
Abstract
Background
Isopentenols, such as prenol and isoprenol, are promising advanced biofuels because of their higher energy densities and better combustion efficiencies compared with ethanol. Microbial production of isopentenols has been developed recently via metabolically engineered E. coli. However, current yields remain low and the underlying pathways require systematic optimization.
Results
In this study, we targeted the E. coli native 2-methyl-(D)-erythritol-4-phosphate (MEP) pathway and its upstream glycolysis pathway for the optimization of isopentenol production. Two codon optimized genes, nudF and yhfR from Bacillus subtilis, were synthesized and expressed in E. coli W3110 to confer the isopentenol production of the strain. Two key enzymes (IspG and Dxs) were then overexpressed to optimize the E. coli native MEP pathway, which led to a significant increase (3.3-fold) in isopentenol production. Subsequently, the glycolysis pathway was tuned to enhance the precursor and NADPH supplies for the MEP pathway by activating the pentose phosphate pathway (PPP) and Entner-Doudoroff pathway (ED), which resulted in additional 1.9 folds of increase in isopentenol production. A 5 L-scale batch cultivation experiment was finally implemented, showing a total of 61.9 mg L−1 isopentenol production from 20 g L−1 of glucose.
Conclusion
The isopentenol production was successfully increased through multi-step optimization of the MEP and its upstream glycolysis pathways. It demonstrated that the total fluxes and their balance of the precursors of the MEP pathway are of critical importance in isopentenol production. In the future, an elucidation of the contribution of PPP and ED to MEP is needed for further optimization of isopentenol production.
Similar content being viewed by others

Background
The growing global demand for energy, continuing concerns about the environment and the increasing cost of fossil fuels have called for the development of new forms of energy [1],[2]. The microbial production of bio-based fuels and chemicals from biomass has recently become as a promising, sustainable solution to this need [3],[4]. Early efforts in biofuel production have focused on improving the yield of ethanol made from the fermentation of plant sugars, and have shown a great success [5],[6].
Recently, microbial production of advanced biofuels, such as butanol and isobutanol, is being increasingly explored due to the facts that advanced biofuels have higher energy contents and, more importantly, are more compatible with existing engines and fuel distribution infrastructures [7] [9]. One class of such advanced biofuels is isopentenols, including prenol and isoprenol, which have better combustion efficiencies than ethanol and better octane numbers that are more similar to that of gasoline compared with ethanol [10].
Isopentenol production using engineered microbes was first reported in 2007 by Withers and co-workers [11]. Through the enrichment of a library of genomic DNA from Bacillus subtilis, the authors discovered two genes (yhfR and nudF) that encode enzymes that function directly on the prenyl diphosphate precursors, isopentenyl pyrophosphate (IPP) and dimethylallyl pyrophosphate (DMAPP) and convert them to isopentenols. In 2012, Chou and Keasling reported another gene nudB which encodes an enzyme capable of converting prenyl diphosphate precursors to isopentenols [10]. Overexpression of these genes along with the introduction of a heterologous mevalonate-dependent pathway (MVA) in E. coli has resulted in successful isopentenol production [10] [12]. In addition to the MVA pathway, the E. coli native MEP pathway also holds the potential to be exploited for isopentenol production. Indeed, several attempts have been made for successful biosynthesis of other forms of isoprenoids including taxadiene through the E. coli native MEP pathway [13] [15]. Up to date, reported isopentenol production via the MVA pathway is much higher than that via the MEP pathway. On the other hand, there are advantages in using the MEP pathway: According to the stoichiometry and redox balance analysis by a previous study [16], MEP is energetically more balanced and theoretically more efficient than MVA in converting sugars or glycerol to isoprenoid. In addition, due to the inherent presence of the MEP pathway, it requires fewer heterologous genes to be introduced.
