
Abstract
Drug delivery systems (DDS) have recently emerged as a promising approach for the unique advantages of drug protection and targeted delivery. However, the access of nanoparticles/drugs to the central nervous system (CNS) remains a challenge mainly due to the obstruction from brain barriers. Immune cells infiltrating the CNS in the pathological state have inspired the development of strategies for CNS foundation drug delivery. Herein, we outline the three major brain barriers in the CNS and the mechanisms by which immune cells migrate across the blood–brain barrier. We subsequently review biomimetic strategies utilizing immune cell-based nanoparticles for the delivery of nanoparticles/drugs to the CNS, as well as recent progress in rationally engineering immune cell-based DDS for CNS diseases. Finally, we discuss the challenges and opportunities of immune cell-based DDS in CNS diseases to promote their clinical development.
Similar content being viewed by others
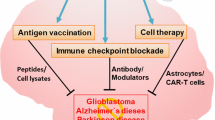

Background
According to global statistics, central nervous system (CNS) diseases, such as gliomas, stroke, Alzheimer’s disease (AD), Parkinson’s disease (PD), multiple sclerosis (MS), epilepsy, and several others diseases, are the leading causes of disability worldwide, accounting for approximately 12% of total deaths. Among these conditions, AD and PD stand out as the most prevalent neurodegenerative diseases [1]; meanwhile, stroke ranks second in terms of mortality rates [2]. Therefore, there is an urgent demand for enhanced management and treatment strategies for CNS diseases. However, due to the presence of CNS barriers, nearly 98% of small-molecule drugs and all large-molecule drugs are routinely excluded from reaching the brain [3]. This predicament has forced researchers to explore more effective treatments. Nevertheless, the lack of success in most preclinical and clinical studies conducted so far has highlighted great challenges in the clinical application of drugs targeting CNS diseases, including inadequate drug selectivity, limited permeability across the low blood–brain barrier (BBB), and/or rapid elimination.
The advantages of drug delivery systems (DDS) in tissue and/or cell-targeted drug delivery, reduction of system cytotoxicity, prolongation of drug half-life, enhancement of dispersion, and biocompatibility of insoluble drugs have received increasing attention for several decades. In CNS diseases, many strategies based on nanoparticles have been developed to enhance the efficacy of delivering therapeutic drugs to the brain, including receptor-mediated transcytosis (utilizing specific ligands such as lactoferrin, transferrin, and certain antibodies), adsorptive-mediated transcytosis (employing cation nanoparticles), and carrier-mediated transport (modified with fatty acids, glucose, galactose, or mannose) [4, 5]. However, there remains much room for improvement (the incomplete specificity of nanoparticles, the toxicity of cation nanoparticles, the breakdown of the BBB, and so on). A deeper understanding of the microenvironment in CNS diseases, coupled with advancements in nanomedicine, has paved the way for bionic nanotherapeutics to emerge as a potential drug delivery strategy for the treatment of CNS disorders. Recently, the immune cells has been observed to migrate specifically to the brain parenchyma in CNS disease, which has sparked interest in develo** immune cell-based nanoparticles for drug delivery to the CNS. The reemergence of intrinsic biological properties of the source cells, such as directed migration and the immune cell-based nanoparticles cloaking, offers the potential for effective drug delivery to the CNS (Fig. 1).

