master TF that regulates these processes. FN1, a key ECM protein, is a direct target of CEBPD mediating these processes. In addition, many other ECM proteins are positively regulated by CEBPD, including TNC, LAMC1, several collagens (COL1A1, COL1A2, COL5A1, COL6A1, COL6A2, and COL6A3) (Fig. 5C) and the subunit of integrin receptors such as ITGAV. Furthermore, most of these genes contained CEBPD binding sites in their promotors (Fig. 6D) and are highly correlated with CEBPD (Fig. 7A), especially TNC and COL6A3. The correlation results are further supported by the correlation analysis in GBM subtypes (Fig. 7A) to exclude the possibility that the correlations were mainly caused by the fact that CEBPD and these genes are up-regulated in mesenchymal subtype. Furthermore, mathematical simulations demonstrated that, if the correlation is mainly caused by different expression level in subtypes, then the correlation in each subtype can be very weak.
Given that ECM-integrin interactions are important outside-in signal pathways, and are promising therapeutic targets [55, 56], our findings uncovered novel up-steam regulatory mechanisms of ECM-integrin interactions. However, the detailed mechanism among these proteins requires future studies. Nevertheless, the finding that CEBPD is a key TF for this process has important theoretical and clinical significance, because ECM-integrin interactions are important activators of the EGFR/PI3K pathway, and the EGFR/PI3K pathway is one of the core pathways altered in GBM [52].
This study was the first reveal that CEBPD is a master TF for hypoxia-induced molecules in both mRNA and protein levels, and this conclusion is consistently obtained from three kinds of independent experimental data (Fig. 1A–C). Although reciprocal regulation is present between CEBPD and HIFs [10, 13, 58], the central role of CEBPD for hypoxia-induced responses uncovered in the present study are not elucidated before. The mechanism behind the central role of CEBPD is that it regulates ECM-integrin proteins, while ECM-integrin interactions are also key components of hypoxia induced responses (Fig. 7F–H). As a hypoxia-induced protein, CEBPD was identified in the list of PANSGs, but not in HRPs and SR-HRGs, because for HRPs, CEBPD is not covered in the proteomic panel in the high-throughput quantification. For SR-HRGs, CEBPD is overlapped in 2 sets out of the 5 sets, while we used genes overlapped in at least 3 sets as our SR-HRGs. Notably, for PANSGs analysis, CEBPD is specifically highly expressed in PAN area (Fig. 2I), consistent with previous report [14].
A notable novel finding of the present study is that FN1 is a direct target of CEBPD. FN1 is also up-regulated by hypoxia (Fig. 5A, D, E, Fig. 6A), consistent with other studies. Nevertheless, FN1 is induced by TGFβ signal [59], rather than by HIFs [60]. Consistently, HIF binding elements were not identified in FN1 promotor in GBM cells when we checked the previously reported Chip-seq studies [61, 62]. Therefore, it is more possible that FN1 up-regulation in hypoxia is mediated through HIF-independent pathways such as TGFβ signal or as reported in present study, by CEBPD. TGFβ signal pathway is closely associated with ECM-integrin outside-in signals [55, 56, 63], and TGFβ signal pathway is activated in hypoxia [6, 59].
Moreover, the critical proteins of TGFβ signal pathway, TGFBI and a component of integrin receptor ITGA5 were up-regulated under hypoxic condition (TGFBI and ITGA5 were up-regulated by 1.11 and 1.12 folds in hypoxia, p < 0.05). TGFBI is important for the activation of TGFβ signal pathway in GBM [64, 65], which can activate integrins [60]. Notably, TGFBI and its receptor subunit ITGAV are potential direct targets of CEBPD supported by the following information: 1. TGFBI and ITGAV proteins are significantly down-regulated after CEBPD KD (Fig. 5A; TGFBI data were not included in Fig. 5C–E, it was down-regulated by 1.49 folds after CEBPD KD, p = 2.10 × 10−5). 2. Many CEBPD binding elements are present in the promotor of TGFBI (Fig. 7H) and ITGAV (Fig. 6D). 3. CEBPD exhibited highly positive correlation with TGFBI and ITGAV (Fig. 7A; the coefficients between TGFBI and CEBPD were 0.58, 0.54, and 0.62 in TCGA, REMBRANDT, and CGGA GBM datasets, respectively). Therefore, one important potential down-stream results of CEBPD expression in hypoxia is the activation of TGFβ signal pathway by directly regulating TGFBI and ITGAV. In addition, this pathway may also contribute to the activation of FN1 [59, 60]. Therefore, CEBPD-induced FN1 expression can activate the EGFR/PI3K pathway by promoting EGFR phosphorylation. However, CEBPD may also induce FN1 up-regulation and the down-stream pathways through other indirect ways, such as through TGFβ signal-induced FN1 expression. Detailed investigation of the interplays among CEBPD, TGFβ signal pathway, ECM and hypoxia will be the focus of future studies.