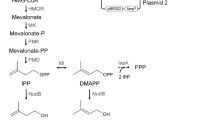
We were therefore motivated in this work to increase the isopentenol production of engineered E. coli by optimizing its inherent MEP pathway (Figure 1). We first synthesized two codon optimized genes, nudF and yhfR from B. subtilis, and expressed them in our expression host E. coli W3110 to confer its production of isopentenols including both prenol and isoprenol. We then overexpressed two key enzymes (IspG and Dxs) to optimize the native E. coli MEP pathway, leading to a significant increase in isopentenol titer. To further maximize the isopentenol productivity, we enhanced the precursor and NADPH supplies needed for the MEP pathway by activating the pentose phosphate pathway (PPP) and Entner-Doudoroff (ED) pathway through the disruption of the E. coli phosphoglucose isomerase gene (pgi). To evaluate the efficacy of our multi-step optimization of isopentenol production, we finally implemented a 5 L-scale laboratory batch fermentation using the optimal strain identified.

The isopentenol biosynthesis pathway in engineered E. coli. Pathway intermediates: G3P, glyceraldehyde-3-phosphate; DXP, 1-deoxy-D-xylulose 5-phosphate; MEP, 2-C-methyl-D-erythritol 4-phosphate; CDP-ME, 4-diphosphocytidyl-2-C-methyl-D-erythritol; CDP-MEP, 4-diphosphocytidyl-2-C-methyl-D-erythritol 2-phosphate; MECPP, 2-C-methyl-D-erythritol 2,4-cyclopyrophosphate; HMBPP, 1-hydroxy-2-methyl-2-(E)-butenyl 4-pyrophosphate; IPP, isopentenyl pyrophosphate; DMAPP, dimethylallyl pyrophosphate; DHAP, dihydroxyacetone 3-phosphate.
Results and discussion
Expression of nudF and yhfR in E. coli W3110
To enable a successful isopentenol production in E. coli, two genes, nudF and yhfR from Bacillus subtilis, were identified as heterologous enzyme genes for direct conversion of IPP and DMAPP into isoprenol and prenol respectively. However, due to the different codon usages of E. coli and B. subtilis, the codon adaption indexes (CAI) of the wild-type yhfR (GeneBank: CP002906.1) and nudF (GeneBank: CP003329.1) genes for the host E. coli W3110 are only 0.31 and 0.25 respectively [17]. To address the potential low expression issue in the E. coli host, the ORF sequences of these two genes were optimized to achieve a CAI of 1.0 for both (See Additional file 1). The optimized yhfR and nudF genes were then synthesized, ligated into the expression vector pACYC-duet, and subsequently introduced into E. coli W3110 (DE3) for isopentenol production. GC analysis results showed that, after 48 h of cultivation in M9 medium containing yeast extract and glucose, the strain E. coli W3110(DE3)/pACYC-yhfR produced a tiny amount of isoprenol and prenol (<1 mg L−1) (Figure 2, EWIP1). In contrast, the strain E. coli W3110 (DE3)/pACYC-nudF produced 2.6 mg L−1 prenol and 2.2 mg L−1 isoprenol (Figure 2, EWIP2). No isoprenol or prenol production was detected in the control strain that harbors the empty vector pACYC-duet (Figure 2, Control). These results supported the earlier shown feasibility of producing isopentenol in E. coli and also suggested that NudF is a more effective enzyme than yhfR in isopentenol production.

Isopentenol production by engineered E. coli strains. A 250 mL-scale shaking flask containing 50 mL of semi-defined medium, consisted of M9 minimum salts, 5 g L−1 yeast extract, 10 g L−1 glucose and 1 mM thiamine pyrophosphate (TPP), was used for the cultivation of the strains. All strains are listed in Table 1.
Optimization of MEP pathway for increased isopentenol production
To elevate isopentenol production, the upstream MEP pathway was targeted for optimization. In a recent study, Zhou et al. showed that overexpression of the E. coli HMBPP synthase gene (ispG) can increase the isoprenoid production by reducing MECPP efflux, i.e., the rate of MECPP transport out of the cell [18]. Based on this finding, the ispG gene was cloned and ligated into pACYC-nudF, resulting in a plasmid denoted as pACYC-ispG-nudF. An overexpression experiment of the E. coli W3110 (DE3)/pACYC-ispG-nudF showed that ispG co-expression led to a 1.6-fold increase in isopentenol production (Figure 2, EWIP 3).
