
Abstract
Background
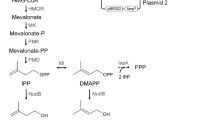
The isopentenols, including isoprenol and prenol, are excellent alternative fuels. However, they are not compounds largely accumulated in natural organism. The need for the next generation of biofuels with better physical and chemical properties impels us to develop biosynthetic routes for the production of isoprenol and prenol from renewable sugar. In this study, we use the heterogenous mevalonate-dependent (MVA) isoprenoid pathway for the synthesis of isopentenyl pyrophosphate (IPP) and dimethylallyl pyrophosphate (DMAPP) intermediates, and then convert IPP and DMAPP to isoprenol and prenol, respectively.
Results
A mevalonate titer of 1.7 g/L was obtained by constructing an efficient MVA upper pathway in engineered E. coli. Different phosphatases and pyrophosphatases were investigated for their abilities in hydrolyzing the IPP and DMAPP. Consequently, ADP-ribose pyrophosphatase was found to be an efficient IPP and DMAPP hydrolase. Moreover, ADP-ribose pyrophosphatase from Bacillus subtilis (BsNudF) exhibited a equivalent substrate specificity towards IPP and DMAPP, while ADP-ribose pyrophosphatase from E. coli (EcNudF) presented a high substrate preference for DMAPP. Without the expression of any phosphatases or pyrophosphatases, a background level of isopentenols was synthesized. When the endogenous pyrophosphatase genes (EcNudF and yggV) that were capable of enhancing the hydrolyzation of the IPP and DMAPP were knocked out, the background level of isopentenols was still obtained. Maybe the synthesized IPP and DMAPP were hydrolyzed by some unknown hydrolases of E. coli. Finally, 1.3 g/L single isoprenol was obtained by blocking the conversion of IPP to DMAPP and employing the BsNudF, and 0.2 g/L ~80% prenol was produced by employing the EcNudF. A maximal yield of 12% was achieved in both isoprenol and prenol producing strains.
Conclusions
To the best of our knowledge, this is the first successful report on high-specificity production of isoprenol and prenol by microbial fermentation. Over 1.3 g/L isoprenol achieved in shake-flask experiments represents a quite encouraging titer of higher alcohols. In addition, the substrate specificities of ADP-ribose pyrophosphatases were determined and successfully applied for the high-specificity synthesis of isoprenol and prenol. Altogether, this work presents a promising strategy for high-specificity production of two excellent biofuels, isoprenol and prenol.
Similar content being viewed by others

Background
With increasing concerns about environmental problems and energy security, research interest has been aroused in the field of microbial production of fuels and chemicals from renewable sources [1–6]. A further yield improvement will be obtained under controlled conditions by using a scalable fermentation system.
Our data also suggests that mevalonate is an important metabolite in determining the production of isoprenol, given that over 80% mevalonate can be converted to isoprenol. It provides another clue to further increase the production of isoprenol.
Compared with the isoprenol production, a much lower titer of prenol was obtained. The lower biomass probably resulted in the decreased prenol production, given the prenol producing strain (YY168) did not grow so well as the strain for isoprenol synthesis (YY159). Though YY168 finally obtained a higher biomass than YY159, YY168 had lower biomasses during the period when the isopentenols were largely produced (Figure 6). To check if the products prenol and isoprenol inhibit the normal growth of E. coli, we measured the growth of E. coli BL21(DE3) in media supplemented with increasing concentrations of exogenous prenol or isoprenol. A slight growth inhibition was indeed observed in the presence of prenol, but the inhibitory effect of prenol on the growth of E. coli was even weaker than that of isoprenol (Figure 7). Moreover, the strain YY168 produced a much lower product titer than YY159, which grew well during the production of isoprenol. Therefore, the growth inhibition of strain YY168 was not caused by the synthesized prenol.

The inhibitory effect of isoprenol and prenol on the growth of E. coli . The E. coli BL21(DE3) was cultured at 37°C in media supplemented with increasing concentrations of exogenous prenol or isoprenol. OD600nm, optical density at 600 nm.