Graphical overview of the process toward immune cell-inspired nanoparticles for the treatment of central nervous system (CNS) diseases. a There are many nervous system diseases, including gliomas, ischemic stroke, Alzheimer’s disease (AD), Parkinson’s disease (PD), and several others. b The delivery of therapeutic drugs is hindered by barriers. c Peripheral immune cells migrate into CNS in the pathological state. d The migration of immune cells promotes the development of immune cell-inspired nanoparticles. e The immune cell-inspired nanoparticles provide a chance for drug delivery to the CNS
The objective of this article is to provide accessible insights into the relationship between immune cells capable of crossing brain barriers and the delivery of therapeutic drugs in the CNS. Following an overview of the anatomy, physiology, and pathology of the brain barrier, we investigate the role of peripheral immune cell involvement in CNS homeostasis and disease as well as mechanisms underlying interactions between immune cells and CNS. We subsequently explore the strategies for using immune cell-inspired DDS on CNS cells and tissues, along with their application in treating CNS diseases. Finally, from a neuroimmunology perspective, we examine current developments in immune cell-based DDS and their potential future regulation of the microenvironment associated with CNS disease. This review aims to summarize various aspects related to migration and roles played by immune cells across different diseases while highlighting promising approaches for leveraging these cells to deliver drugs to the CNS.
The infiltration of immune cells into the brain
Enhanced understanding of the fundamental physiological mechanisms of the CNS is imperative for the advancement of immune cell-based CNS DDS. This section describes the physical structure of the CNS, including complex barrier systems responsible for maintaining homeostasis within this vital region, as well as the mechanisms through which immune cells migrate across the CNS.
CNS structure
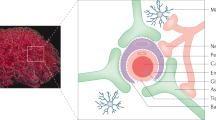
The CNS has traditionally been considered a site of immune privilege owing to its distinctive anatomical features. The meninges and cerebrospinal fluid (CSF) serve as protective barriers for the brain and spinal cord. There are three main physiological barriers between the peripheral blood and CNS, namely, the blood-leptomeningeal barrier, the blood-CSF barrier, and the BBB. These barriers play a crucial role in regulating the transportation of cells and molecules (Fig. 2). The meninges are anatomically divided into three layers: dura mater, arachnoid mater, and pia mater (Fig. 2a) [6, 7]. The dura mater contains lymphatics and fenestrated blood vessels without tight junctions (TJ), facilitating the entry of peripheral material and cells [6]. The arachnoid mater acts as an epithelial layer between the dura mater and the subarachnoid space. Its TJ and efflux pumps establish a barrier that separates the peripheral blood vessels of dura mater from CSF while filling the subarachnoid space [6]. The pia mater, which constitutes the innermost layer covering the surface of the brain and spinal cord, exhibits semipermeable to CSF allowing for soluble substances to pass through along with immune cells. This characteristic is crucial for CSF mixing with brain interstitial fluid, thereby exposing immune cells to CNS antigens [8]. The component of CSF is related to the choroid plexus, which is the primary site for CSF production. Like the dura mater, the vessels in the choroid plexus are fenestrated and without TJ. During homeostasis, the choroid plexus contains resident immune cells, including dendritic cells (DCs), macrophages, innate lymphoid cells, and CD4+ T cells, as well [7]. Similar to the arachnoid mater, ependymal cells surrounding the choroid plexus form a blood-CSF barrier between blood vessels and CSF (Fig. 2b), regulating the entry of immune cells and soluble factors into CSF through TJ [6]. Lastly, in CNS parenchyma, there exists a BBB that directly separates blood vessels from adjacent CNS tissue (Fig. 2c) [6]. The BBB is comprised of specialized endothelial cells, endothelial basement membrane, pericytes, astrocyte basement membrane, and astrocyte end feet. These specialized endothelial cells are tightly connected by claudins, occludins, annexin-1, and junction adhesives to form TJ, which are distinct from peripheral vessels. The BBB functions as the natural selective biochemical barrier between blood and CNS. It facilitates the transport of essential nutrients such as glucose, amino acids, vitamins, free fatty acids, minerals, and electrolytes from the bloodstream to the CNS by special transporters, maintaining CNS homeostasis by utilizing efflux pumps like p-glycoprotein to prevent harmful agents such as pathogens from entering the CNS. Within postcapillary microvessels, the endothelial basement membrane separates from the glial cell boundary, resulting in the formation of a perivascular space alongside a limited presence of antigen-presenting cells. The cerebral cortex exhibits a tight association between the endothelial basement membrane, glial cell boundaries, and specialized endothelial cells, with no presence of perivascular space. Under steady-state conditions, immune cells face challenges in accessing the brain parenchyma from the bloodstream due to the requirement of crossing both the vascular endothelium and glial junctions. Additionally, low expression levels of endothelial cell adhesion molecules further restrict peripheral immune cell entry into the CNS parenchyma. These inherent physiological barriers have evolved specialized mechanisms to ensure precise transmission of electrochemical signals within the CNS and maintain a balanced physical environment by regulating substance entry and exit. However, these tightly integrated barriers pose significant difficulties for drugs to reach the CNS.