One of the key signal feature of GBM is the activation of receptor tyrosine kinase (RTK), especially the EGFR-related activation of PI3K/AKT pathways [52]. Alterations in canonical PI3K/MAPK pathways are identified in more than 90% GBM samples [52, 66], and the most common and important gene alteration in this pathway is EGFR, whose amplification or mutation is identified in more than half (57%) of GBM samples [66]. Specifically, EGFR is responsible for cancer cell survival and invasiveness under hypoxic condition [67]. However, treatment strategies that target EGFR have thus far failed in clinical trials [17]. Therefore, the mechanisms underlying sustained EGFR pathway activation have been important issues for long time and need to be further explored [16]. In the present study, the EGFR/PI3K/Akt pathway activation was increased under hypoxic condition, and is dependent on CEBPD expression and its induction of ECM-integrin activation, especially direct transcriptional regulation of FN1. In terms of mechanisms, the activation of this pathway is mainly dependent on the phosphorylation rather than the transcriptional regulation of EGFR. Other possible downstream targets of CEBPD may contribute to the activation of the PI3K/AKT pathway, such as FGF1 (Fig. 5A, C), or other unidentified growth factors. The hypoxia-induced activation of EGFR/PI3K pathway activity in the present study (Fig. 6A) is consistent with previous reports [68]. Notably, EGFR activation also promoted HIF-induced signaling [69, 70], indicating a more complicated network interaction underlining EGFR/PI3K pathway activation in GBM. In addition, EGFR activity can promote CEBPD expression [71], implying a positive feedback between them, and targeting CEBPD would be promising strategies to break the signal loops. Collectively, our data deepens the understanding of the activation mechanisms of EGFR/PI3K pathways, especially in hypoxia, and provide novel potential therapeutic avenues to target this pathway.
Conclusion
Our study revealed novel features of the HRGs-MINW, in which CEBPD functions as a key TF controlling hypoxia-induced tumorigenic potential of GBM. This process mainly occurs through ECM-integrin signal-mediated EGFR phosphorylation and subsequent activation of EGFR/PI3K pathway, which ultimately increases the invasion capacity of GBM. FN1, a key ECM protein, is a direct transcriptional target of CEBPD that mediates these processes.
References
Tan AC, Ashley DM, Lopez GY, Malinzak M, Friedman HS, Khasraw M. Management of glioblastoma: State of the art and future directions. CA Cancer J. Clin. 2020;70:299–312.
Mao XG, Xue XY, Wang L, Wang L, Li L, Zhang X. Hypoxia regulated gene network in glioblastoma has special algebraic topology structures and revealed communications involving warburg effect and immune regulation. Cell Mol Neurobiol. 2019;39:1093–114.
Colwell N, Larion M, Giles AJ, Seldomridge AN, Sizdahkhani S, Gilbert MR, et al. Hypoxia in the glioblastoma microenvironment: sha** the phenotype of cancer stem-like cells. Neuro Oncol. 2017;19:887–96.
Joseph JV, Conroy S, Pavlov K, Sontakke P, Tomar T, Eggens-Meijer E, et al. Hypoxia enhances migration and invasion in glioblastoma by promoting a mesenchymal shift mediated by the HIF1alpha-ZEB1 axis. Cancer Lett. 2015;359:107–16.
Li Z, Bao S, Wu Q, Wang H, Eyler C, Sathornsumetee S, et al. Hypoxia-inducible factors regulate tumorigenic capacity of glioma stem cells. Cancer Cell. 2009;15:501–13.