Additionally, two recent studies have shown that overexpression of the DXP synthase gene dxs is able to enhance the metabolic flux of the MEP pathway and hence isoprenoid production [19],[20]. Encouraged by these results, we further over-expressed the dxs gene along with ispG and nudF in E. coli W3110 (DE3). Encouragingly, the new strain resulted in an additional 2.1-fold increase in isopentenol production (Figure 2, EWIP 4).
Tuning the glycolysis pathway to maximize the isopentenol production
To further increase the isopentenol production of our engineered strains, we traced back to the precursor supplies of the MEP pathway and examined their roles in influencing the overall isopentenol production.
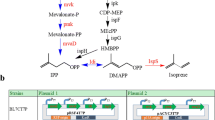
As shown in the first step of MEP pathway in Figure 1, one mole of deoxyxylulose-5-phosphate (DXP) is formed from the equimolar condensation of glyceraldehyde-3-phosphate (G3P) and pyruvate. This suggests that limitations in G3P or pyruvate or their imbalance may reduce DXP formation and hence hinder isopentenol production [21]. In wild-type E. coli strains, glucose is metabolized mainly through the Embden-Meyerhof-Parnas pathway (~70%) and partially through Pentose Phosphate Pathway (~30%) as depicted in Figure 3 (panel A). Both of the pathways lack the control of pyruvate and G3P productions, which may imply an imbalanced G3P-pyruvate ratio. Indeed, a couple of previous studies have supported this speculation [22],[23]: They showed that re-balancing the pyruvate and G3P pools via the modification of the EMP pathway can increase the isoprenoid production significantly.

Thee glycolytic pathways present in E. coli . (A), The native EMP and PPP pathways are active in the E. coli strains EWIP1-4. (B) The PPP and ED pathways are activated in the strain EWIP5 by disrupting the pgi gene. EMP, Embden-Meyerhof pathway; PPP, pentose phosphate pathway; ED, Entner-Doudoroff.
Meanwhile, there is an additional glycolytic pathway responsible for glucose metabolism in E. coli the Enter-Doudoroff pathway [24],[25]. The ED pathway is silent in native E. coli strains consuming glucose but it is capable of simultaneous production of pyruvate and G3P because the pathway generates the two precursors concurrently with a common cleavage reaction (Figure 3, panel B). Therefore, compared with the EMP pathway, the ED pathway is a potentially more appealing route for supplying the precursors of the MEP pathway.
In addition, NADPH supply is another factor that influences the efficiency of the MEP pathway. This is because two moles of NADPH are needed in order to produce one mole of isoprenoid unit (Figure 1). A recent study has indeed proven that enhancing the carbon flux to the PPP pathway, the major NADPH producing system in E. coli, can increase isoprenoid production [26].
Taking into account both the precursor balance and NADPH supply, we decided to shunt the glucose metabolism from EMP to PPP and ED to maximize our isopentenol production. To this end, we blocked the EMP by disrupting the pgi gene in E. coli W3110, out of the reason that pgi disruption can completely channel the glucose metabolic flux to PPP (~90%) and ED (~10%) from EMP [27] [29]. Shaking flask cultivation experiments showed that it significantly increased the isopentenol production to 30.0 mg L−1 (Figure 2, EWIP5), 1.9 fold higher than the strain EWIP4.