Martin et al. found that, using mevalonate as substrate, the expression of partial MVA lower pathway (MK, MKK and MVD) resulted in the highest concentration of IPP, the expression of entire MVA lower pathway (MK, MKK, MVD and IDI) led to a decreased concentration of IPP, and the expression of an additional synthetic pathway that converts IPP and DMAPP to other metabolites further decreased the intracellular concentration of IPP [7]. That is to say, IPP will largely accumulate in engineered E. coli if it can’t be efficiently converted to other metabolites. In the meanwhile, compared to BsNudF, EcNudF has a much poorer substrate specificity towards IPP, making IPP can’t be efficiently hydrolyzed. Therefore, the lower cell density of YY168 may be attributed to the accumulation of IPP, which is toxic and can inhibit normal cell growth [7].
Conclusions
The engineering strategy described above inaugurates a new realm for the production of alcoholic biofuels. The substrate specificities of ADP-ribose pyrophosphatases were determined and successfully applied for the high-specificity synthesis of isoprenol and prenol. The desired isoprenol and prenol were finally produced with high specificities, being the first successful report on high-specificity production of isoprenol and prenol from renewable sugar. The achieved over 1.3 g/L isoprenol represents an encouraging isopentenol titer. In addition, the isopentenols can be isolated with the well-developed butanol recovery techniques, such as gas strip**. Therefore, this strategy provides the potential and possibility for scale-up production of isopentenols by utilizing the butanol production line without too much reconstruction. Altogether, this work presents a promising strategy for high-specificity production of two excellent biofuels, isoprenol and prenol.
Methods
Plasmid construction
The HMGS [Genbank: NM_001182489] was polymerase chain reaction (PCR)-amplified from genomic DNA of S. cerevisiae (ATCC 204508) with the primer set HMGS-F and HMGS-R. The PCR product digested with NdeI and XhoI was cloned into pACYCDuet-1 (Novagen, Darmstadt, Germany) cut with the same restriction enzymes, creating pISP211. The tHMGR (a truncated version of HMGR) was PCR-amplified from genomic DNA of S. cerevisiae (ATCC 204508) with the primer set tHMGR-F and tHMGR-R [7, 12]. The PCR product digested with NcoI and BamHI was cloned into pISP211 cut with the same restriction enzymes, creating pISP212.
The mvaE [Genbank: AF290092] was PCR-amplified from genomic DNA of Enterococcus faecalis (ATCC 700802D-5) with the primer set mvaE-F and mvaE-R. The PCR product digested with NcoI and BamHI was cloned into pACYCDuet-1 cut with the same restriction enzymes, creating pISP213. The mvaS [Genbank: AF290092] was PCR-amplified from genomic DNA of E. faecalis (ATCC 700802D-5) with the primer set mvaS-F and mvaS-R. The PCR product digested with SacI and PstI was cloned into pISP213 cut with the same restriction enzymes, creating pISP214.
The ERG12, ERG8, ERG19 and IDI1 genes from S. cerevisiae (ATCC 204508) were cloned into pTrcHis2B (Invitrogen, Carlsbad, CA) using a method of successive hybridization to yield pTrcLower [19].
To delete the IDI1 gene from pTrcLower, a fragment containing ERG19, ERG8 and a part of ERG12 was first PCR-amplified from pTrcLower with the primers IDIKO-F and IDIKO-R, then digested with BamHI and PstI, and finally cloned into the pTrcLower cut with the same restriction enzymes. The resultant recombinant plasmid designated pISP9.
The ‘phoA (a leaderless version of phoA [Genbank: NC_000913.2:400971..402386]) was PCR-amplified from genomic DNA of E. coli K12 with the primer set phoA-F2 and phoA-R. The PCR product digested with BglII and XhoI was cloned into pCOLADuet-1 (Novagen, Darmstadt, Germany) cut with the same restriction enzymes, creating pYY11.