Kaur B, Khwaja FW, Severson EA, Matheny SL, Brat DJ, Van, et al. Hypoxia and the hypoxia-inducible-factor pathway in glioma growth and angiogenesis. Neuro Oncol. 2005;7:134–53.
Wang P, Yan Q, Liao B, Zhao L, **ong S, Wang J, et al. The HIF1alpha/HIF2alpha-miR210-3p network regulates glioblastoma cell proliferation, dedifferentiation and chemoresistance through EGF under hypoxic conditions. Cell Death Dis. 2020;11:992.
**g X, Yang F, Shao C, Wei K, **e M, Shen H, et al. Role of hypoxia in cancer therapy by regulating the tumor microenvironment. Mol Cancer. 2019;18:157.
Choudhry H, Harris AL. Advances in hypoxia-inducible factor biology. Cell Metab. 2018;27:281–98.
Balamurugan K, Wang JM, Tsai HH, Sharan S, Anver M, Leighty R, et al. The tumour suppressor C/EBPdelta inhibits FBXW7 expression and promotes mammary tumour metastasis. EMBO J. 2010;29:4106–17.
Jiang C, Sun J, Dai Y, Cao P, Zhang L, Peng S, et al. HIF-1A and C/EBPs transcriptionally regulate adipogenic differentiation of bone marrow-derived MSCs in hypoxia. Stem Cell. Res. Ther. 2015;6:21.
Min Y, Ghose S, Boelte K, Li J, Yang L, Lin PC. C/EBP-delta regulates VEGF-C autocrine signaling in lymphangiogenesis and metastasis of lung cancer through HIF-1alpha. Oncogene. 2011;30:4901–9.
Balamurugan K, Mendoza-Villanueva D, Sharan S, Summers GH, Dobrolecki LE, Lewis MT, et al. C/EBPdelta links IL-6 and HIF-1 signaling to promote breast cancer stem cell-associated phenotypes. Oncogene. 2019;38:3765–80.
Cooper LA, Gutman DA, Chisolm C, Appin C, Kong J, Rong Y, et al. The tumor microenvironment strongly impacts master transcriptional regulators and gene expression class of glioblastoma. Am J Pathol. 2012;180:2108–19.
Chen JC, Alvarez MJ, Talos F, Dhruv H, Rieckhof GE, Iyer A, et al. Identification of causal genetic drivers of human disease through systems-level analysis of regulatory networks. Cell. 2014;159:402–14.
Liu Y, Li Z, Zhang M, Zhou H, Wu X, Zhong J, et al. Rolling-translated EGFR variants sustain EGFR signaling and promote glioblastoma tumorigenicity. Neuro Oncol. 2021;23:743–56.
Eskilsson E, Rosland GV, Solecki G, Wang Q, Harter PN, Graziani G, et al. EGFR heterogeneity and implications for therapeutic intervention in glioblastoma. Neuro Oncol. 2018;20:743–52.
Barnes JM, Kaushik S, Bainer RO, Sa JK, Woods EC, Kai F, et al. A tension-mediated glycocalyx-integrin feedback loop promotes mesenchymal-like glioblastoma. Nat Cell Biol. 2018;20:1203–14.
Comba A, Faisal SM, Dunn PJ, Argento AE, Hollon TC, Al-Holou WN, et al. Spatiotemporal analysis of glioma heterogeneity reveals COL1A1 as an actionable target to disrupt tumor progression. Nat. Commun. 2022;13:3606.
Lugano R, Vemuri K, Yu D, Bergqvist M, Smits A, Essand M, et al. CD93 promotes beta1 integrin activation and fibronectin fibrillogenesis during tumor angiogenesis. J Clin Invest. 2018;128:3280–97.
Verhaak RG, Hoadley KA, Purdom E, Wang V, Qi Y, Wilkerson MD, et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell. 2010;17:98–110.
Madhavan S, Zenklusen JC, Kotliarov Y, Sahni H, Fine HA, Buetow K. Rembrandt: hel** personalized medicine become a reality through integrative translational research. Mol Cancer Res. 2009;7:157–67.
Lee Y, Scheck AC, Cloughesy TF, Lai A, Dong J, Farooqi HK, et al. Gene expression analysis of glioblastomas identifies the major molecular basis for the prognostic benefit of younger age. BMC Med Genomics. 2008;1:52.