We also replaced the native promoter of edd-eda operon (eddp1) by a strong inducible Trc promoter in E. coli W3110Δpgi (DE3) chromosome with the goal of altering the carbon flux between PPP and ED. However, this modification severely affected cellular growth and glucose consumption and caused a decrease in isopentenol production (Figure 2, EWIP6). One possible reason is the reduction of total NADPH production two moles of NADPH production from the PPP pathway compared with one mole of NADPH via ED from one mole of glucose. Future studies in the impacts of EMP blockage and eddp1 replacement are needed for further optimization of isopentenol production. In addition, a comprehensive study on the alteration of metabolic flux upon EMP blockage and eddp1 replacement will be also valuable in order to elucidate the PPP and ED contributions to MEP pathway and isopentenol production.
Lab-scale batch production of isopentenols
To evaluate the outcome of our multi-step optimization of isopentenol production, a 5 L-scale laboratory batch reactor was used for fermentation. Our most optimized strain, EWIP5, was grown in 2 L M9 minimal salts medium containing 5 g L−1 yeast extract and 20 g L−1 glucose. Our fermentation results (Figure 4) showed that glucose was completely consumed at hour 24 and 23.7 mg L−1 of prenol and 16.2 mg L−1 of isoprenol were produced. However, the prenol and isoprenol levels continued to increase and reached 35.2 mg L−1 and 26.7 mg L−1 at hour 48. We speculate that the additional isopentenol production after glucose depletion was due to the consumption of yeast extract and/or other secondary metabolites (such as acetic acid) produced from glucose.

Time course of isopentenol production, glucose consumption and biomass formation of the engineered strain E. coli EWIP5 during batch fermentation.
Conclusions
In this paper, we have successfully exploited the E. coli native MEP pathway for the production of isopentenols including prenol and isoprenol. We chose MEP instead of MVA as the optimization target because the former is more energetically balanced and theoretically more efficient than the latter in converting sugars or glycerol to isoprenoid according to the stoichiometry and redox balance analysis [16]. To achieve our goal, multi-step optimizations were employed to increase isopentenol production: The expression of codon-optimized nudF in E. coli W3110 led to the production of both isoprenol and prenol; overexpression of the HMBPP synthase gene ispG and DXP synthase gene dxs resulted in an additional 3.3-fold increase in isopentenols production; and shunting the glycolysis from EMP to PPP and ED led to another 1.9-fold increase. Through the multi-step optimizations, the resulting strain was able to produce isopentenols at a level of 61.9 mg L−1.
It is also worthy to notice that the blocking of EMP to channel glucose metabolism via PPP and ED explored here provided a more balanced precursor pool for the MEP pathway. Although further studies are required to elucidate the exact contributions of PPP and ED to isopentenol production, this strategy may serve as a novel approach for further optimization of MEP-dependent isoprenoid biosynthesis. In the future, additional efforts, such as those on precursor balance and NADPH supply, are also needed for further improvement of the titer of isopentenols.
Methods
Strains and plasmids
All strains and plasmids used in this study are listed in Table 1. E. coli W3110 was purchased from ATCC (No. 27325), whereas E. coli W3110 Δpgi was constructed by deleting pgi gene in it. The pgi gene disruption cassette was amplified with primers Pgi-KF and Pgi-KR (Table 2) using pKD3 as template. The eddp1 replacement with pTrc promoter was carried out using the cassette amplified with primers Eddp1-KF and Eddp1-KR and pKD4 as template. The plasmid pKD46 was used as the Red recombinase expression vector while pCP20 was used to eliminate the resistance gene. Gene disruption and resistant gene elimination experiments were performed according to the protocols obtained from OPENWETWARE [30]. To express the relevant genes under the control of the T7 promoter, the 69734 λDE3 Lysogenization Kit (Novagen, EMD Millipore, U.S.) was used to integrate an λDE3 prophage into the E. coli host chromosome.
The codon adaption indices (CAI) of the B. subtilis yhfR and nudF genes were analyzed using Optimizer [17]. The codon-optimized sequences of yhfR and nudF were synthesized and ligated into pACYCDuet-1 by Nde I and BglII to construct pACYC-yhfR and pACYC-nudf, respectively. The DXP synthase gene (dxs) was ligated into pACYC-nudf using NcoI and Bam HI, followed by further ligation of the ispG with HindIII. The final plasmid was denoted as pACYC-dxs-ispG-nudF. All primers used for the construction of plasmids are listed in Table 2.