The ‘DPP1 (a leaderless version of DPP1 [Genbank: NM_001180592]) was PCR-amplified from genomic DNA of S. cerevisiae (ATCC 204508) with the primer set DPP1-F2 and DPP1-R. The PCR product digested with BglII and XhoI was cloned into pCOLADuet-1 cut with the same restriction enzymes, creating pYY12.
The ‘LPP1 (a leaderless version of LPP1 [Genbank: NM_001180811]) was PCR-amplified from genomic DNA of S. cerevisiae (ATCC 204508) with the primer set LPP1-F2 and LPP1-R. The PCR product digested with BglII and XhoI was cloned into pCOLADuet-1 cut with the same restriction enzymes, creating pYY13.
The phoE [Genbank: AL009126] was PCR-amplified from genomic DNA of Bacillus subtilis (ATCC 23857) with the primer set phoE-F and phoE-R. The PCR product digested with BglII and XhoI was cloned into pCOLADuet-1 cut with the same restriction enzymes, creating pYY14.
The BsNudF [Genbank: AL009126] was PCR-amplified from genomic DNA of B. subtilis (ATCC 23857) with the primer set BsNudF-F and BsNudF-R. The PCR product digested with NcoI and BamHI was cloned into pCOLADuet-1 cut with the same restriction enzymes, creating pYY15.
The EcNudF [Genbank: NC_010473.1:3273048..3273677] was PCR-amplified from genomic DNA of E. coli K12 with the primer set EcNudF-F and EcNudF-R. The PCR product digested with NcoI and BamHI was cloned into pCOLADuet-1 cut with the same restriction enzymes, creating pYY16.
The nudC, yggV, hisl, lpxH, ppa and cdh were PCR-amplified from genomic DNA of E. coli K12 with the primer set nudC-F/nudC-R, yggV-F/yggV-R, hisl-F/hisl-R, lpxH-F/lpxH-R, ppa-F/ppa-R and cdh-F/cdh-R, respectively. The PCR products digested with NcoI and BamHI were cloned into pCOLADuet-1 cut with the same restriction enzymes, creating pYY26, pYY36, pYY46, pYY56, pYY66 and pYY76, respectively.
All the plasmids and strains used in this work are listed in Table 1, and the oligonucleotide primers are given in Table 2.
Site-directed mutagenesis
A method based on the amplification of the entire plasmid using primers that include the desired changes was employed for the site-directed mutagenesis [25]. The Ala-110 of HMG-CoA synthase MvaS was mutated to Gly by replacing a nucleotide C with G using the mutant primers A110G-F and A110G-R.
The PrimeSTAR HS DNA polymerase (Takara) was used for PCR. The PCR conditions were as follows: 98°C for 30 sec; 12 × (98°C for 5 sec; 63°C for 5 sec; 72°C for 8 min); 72°C for 10 min. The mutant was verified by sequencing (BGI).
Gene knockout and λDE3 lysogenization
The EcNudF gene of E. coli was knocked out using the one-step inactivation method previously reported [26]. This method includes following steps: amplification of the kanamycin-resistant (KanR) gene with PCR using pKD4 as template. The PCR product was transformed into the cells by electro-poration after gel purification. Transformants were selected with kanamycin-resistance plate. The mutant was verified by PCR using the test primers KOnudF-VF/KOnudF-VR. The kanamycin cassette was removed with the helper plasmid pCP20 that expresses FLP. The E. coli DNF was finally obtained after removing the helper plasmids.
To express target genes cloned in vectors under the control of the T7 promoter, λDE3 prophage was integrated into the E. coli DNF chromosome using the λDE3 lysogenization kit (Novagen, Darmstadt, Germany) according to the manufacturer’s instructions. The obtained strain was designated as E. coli DNF (DE3). The yggV gene was then deleted with the same method using E. coli DNF (DE3) as the starting strain. The resultant double-gene knockout mutant was designated as E. coli DNFYV (DE3).