Gravendeel LA, Kouwenhoven MC, Gevaert O, de Rooi JJ, Stubbs AP, Duijm JE, et al. Intrinsic gene expression profiles of gliomas are a better predictor of survival than histology. Cancer Res. 2009;69:9065–72.
Mao XG, Xue XY, Wang L, Zhang X, Yan M, Tu YY, et al. CDH5 is specifically activated in glioblastoma stem like cells and contributes to vasculogenic mimicry induced by hypoxia. Neuro Oncol. 2013;15:865–79.
Puchalski RB, Shah N, Miller J, Dalley R, Nomura SR, Yoon JG, et al. An anatomic transcriptional atlas of human glioblastoma. Science. 2018;360:660–3.
Zhen HN, Zhang X, Hu PZ, Yang TT, Fei Z, Zhang JN, et al. Survivin expression and its relation with proliferation, apoptosis, and angiogenesis in brain gliomas. Cancer. 2005;104:2775–83.
Nissou MF, El Atifi M, Guttin A, Godfraind C, Salon C, Garcion E, et al. Hypoxia-induced expression of VE-cadherin and filamin B in glioma cell cultures and pseudopalisade structures. J Neurooncol. 2013;113:239–49.
Kucharzewska P, Christianson HC, Welch JE, Svensson KJ, Fredlund E, Ringner M, et al. Exosomes reflect the hypoxic status of glioma cells and mediate hypoxia-dependent activation of vascular cells during tumor development. Proc Natl Acad Sci USA 2013;110:7312–7.
Koh MY, Lemos R Jr., Liu X, Powis G. The hypoxia-associated factor switches cells from HIF-1alpha- to HIF-2alpha-dependent signaling promoting stem cell characteristics, aggressive tumor growth and invasion. Cancer Res. 2011;71:4015–27.
Mao XG, Xue XY. General hypergeometric distribution: a basic statistical distribution for the number of overlapped elements in multiple subsets drawn from a finite population. ar**v:1808.06924 [Preprint]. 2018. Available from: https://arxiv.org/abs/1808.06924.
Mao XG, Xue XY. Mean, Variance and asymptotic property for general hypergeometric distribution. ar**v:2208.14939 [Preprint]. 2022. Available from: https://arxiv.org/abs/2208.14939.
Lambert SA, Jolma A, Campitelli LF, Das PK, Yin Y, Albu M, et al. The human transcription factors. Cell. 2018;172:650–65.
Margolin AA, Wang K, Lim WK, Kustagi M, Nemenman I, Califano A. Reverse engineering cellular networks. Nat Protoc. 2006;1:662–71.
Zhang Q, Liu W, Zhang HM, **e GY, Miao YR, **a M, et al. hTFtarget: a comprehensive database for regulations of human transcription factors and their targets. Genom Proteom Bioinf. 2020;18:120–8.
Li ZZ, Han WJ, Sun ZC, Chen Y, Sun JY, Cai GH, et al. Extracellular matrix protein laminin beta1 regulates pain sensitivity and anxiodepression-like behaviors in mice. J Clin Invest. 2021;131:e146323.
Robinson JT, Thorvaldsdottir H, Winckler W, Guttman M, Lander ES, Getz G, et al. Integrative genomics viewer. Nat Biotechnol. 2011;29:24–6.
Hoadley KA, Yau C, Hinoue T, Wolf DM, Lazar AJ, Drill E, et al. Cell-of-origin patterns dominate the molecular classification of 10,000 tumors from 33 types of cancer. Cell. 2018;173:291–304. e6
Ceccarelli M, Barthel FP, Malta TM, Sabedot TS, Salama SR, Murray BA, et al. Molecular profiling reveals biologically discrete subsets and pathways of progression in diffuse glioma. Cell. 2016;164:550–63.
Zhao Z, Meng F, Wang W, Wang Z, Zhang C, Jiang T. Comprehensive RNA-seq transcriptomic profiling in the malignant progression of gliomas. Sci Data. 2017;4:170024.
Ayuso JM, Monge R, Martinez-Gonzalez A, Virumbrales-Munoz M, Llamazares GA, Berganzo J, et al. Glioblastoma on a microfluidic chip: Generating pseudopalisades and enhancing aggressiveness through blood vessel obstruction events. Neuro Oncol. 2017;19:503–13.