Culture conditions
A 250 mL shaking flask containing 50 mL of semi-defined medium, consisted of M9 minimum salts, 5 g L−1 yeast extract, 10 g L−1 glucose and 1 mM thiamine pyrophosphate (TPP), was utilized for the cultivation of the isopentenol producing strains, chloramphenicol (35 mg mL−1) was also added into the medium. The shaking flask was inoculated with 1 mL of overnight culture and incubated with 150 rpm agitation at 37 C. 0.5 mM Isopropyl D-1-thiogalactopyranoside (IPTG) was added when the optical density (OD600) of the culture reached 0.3 AU. After IPTG addition, the culture was transferred into 30 C shaking incubator (150 rpm) for 48 h of cultivation.
For fermenter scale isopentenol production, a 5 L laboratory reactor containing 2 L medium was used. The 2 L fermentation medium contained M9 minimum salts, 5 g L−1 yeast extract, 20 g L−1 glucose and 1 mM TPP. Chloramphenicol was also added into the fermentation medium at the same time. Inoculant was prepared by introducing a single colony picked from an agar plate into 5 mL of LB medium containing chloramphenicol. The culture was grown at 37 C with 150 rpm agitation. After 12 h, the culture was transferred to 100 mL fresh LB medium containing chloramphenicol and was further cultured for another 12 h. The inoculant was then transferred into the fermentation vessel to initiate the batch fermentation (t = 0 h). When OD600 value reached 3.0 AU, 0.5 mM IPTG was added and the temperature was decreased to 34 C. The agitation speed was controlled by PID to maintain the dissolved oxygen (D.O.) at 20% air saturation. Foaming was controlled by addition of antifoam A whereas the pH was maintained at 7.0 by addition of 2 N H2SO4 or NH4OH (28%).
Biomass analysis
Cellular growth was measured in terms of OD600. For the calculation of biomass production, a standard curve of dry cell weight was correlated with OD600. Samples were collected in 2 mL pre-weighed pre-dried centrifuge tubes and were pelleted at 8,000 g for 10 minutes. After discarding the supernatant, the pellets were washed twice with distilled water and dried at 105 C. One OD600 unit was equivalent to 0.29 g L−1 of dry cell weight.
Metabolite analysis
For glucose analysis, each culture sample was pelleted by centrifugation and the collected supernatant was analyzed in Waters HPLC equipped with a Bio-Rad Aminex HPX-87H Column (300 × 7.8 mm). The eluent (5 mM H2SO4) was pumped at a flow rate of 0.4 mL min−1. The column temperature was maintained at 55 C and the peaks were detected using a Waters 2414 refractive index detector.
For isopentenol analysis, each sample was prepared by ethyl acetate extraction and only the upper layer was subject to GC analysis. The GC equipped with a flame ionization detector (FID) and a CP-FFAP CB capillary column (50 m × 0.25 mm; 0.2 μm film thickness) was applied. The oven temperature was initially held at 50 C for 1 min, then raised with a gradient of 5 C/min until reaching 100 C, and finally programmed to 150 C at 25 C/min, then hold for 5 min. Nitrogen was used as the carrier gas. The injector and detector were held at 250 C and 270 C, respectively. This method can separate isoprenol and prenol efficiently, the isoprenol peak appeared at 13.9 min while the prenol peak appeared at 15.3 min.
Authors contributions
TL, WC and HL designed the study, HL and YW carried out the strain and plasmid construction and the culture experiments, QT and WK participated in the plasmid construction, and HL and TL wrote the manuscript. All authors read and approved the final manuscript.