Bacterial strains, media and growth conditions
The bacterial strains used in this study are listed in Table 1. E. coli BL21(DE3) (Invitrogen, Carlsbad, CA) was used as the host to overproduce proteins. During strain construction, cultures were grown aerobically at 37°C in Luria Broth (10 g/L tryptone, 10 g/L NaCl, and 5 g/L yeast extract). Kanamycin (50 mg/L), Ampicillin (100 mg/L) or chloramphenicol (34 mg/L) was added if necessary. For initial production experiments in shake flasks, strains were grown in a medium consisted of the following: 7.5 g/L K2HPO4 · 3H2O, 2.1 g/L citric acid monohydrate, 0.3 g/L ferric ammonium citrate, 2.92 g/L (NH4)2SO4, 30 g/L of glucose, 9 g/L beef extract, 4 mM MgSO4, trace metals mix (2.86 mg/L H3BO3, 1.81 mg/L MnCl2 · 4H2O, 0.222 mg/L ZnSO4 · 7H2O, 0.39 mg/L Na2MoO4 · 2H2O, 0.079 mg/L CuSO4 · 5H2O, 49.4 μg/L Co(NO3)2 · 6H2O). The residual glucose was measured by the SBA-40D Biosensor equipped with glucose oxidase membrane electrodes (Shandong Academy of Sciences, **an, China). Protein production was induced with 0.5 mM isopropyl β-D-thiogalactoside (IPTG) at 30°C.
Determination of mevalonate
Ten milliliter cell free cultures were collected, adjusted to pH 2.0 using hydrochloric acid, and incubated at 45°C for 1 hour. Then 5 g of anhydrous granular sodium sulphate was added to each vial followed by 10 ml of ethyl acetate. The vials were mixed on a vortexer for 5 min and the phases were allowed to separate naturally. One milliliter supernatant was collected for gas chromatograph (GC) analysis. The separation of mevolonatone lactone was performed using a CP-FFAP CB capillary column (25 m × 0.25 mm; 0.2 μm film thickness) purchased from Agilent Technologies (Santa Clara, CA). The oven temperature was initially held at 150°C for 1 min, then raised with a gradient of 10°C/min until reaching 250°C, and finally held at 250°C for 10 min. Nitrogen was used as the carrier gas. The injector and detector were held at 250°C and 270°C, respectively.
Analysis of isoprenol and prenol by GC-MS
Isoprenol and prenol produced by the engineered strains were identified by GC–MS. The system consisted of model 7890A network GC system (Agilent Technologies) and a model 5975C network mass selective detector (Agilent Technologies, Santa Clara, CA). A HP-INNOWAX capillary column (30 m × 0.25 mm; 0.25 μm film thickness; Agilent Technologies) was used, with helium as the carrier gas. The following oven temperature program was carried out: 100°C for 1 min, increase of 5°C/min to 100°C, then programmed from 100°C to 200°C at 25°C/min. The injector was maintained at 250°C. Alcohol compounds were isolated by ethyl acetate extraction. A 1 μl sample was injected in split injection mode with a 20:1 split ratio.
Analysis of isoprenol and prenol by GC–FID
The produced isoprenol and prenol were quantified by a GC equipped with flame ionization detector (FID). The separation of isoprenol and prenol was performed using a CP-FFAP CB capillary column (25 m × 0.25 mm; 0.2 μm film thickness) purchased from Agilent Technologies (Santa Clara, CA). The oven temperature was initially held at 50°C for 1 min, then raised with a gradient of 5°C/min until reaching 100°C, and finally programmed to 150°C at 25°C/min. Nitrogen was used as the carrier gas. The injector and detector were held at 250°C and 270°C, respectively. Samples were prepared by ethyl acetate extraction. Isoamyl alcohol was added into the samples as the internal standard before solvent extraction.
Abbreviations
- IPP:
-
Isopentenyl pyrophosphate
- DMAPP:
-
Dimethylallyl pyrophosphate
- IPTG:
-
Isopropyl β-D-thiogalactoside
- PCR:
-
Polymerase chain reaction
- GC:
-
Gas chromatography
- FID:
-
Flame ionization detector
- GC-MS:
-
Gas chromatography–mass spectrometry.