Brat DJ, Castellano-Sanchez AA, Hunter SB, Pecot M, Cohen C, Hammond EH, et al. Pseudopalisades in glioblastoma are hypoxic, express extracellular matrix proteases, and are formed by an actively migrating cell population. Cancer Res. 2004;64:920–7.
Fornes O, Castro-Mondragon JA, Khan A, van der Lee R, Zhang X, Richmond PA, et al. JASPAR 2020: update of the open-access database of transcription factor binding profiles. Nucleic Acids Res. 2020;48:D87–D92.
Feng H, Li Y, Yin Y, Zhang W, Hou Y, Zhang L, et al. Protein kinase A-dependent phosphorylation of Dock180 at serine residue 1250 is important for glioma growth and invasion stimulated by platelet derived-growth factor receptor alpha. Neuro Oncol. 2015;17:832–42.
Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4:44–57.
Yu S, Yu X, Sun L, Zheng Y, Chen L, Xu H, et al. GBP2 enhances glioblastoma invasion through Stat3/fibronectin pathway. Oncogene. 2020;39:5042–55.
Miroshnikova YA, Mouw JK, Barnes JM, Pickup MW, Lakins JN, Kim Y, et al. Tissue mechanics promote IDH1-dependent HIF1alpha-tenascin C feedback to regulate glioblastoma aggression. Nat Cell Biol. 2016;18:1336–45.
Sun S, Wang Y, Wu Y, Gao Y, Li Q, Abdulrahman AA, et al. Identification of COL1A1 as an invasionrelated gene in malignant astrocytoma. Int J Oncol. 2018;53:2542–54.
Fang R, Chen X, Zhang S, Shi H, Ye Y, Shi H, et al. EGFR/SRC/ERK-stabilized YTHDF2 promotes cholesterol dysregulation and invasive growth of glioblastoma. Nature communications. 2021;12:177.
Liu SL, Chen MH, Wang XB, You RK, An XW, Zhao Q, et al. LncRNA PCGEM1 contributes to malignant behaviors of glioma by regulating miR-539-5p/CDK6 axis. Aging. 2021;13:5475–84.
Schuster A, Klein E, Neirinckx V, Knudsen AM, Fabian C, Hau AC, et al. AN1-type zinc finger protein 3 (ZFAND3) is a transcriptional regulator that drives Glioblastoma invasion. Nature communications. 2020;11:6366.
TCGA. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008;455:1061–8.
Vitucci M, Karpinich NO, Bash RE, Werneke AM, Schmid RS, White KK, et al. Cooperativity between MAPK and PI3K signaling activation is required for glioblastoma pathogenesis. Neuro Oncol. 2013;15:1317–29.
Cabodi S, Moro L, Bergatto E, Boeri Erba E, Di Stefano P, Turco E, et al. Integrin regulation of epidermal growth factor (EGF) receptor and of EGF-dependent responses. Biochem Soc Trans. 2004;32:438–42.
Cooper J, Giancotti FG. Integrin signaling in cancer: mechanotransduction, stemness, epithelial plasticity, and therapeutic resistance. Cancer Cell. 2019;35:347–67.
Hamidi H, Ivaska J. Every step of the way: integrins in cancer progression and metastasis. Nat Rev Cancer. 2018;18:533–48.
Hajizadeh F, Okoye I, Esmaily M, Ghasemi Chaleshtari M, Masjedi A, Azizi G, et al. Hypoxia inducible factors in the tumor microenvironment as therapeutic targets of cancer stem cells. Life Sci. 2019;237:116952.
Balamurugan K, Sharan S, Klarmann KD, Zhang Y, Coppola V, Summers GH, et al. FBXW7alpha attenuates inflammatory signalling by downregulating C/EBPdelta and its target gene Tlr4. Nature communications. 2013;4:1662.
Wang WY, Twu CW, Liu YC, Lin HH, Chen CJ, Lin JC. Fibronectin promotes nasopharyngeal cancer cell motility and proliferation. Biomed Pharmacother. 2019;109:1772–84.
Rana MK, Srivastava J, Yang M, Chen CS, Barber DL. Hypoxia increases the abundance but not the assembly of extracellular fibronectin during epithelial cell transdifferentiation. J Cell Sci. 2015;128:1083–9.