Interactions between the peripheral immune cells and CNS
The CNS is considered to be an immune-privileged organ, where the BBB, blood-CSF, and blood-leptomeningeal barriers tightly regulate the entry of immune cells into different compartments of the CNS. Under normal conditions, leukocytes such as granulocytes, T cells, and B cells remain within the blood vessels and typically do not penetrate healthy brain tissue. The primary immune defense during homeostasis is provided by innate immune cells, including parenchymal microglia and non-parenchymal macrophages [9].
In pathological states, the CNS microenvironment changes, leading to the production of pro-inflammatory cytokines, chemokines, and adhesion molecules, which facilitate the recruitment of circulating leukocytes across the BBB [10]. Concurrently, the BBB becomes disrupted, allowing peripheral immune cells to access the diseased brain [10]. There are three pathways through which immune cells can reach the CNS: CNS parenchymal blood vessels, leptomeningeal blood vessels, and the choroid plexus [11]. The migration of immune cells across the BBB is a complex process involving various molecules, including adhesion, activation, and migration proteins expressed on the BBB endothelial cells and/or migrating immune cells. Specifically, adhesion molecules such as intracellular adhesion molecule (ICAM)-1, ICAM-2, vascular cell adhesion molecule-1 (VCAM-1), and P-selectin which are expressed on endothelial cells, and their ligands like lymphocyte function-associated antigen-1 (LFA-1), very late antigen-4 (VLA-4), P-selectin glycoprotein ligand-1 (PSGL-1) play central roles in facilitating immune cell migration across the BBB. Additionally, chemokines such as C–C motif chemokine ligand (CCL) 2, CCL4, CCL5, and C-X-C motif chemokine ligand 12 (CXCL12) and their respective receptors CC-chemokine receptor (CCR) 1, CCR2, CCR5, and chemokine (C-X-C motif) receptor 4/7 (CXCR4/7) are also involved in the migration process [12].
The process of recruiting immune cells can be broken down into four distinct steps, as illustrated in Fig. 3 [13]. (1) The first step involves a process known as rolling, which serves to decrease the speed of immune cells to facilitate their recognition of proteoglycans expressed in endothelial cells [14]. The upregulation of E/P-selectin on endothelial cells facilitates interaction with PSGL-1 on immune cells, although this step is not essential for immune cell migration across the inflamed BBB. (2) The second step, arrest, is initiated by G-protein-coupled receptor signaling and involves the binding of integrins LFA-1, vVLA-4, and α4β1 integrin to their endothelial ligands, ICAM-1 and VCAM-1 [15]. (3) The third step, polarization and crawling, entails activated immune cells crawling against the flow direction on ICAM-1-coated surfaces to locate rare permissive sites for diapedesis across the BBB endothelium. (4) Finally, the fourth step, diapedesis, involves immune cells promptly crossing the endothelium through endothelial junctions, known as paracellular diapedesis, as well as transcellular immune cell diapedesis in the migration of immune cells across the BBB.