Additional file
Abbreviations
- pgi:
-
Phosphoglucose isomerase gene
- ispG:
-
HMBPP synthase gene
- dxs:
-
DXP synthase gene
- MEP:
-
2-methyl-(D)-erythritol-4-phosphate
- PPP:
-
Pentose phosphate pathway
- ED:
-
Entner-Doudoroff
- EMP:
-
Embden-Meyerhof-Parnas
- IPP:
-
Isopentenyl pyrophosphate
- DMAPP:
-
Dimethylallyl pyrophosphate
- MVA:
-
Mevalonate-dependent pathway
- G3P:
-
Glyceraldehyde-3-phosphate
- CAI:
-
Codon adaption index
- TPP:
-
Thiamine pyrophosphate
- IPTG:
-
D-1-thiogalactopyranoside
- FID:
-
Flame ionization detector
References
Caspeta L, Nielsen J: Economic and environmental impacts of microbial biodiesel. Nat Biotechnol 2013, 31: 789-793. 10.1038/nbt.2683
Wargacki AJ, Leonard E, Win MN, Regitsky DD, Santos CN, Kim PB, Cooper SR, Raisner RM, Herman A, Sivitz AB, Lakshmanaswamy A, Kashiyama Y, Baker D, Yoshikuni Y: An engineered microbial platform for direct biofuel production from brown macroalgae. Science 2012, 335: 308-313. 10.1126/science.1214547
Heeres AS, Picone CS, van der Wielen LA, Cunha RL, Cuellar MC: Microbial advanced biofuels production: overcoming emulsification challenges for large-scale operation. Trends Biotechnol 2014, 32: 221-229. 10.1016/j.tibtech.2014.02.002
Zeng AP, Sabra W: Microbial production of diols as platform chemicals: recent progresses. Curr Opin Biotechnol 2011, 22: 749-757. 10.1016/j.copbio.2011.05.005
Kim SR, Park YC, ** YS, Seo JH: Strain engineering of Saccharomyces cerevisiae for enhanced xylose metabolism. Biotechnol Adv 2013, 31:851-861.
Nielsen J, Larsson C, Van Maris A, Pronk J: Metabolic engineering of yeast for production of fuels and chemicals. Curr Opin Biotechnol 2013, 24: 398-404. 10.1016/j.copbio.2013.03.023
Buijs NA, Siewers V, Nielsen J: Advanced biofuel production by the yeast Saccharomyces cerevisiae . Curr Opin Chem Biol 2013, 17: 480 488.
Smith KM, Liao JC: An evolutionary strategy for isobutanol production strain development in Escherichia coli . Metab Eng 2011, 13: 674-681.
Matsuda F, Ishii J, Kondo T, Ida K, Tezuka H, Kondo A: Increased isobutanol production in Saccharomyces cerevisiae by eliminating competing pathways and resolving cofactor imbalance. Microb Cell Fact 2013, 12: 119. doi:10.1186/1475-2859-12-119.
Chou HH, Keasling JD: Synthetic pathway for production of five-carbon alcohols from isopentenyl diphosphate. Appl Environ Microbiol 2012, 78: 7849-7855. 10.1128/AEM.01175-12
Withers ST, Gottlieb SS, Lieu B, Newman JD, Keasling JD: Identification of isopentenol biosynthetic genes from Bacillus subtilis by a screening method based on isoprenoid precursor toxicity. Appl Environ Microbiol 2007, 73: 6277-6283.
Zheng Y, Liu Q, Li L, Qin W, Yang J, Zhang H, Jiang X, Cheng T, Liu W, Xu X, **an M: Metabolic engineering of Escherichia coli for high-specificity production of isoprenol and prenol as next generation of biofuels. Biotechnol Biofuels 2013, 6: 54.
Ajikumar PK, **ao WH, Tyo KE, Wang Y, Simeon F, Leonard E, Mucha O, Phon TH, Pfeifer B, Stephanopoulos G: Isoprenoid pathway optimization for Taxol precursor overproduction in Escherichia coli . Science 2010, 330: 70-74.
Misawa N: Pathway engineering for functional isoprenoids. Curr Opin Biotechnol 2011, 22: 627-633. 10.1016/j.copbio.2011.01.002
Zurbriggen A, Kirst H, Melis A: Isoprene production via the Mevalonic acid pathway in Escherichia coli (Bacteria). Bioenerg Res 2012, 5: 814 828.