**a X, Kung AL. Preferential binding of HIF-1 to transcriptionally active loci determines cell-type specific response to hypoxia. Genome Biol. 2009;10:R113.
Mole DR, Blancher C, Copley RR, Pollard PJ, Gleadle JM, Ragoussis J, et al. Genome-wide association of hypoxia-inducible factor (HIF)-1alpha and HIF-2alpha DNA binding with expression profiling of hypoxia-inducible transcripts. J Biol Chem. 2009;284:16767–75.
Shaim H, Shanley M, Basar R, Daher M, Gumin J, Zamler DB, et al. Targeting the alphav integrin/TGF-beta axis improves natural killer cell function against glioblastoma stem cells. J Clin Invest. 2021;131:e142116.
Peng P, Zhu H, Liu D, Chen Z, Zhang X, Guo Z, et al. TGFBI secreted by tumor-associated macrophages promotes glioblastoma stem cell-driven tumor growth via integrin alphavbeta5-Src-Stat3 signaling. Theranostics. 2022;12:4221–36.
Zhu Z, Mesci P, Bernatchez JA, Gimple RC, Wang X, Schafer ST, et al. Zika virus targets glioblastoma stem cells through a SOX2-integrin alphavbeta5 axis. Cell Stem Cell. 2020;26:187–204.e10.
Brennan CW, Verhaak RG, McKenna A, Campos B, Noushmehr H, Salama SR, et al. The somatic genomic landscape of glioblastoma. Cell. 2013;155:462–77.
Shen J, **a W, Khotskaya YB, Huo L, Nakanishi K, Lim SO, et al. EGFR modulates microRNA maturation in response to hypoxia through phosphorylation of AGO2. Nature. 2013;497:383–7.
Wang P, Zhao L, Gong S, **ong S, Wang J, Zou D, et al. HIF1alpha/HIF2alpha-Sox2/Klf4 promotes the malignant progression of glioblastoma via the EGFR-PI3K/AKT signalling pathway with positive feedback under hypoxia. Cell Death Dis. 2021;12:312.
Li X, Fan Z. The epidermal growth factor receptor antibody cetuximab induces autophagy in cancer cells by downregulating HIF-1alpha and Bcl-2 and activating the beclin 1/hVps34 complex. Cancer Res. 2010;70:5942–52.
Wouters A, Boeckx C, Vermorken JB, Van den Weyngaert D, Peeters M, Lardon F. The intriguing interplay between therapies targeting the epidermal growth factor receptor, the hypoxic microenvironment and hypoxia-inducible factors. Curr Pharm Des. 2013;19:907–17.
Wang WJ, Li CF, Chu YY, Wang YH, Hour TC, Yen CJ, et al. Inhibition of the EGFR/STAT3/CEBPD axis reverses cisplatin cross-resistance with paclitaxel in the urothelial carcinoma of the urinary bladder. Clin Cancer Res. 2017;23:503–13.
Acknowledgements
This research was supported by the National Natural Science Foundation of China (81972359, 82273978, 81502143) and the Key Research and Development Program of Shaanxi Province (No. 2023-YBSF-629).
Author information
Authors and Affiliations
Contributions
XGM, XFJ, and XZ developed the hypothesis. XGM and XYX designed the experiments and developed the methodology. XGM, XYX, and TYS performed all the experiments; AJ, RL, and XYC performed IHC, animal models, cell culture and WB assays; XGM, XYX collected and analyzed the data. XGM performed bioinformatics analysis. XGM, XYX, and TYS wrote the manuscript. XFJ and XZ supervised the project.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics statement
All animal experiments were conducted in accordance with the guidelines of Animal Experiments guidelines in force at the Fourth Military Medical University.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Edited by Dr Maria Victoria Niklison Chirou
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Mao, Xg., Xue, Xy., Lv, R. et al. CEBPD is a master transcriptional factor for hypoxia regulated proteins in glioblastoma and augments hypoxia induced invasion through extracellular matrix-integrin mediated EGFR/PI3K pathway. Cell Death Dis 14, 269 (2023). https://doi.org/10.1038/s41419-023-05788-y
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41419-023-05788-y
- Springer Nature Limited
This article is cited by
-
Bioinformatics Analysis and Experimental Validation of Differential Genes and Pathways in Bone Nonunions
Biochemical Genetics (2024)