The process of immune cell migration across the blood–brain barrier (BBB) can be delineated as a series of consecutive stages. a Rolling. Immune cells engage with the endothelium through the interaction between PSGL-1 and P-selectin in the inflamed endothelial cells. b Arrest. The immune cells are activated upon transient contact with the endothelium, resulting in their firm adhesion to the endothelium. c Crawling. The immune cells polarize and crawl to locate permissive sites. d Diapedesis. The ultimate step is in which the immune cells traverse the barrier through either a paracellular (across the tight junctions) or transcellular pathway. This figure is a modification of Fig. 4 by Mastorakos et al. [6]. CXCL12 C-X-C motif chemokine ligand 12, CXCR4 chemokine (C-X-C motif) receptor 4, ICAM intracellular adhesion molecule, IL-1β interleukin-1β, LFA lymphocyte function-associated antigen, PSGL-1 P-selectin glycoprotein ligand-1, TNF-α tumor necrosis factor-α, VCAM vascular cell adhesion molecule, VLA very late antigen
In a variety of CNS disorders, distinct brain microenvironments give rise to diverse molecular mechanisms for the attraction of peripheral immune cells to the CNS. For instance, the adhesion of neutrophils to endothelial cells is typically mediated by P-selectin, but in the context of cerebral ischemia, the involvement of PSGL-1 in the accumulation of neutrophils in the CNS is disregarded. Furthermore, the quantity and nature of the recruited peripheral immune cells are intricately linked to the specific diseases and will be detailed in the subsequent section.
Immune-inspired nanoparticle delivery to the CNS
The application of immune cell components in DDS
Cells
Biological cells have been recognized as “living drugs” due to their inherent characteristics, such as strong biocompatibility and dynamic response to disease, including immunomodulation, tissue regeneration, and tumor destruction. Numerous cell therapy products, such as T cells, stem cells, and DCs, have been identified and approved for clinical use [16], with at least 1700 cell therapy products currently undergoing clinical trials [16]. Immune cell-based clinical trials account for over 60% of active cell therapy clinical trials. cells have been utilized as carriers to transport pharmaceutical cargo, such as drugs, mRNA, and peptides, to targeted sites, leveraging their natural capacities to target tissues, transport biological barriers, and reduce immunogenicity for mononuclear phagocyte system escape [17]. The mechanisms by which cells act as carriers include backpacks, hitchhiking, and Trojan horses (Fig. 4a) [18].

The utilization of immune cells as a delivery system for central nervous system drugs has been explored through various approaches. These include the direct use of immune cells as transport vehicles (a), the development of immune cell membrane-camouflaged nanoparticles (b), and the creation of exosome-loaded nanoparticles/drugs (c). NPs nanoparticles
Depending on their physical characteristics, cellular backpacks can be categorized into two types: spherical structures and anisotropic shapes. Phagocytosis is a natural behavior of cells such as macrophages, neutrophils, and monocytes, and the geometry of particles plays a decisive role in their phagocytic fate. Anisotropically shaped particles can resist phagocytosis for longer periods compared to spherical structures [19]. Typically, cellular backpacks are polymer layers with diameters ranging from 7 to 10 μm and exhibit anisotropic shapes, enabling them to adhere to cells (Fig. 4a) [20,21,22,23]. The anisotropic shapes of cellular backpacks can hinder their cellular uptake by inhibiting the formation of actin structures necessary for phagocytosis, thereby decreasing the endosomal degradation of polymeric particles [19]. Preserving the biological functions of the cell-carriers, including their tropism, metabolic activities, and responsiveness to disease, is of paramount importance [24]. The assembled heterostructure is comprised of a cell-adhesive region, a payload region, and a release region [25]. It is important for the release region to be rapidly degraded under specific conditions, such as low pH and special temperatures [25]. Once the release region is degraded, the payload layers are exposed directly to the cellular environment, allowing the unloading of cargo such as drugs, proteins, or nanoparticles. The cell-adhesive region is responsible for anchoring the assembled heterostructure to the cell membrane, so it is essential to consider the surface features of the attached cells. Furthermore, it is imperative that the desirable cellular backpacks do not impact the functions of the attached cells, including their ability to migrate into the brain and their immunoregulation functions [20].