Yadav VG, De Mey M, Lim CG, Ajikumar PK, Stephanopoulos G: The future of metabolic engineering and synthetic biology: towards a systematic practice. Metab Eng 2012, 14: 233-241. 10.1016/j.ymben.2012.02.001
Puigb P, Guzm n E, Romeu A, Garcia-Vallv S: OPTIMIZER: a web server for optimizing the codon usage of DNA sequences. Nucleic Acids Res 2007, 35: W126-W131. 10.1093/nar/gkm219
Zhou K, Zou R, Stephanopoulos G, Too HP: Metabolite profiling identified methylerythritol cyclodiphosphate efflux as a limiting step in microbial isoprenoid production. PLoS ONE 2012, 7: e47513. 10.1371/journal.pone.0047513
Lv X, Xu H, Yu H: Significantly enhanced production of isoprene by ordered coexpression of genes dxs , dxr , and idi in Escherichia coli . Appl Microbiol Biotechnol 2013, 97: 2357-2365.
Xue J, Ahring BK: Enhancing isoprene production by genetic modification of the 1-deoxy-d-xylulose-5-phosphate pathway in Bacillus subtilis . Appl Environ Microbiol 2011, 77: 2399 2405.
Farmer WR, Liao JC: Precursor balancing for metabolic engineering of lycopene production in Escherichia coli . Biotechnol Prog 2007, 17: 57 61.
Choi HS, Lee SY, Kim TY, Woo HM: In silico identification of gene amplification targets for improvement of lycopene production. Appl Environ Microbiol 2010, 76: 3097-3105. 10.1128/AEM.00115-10
Alper H, ** YS, Moxley JF, Stephanopoulos G: Identifying gene targets for the metabolic engineering of lycopene biosynthesis in Escherichia coli . Metab Eng 2005, 7: 155 164.
Flamholz A, Noor E, Bar-Even A, Liebermeister W, Milo R: Glycolytic strategy as a tradeoff between energy yield and protein cost. Proc Natl Acad Sci U S A 2013, 110: 10039-10044. 10.1073/pnas.1215283110
Peekhaus N, Conway T: What s for dinner?: Entner-Doudoroff metabolism in Escherichia coli . J Bacteriol 1998, 180: 3495 3502.
Zhao J, Li Q, Sun T, Zhu X, Xu H, Tang J, Zhang X, Ma Y: Engineering central metabolic modules of Escherichia coli for improving β-carotene production. Metab Eng 2013, 17: 42 50.
Fong SS, Nanchen A, Palsson BO, Sauer U: Latent pathway activation and increased pathway capacity enable Escherichia coli adaptation to loss of key metabolic enzymes. J Biol Chem 2006, 281: 8024 8033.
Fraenkel DG, Levisohn SR: Glucose and gluconate metabolism in an Escherichia coli mutant lacking phosphoglucose isomerase. J Bacteriol 1967, 93: 1571 1578.
Charusanti P, Conrad TM, Knight EM, Venkataraman K, Fong NL, **e B, Gao Y, Palsson B : Genetic basis of growth adaptation of Escherichia coli after deletion of pgi a major metabolic gene. PLoS Genet 2010, 6: e1001186.
Jiang X, Malley K, Jensen HM, Nanobio: Protocol for gene knockout. URL: ()., [http://openwetware.org/index.php?title=NanoBio:Protocol_for_gene_knockout&oldid=339609]
Acknowledgements
This work was supported by TL s startup fund from the University of Illinois at Urbana-Champaign.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Electronic supplementary material
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Cite this article
Liu, H., Wang, Y., Tang, Q. et al. MEP pathway-mediated isopentenol production in metabolically engineered Escherichia coli . Microb Cell Fact 13, 135 (2014). https://doi.org/10.1186/s12934-014-0135-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12934-014-0135-y