References
Atsumi S, Hanai T, Liao JC: Non-fermentative pathways for synthesis of branched-chain higher alcohols as biofuels. Nature 2008, 451: 86-89. 10.1038/nature06450
Steen EJ, Kang Y, Bokinsky G, Hu Z, Schirmer A, McClure A, del Cardayre SB, Keasling JD: Microbial production of fatty-acid-derived fuels and chemicals from plant biomass. Nature 2010, 463: 559-562. 10.1038/nature08721
Bond-Watts BB, Bellerose RJ, Chang MCY: Enzyme mechanism as a kinetic control element for designing synthetic biofuel pathways. Nat Chem Biol 2011, 7: 222-227. 10.1038/nchembio.537
Zheng Y, Li L, Liu Q, Yang J, Cao Y, Jiang X, Zhao G, **an M: Boosting the free fatty acid synthesis of Escherichia coli by expression of a cytosolic Acinetobacter baylyi thioesterase. Biotechnol Biofuels 2012, 5: 76. 10.1186/1754-6834-5-76
Koppram R, Nielsen F, Albers E, Lambert A, Wännström S, Welin L, Zacchi G, Olsson L: Simultaneous saccharification and co-fermentation for bioethanol production using corncobs at lab, PDU and demo scales. Biotechnol Biofuels 2013, 6: 2. 10.1186/1754-6834-6-2
Connor MR, Cann AF, Liao JC: 3-Methyl-1-butanol production in Escherichia coli : random mutagenesis and two-phase fermentation. Appl Microbiol Biotechnol 2010, 86: 1155-1164. 10.1007/s00253-009-2401-1
Martin VJ, Pitera DJ, Withers ST, Newman JD, Keasling JD: Engineering a mevalonate pathway in Escherichia coli for production of terpenoids. Nat Biotechnol 2003, 21: 796-802. 10.1038/nbt833
Ajikumar PK, **ao WH, Tyo KEJ, Wang Y, Simeon F, Leonard E, Mucha O, Phon TH, Pfeifer B, Stephanopoulos G: Isoprenoid pathway optimization for taxol precursor overproduction in Escherichia coli . Science 2010, 330: 70-74. 10.1126/science.1191652
Bonanno JB: Structural genomics of enzymes involved in sterol/isoprenoid biosynthesis. Proc Natl Acad Sci USA 2001, 98: 12896-12901. 10.1073/pnas.181466998
Wu S, Schalk M, Clark A, Miles RB, Coates R, Chappell J: Redirection of cytosolic or plastidic isoprenoid precursors elevates terpene production in plants. Nat Biotechnol 2006, 24: 1441-1447. 10.1038/nbt1251
Agranoff BW, Eggerer H, Henning U, Lynen F: Biosynthesis of terpenes. VII. Isopentenyl pyrophosphate isomerase. J Biol Chem 1960, 235: 326-332.
Polakowski T, Stahl U, Lang C: Overexpression of a cytosolic hydroxymethylglutaryl-CoA reductase leads to squalene accumulation in yeast. Appl Microbiol Biotechnol 1998, 49: 66-71. 10.1007/s002530051138
Tabata K, Hashimoto S: Production of mevalonate by a metabolically-engineered Escherichia coli . Biotechnol Lett 2004, 26: 1487-1491.
Wilding EI, Brown JR, Bryant AP, Chalker AF, Holmes DJ, Ingraham KA, Iordanescu S, So CY, Rosenberg M, Gwynn MN: Identification, evolution, and essentiality of the mevalonate pathway for isopentenyl diphosphate biosynthesis in gram-positive cocci. J Bacteriol 2000, 182: 4319-4327. 10.1128/JB.182.15.4319-4327.2000
Campos N, Rodríguez-Concepción M, Sauret-Güeto S, Gallego F, Lois LM, Boronat A: Escherichia coli engineered to synthesize isopentenyl diphosphate and dimethylallyl diphosphate from mevalonate : a novel system for the genetic analysis of the 2-C-methyl-D-erythritol 4-phosphate pathway for isoprenoid biosynthesis. Biochem J 2001, 353: 59-67.