The term “hitchhiking nanoparticles” refers to the nanoparticles that directly attach to the membrane of carrier cells (Fig. 4a) [26, 27]. Similar to cellular backpacks, these hitchhiking nanoparticles are secured to the cell surface through receptor–ligand recognition, chemical bonding, or physical adhesion [27]. Different attachment methods offer distinct characteristics. For instance, hitchhiking nanoparticles attached to the cell-carriers through physical adhesion, such as electrostatic interactions, hydrophobic interaction, van der Waals forces, and hydrogen bonding [28,29,30,31], require minimal modification. While the weak interaction between hitchhiking nanoparticles and cell-carriers results in limited anchoring stability in circulation, the abundance of receptors on the cell-carriers provides a reliable, reproducible, and straightforward attachment method for the nanoparticles. Moreover, by modifying the ligands on the hitchhiking nanoparticles, multiple cells can be used as potential carriers. However, the numerous receptors on various cells can lead to non-specific attachment [17]. Additionally, the interaction between receptor-ligand may disrupt the biological functions of the cell-carriers [27]. The plentiful proteins on the cell surface offer numerous active groups, such as amines and thiols, for hitchhiking nanoparticles [32, 72, 73]. For example, the magnetic beads enrichment method employs magnetic nanoparticles attached to antibodies targeting exosomal biomarkers to separate exosomes with high efficiency and low cost [64]. The advantages and disadvantages of different isolation methods are summarized in Table 1 [74].
Exosomes, with their distinctive structure comprising an aqueous core and a lipid bilayer, have the capability to encapsulate both hydrophilic drugs and hydrophobic molecules via pre-loading or post-loading methods (Fig. 4c). Pre-loading can be achieved through intracellular expression of biological cargos, such as peptides and proteins, via cell transfection, or by uptake of therapeutic molecules into the originating cells by pre-incubation [75]. These cargos and drugs can be integrated into exosomes during the biogenesis process [75]. In addition, post-loading involves a simple and direct loading method through incubation, depending on passive diffusion to incorporate the target drug into the exosomes [67, 76]. The efficiency of loading is contingent upon drug characteristics such as solubility and pH and is constrained by the cellular tolerance of the dose. Furthermore, the separation of unloaded drugs poses a hindrance to its application. To enhance drug loading efficiency, mechanical or chemical techniques such as sonication, extrusions, electroporation, freeze–thaw cycles, and permeabilization (Saponin) are used to open the exosome membranes, allowing for increased drug diffusion into the purified exosomes [77]. While these technologies indeed improve drug loading efficiency, they may compromise the structural integrity of the exosome membrane, potentially leading to drug leakage in vivo. Meanwhile, compared to pre-loading and incubation methods, the complex technologies involved in the post-loading method lead to the introduction of more uncontrollable factors in the construction process.
Exosomes sourced from immune cells and stem cells, among others, are applied in CNS diseases [78, 79]. The exosomes derived from different cells exhibit distinct distribution characteristics in the brains of mice due to variations in the molecules expressed on their surface. To identify superior carriers for treating neurodegenerative disorders, the physicochemical properties, ability to cross the BBB, and accumulation in neuronal cells of exosomes from three types of origin cells, including macrophages (mEVs), neurons (nEVs), and astrocytes (aEVs), were investigated. Among these, the brain accumulation levels of mEVs in a transgenic mouse model of PD were significantly higher than those of nEVs or aEVs, potentially attributed to the highest levels of tetraspanins and integrins in mEVs compared to nEVs and aEVs [80]. Consequently, mEVs were proposed as the most promising nanocarrier system for drug delivery to the brain [80, 81]. However, there are challenges to be addressed for the future application of exosomes in the clinic, including the standardization of isolation techniques with low-cost and efficient drug-loading methodologies.