Toth MJ, Huwyler L: Molecular cloning and expression of the cDNAs encoding human and yeast mevalonate pyrophosphate decarboxylase. J Biol Chem 1996, 271: 7895-7898. 10.1074/jbc.271.14.7895
Tsay YH, Robinson GW: Cloning and characterization of ERG8 , an essential gene of Saccharomyces cerevisiae that encodes phosphomevalonate kinase. Mol Cell Biol 1991, 11: 620-631.
Oulmouden A, Karst F: Nucleotide sequence of the ERG12 gene of Saccharomyces cerevisiae encoding mevalonate kinase. Curr Genet 1991, 19: 9-14. 10.1007/BF00362081
Jiang X, Yang J, Zhang H, Zou H, Wang C, **an M: In vitro assembly of multiple DNA fragments using successive hybridization. PLoS One 2012, 7: e30267. 10.1371/journal.pone.0030267
Tokuhiro K, Muramatsu M, Ohto C, Kawaguchi T, Obata S, Muramoto N, Hirai M, Takahashi H, Kondo A, Sakuradani E, Shimizu S: Overproduction of geranylgeraniol by metabolically engineered Saccharomyces cerevisiae . Appl Environ Microbiol 2009, 75: 5536-5543. 10.1128/AEM.00277-09
Kim EE, Wyckoff HW: Reaction mechanism of alkaline phosphatase based on crystal structures: Two-metal ion catalysis. J Mol Biol 1991, 218: 449-464. 10.1016/0022-2836(91)90724-K
Withers ST, Gottlieb SS, Lieu B, Newman JD, Keasling JD: Identification of isopentenol biosynthetic genes from Bacillus subtilis by a screening method based on isoprenoid precursor toxicity. Appl Environ Microb 2007, 73: 6277-6283. 10.1128/AEM.00861-07
Steussy CN, Robison AD, Tetrick AM, Knight JT, Rodwell VW, Stauffacher CV, Sutherlin AL: A structural limitation on enzyme activity: the case of HMG-CoA synthase. Biochemistry 2006, 45: 14407-14414. 10.1021/bi061505q
Hayashi K, Morooka N, Yamamoto Y, Fujita K, Isono K, Choi S, Ohtsubo E, Baba T, Wanner BL, Mori H, Horiuchi T: Highly accurate genome sequences of Escherichia coli K-12 strains MG1655 and W3110. Mol Syst Biol 2006, 2: 2006. 0007
Hemsley A, Arnheim N, Toney MD, Cortopassi G, Galas DJ: A simple method for site-directed mutagenesis using the polymerase chain reaction. Nucleic Acids Res 1989, 17: 6545-6551. 10.1093/nar/17.16.6545
Datsenko KA, Wanner BL: One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci U S A 2000, 97: 6640-6645. 10.1073/pnas.120163297
Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (Grant No. 21202179; Grant No. 21206185), the Knowledge Innovation Program of the Chinese Academy of Sciences (Grant No. Y112131105), the National High-tech R&D Program of China (863 Program) (Grant No. SS2013AA050703-2), and the National Science and Technology Development Program for Rural Areas during the 12th Five-Year Plan Period (Grant No. 2012BAD32B06-2).
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
YZ designed the research and prepared the manuscript. MX and WQ helped to revise the manuscript. YZ, QL and LL did the lab work, plasmid construction, site-directed mutagenesis, strain cultivation and product detection. JY, HZ, XJ and TC did some work in plasmid construction. WL and XX did some work in product detection. All authors read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Zheng, Y., Liu, Q., Li, L. et al. Metabolic engineering of Escherichia coli for high-specificity production of isoprenol and prenol as next generation of biofuels. Biotechnol Biofuels 6, 57 (2013). https://doi.org/10.1186/1754-6834-6-57
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1754-6834-6-57