Immune-inspired nanoparticles in CNS disease
Glioma
Gliomas represent the most prevalent primary malignant tumors of the CNS. While surgical resection is a standard therapeutic approach, complete removal of tumor cells infiltrating the normal brain tissue is unattainable. Consequently, residual tumor cells are typically managed with pharmacotherapy. However, the BBB or the blood–brain tumor barrier hinders the effective delivery of chemotherapy agents, leading to tumor recurrence. The glioma microenvironment significantly influences tumor initiation and progression. Immunohistochemical analysis of human gliomas has revealed substantial infiltration of immune cells [82]. Tumor cells, endothelial cells, immune cells, and various cytokines collectively constitute the glioma tumor microenvironment (TME). Infiltrating immune cells, such as macrophages, microglia, neutrophils, regulatory T cells (Tregs), myeloid-derived suppressor cells, T lymphocytes, natural killer (NK) cells, DCs, etc., play an important role in regulating immune responses within the microenvironment (Fig. 5) [83,84,85] and have spurred the development of immune cell-related biomimetic DDS (Table 2) [86,87,88,89,90,91,92,93,94,95,96,97,98,99,100,101,102,103,104,105,106,107,108,109,110,111,112].

The involvement of immune cells in glioma and their roles as well as potential applications in immune cell-based drug delivery systems (DDS). This includes macrophage and microglia (a), neutrophil (b), natural killer (NK) cell (c), and dendritic cell (DC, d)
Macrophage
An abundance of tumor-associated macrophages (TAMs), including macrophages, monocytes, and microglia, are observed in the gliomas of experimental animals and patients’ biopsies. Microglia and macrophages have distinct cellular origins. Microglia originates from immature yolk sac progenitors and expresses CCR2−, CX3CR1high, CD11b+, F4/80+, and CD45low markers. While monocytes are generated from hematopoietic stem cells that differentiate into granulocyte macrophage progenitors, and then into monocyte-DC progenitors. Subsequently, mature Ly6Chigh, CCR2+ CX3CR1low/int inflammatory monocytes are released into circulation to colonize peripheral organs under both normal and inflammatory conditions. In gliomas, these monocytes infiltrate into the CNS and differentiate into TAMs, expressing CX3CR1, CCR2, CD45high, F4/80+, and CD11b+ [113].
In the 1990s, researchers found that exogenous macrophages would migrate toward inflammatory body regions or reticuloendothelial organs for elimination [114]. Additionally, a study suggested that the migratory capacity of paramagnetic nanoparticles ingested by monocytes to cross a brain endothelial monolayer was unaffected [115]. Subsequently, Valable et al. [116] demonstrated that intravenously injected micrometer-sized particles of iron-oxide-labeled Mo/Ma could target a brain tumor by magnetic resonance imaging tracking in vivo, leading to the development of TAMs as carriers of drugs, nanoparticles, or photosensitizers to target tumors and tissues surrounding tumor by the Trojan horse strategy [86,87,88,89,90,91,92,93]. The uptake capacity of TAMs is critical to drug delivery, determined by the surface properties and morphology of nanoparticles (cargos) [117, 118]. For example, the uptake efficiency of bare gold–silica nanoshells by macrophages was 4 times that of PEGylated gold–silica nanoshells [117]. Furthermore, gold–silica nanorods were more likely to be ingested by macrophages than gold–silica nanoshells. Moreover, the uptake efficiency of macrophages was associated with its phenotype [86]. It is crucial to improve therapeutic effects on glioma through the promotion of drug offloading from macrophage carriers and the absorption of drugs by tumor cells after the migration of TAM carriers into CNS employing the driver of inflammatory cytokines [89, 90].
Utilizing the Trojan horse strategy, the nanoparticles can enter the brain. Miao et al. [91] prepared a prodrug by attaching an anticancer drug (temozolomide) to β-glucans through a disulfide-containing linker. The self-assembled nanoparticles from this prodrug specifically target intestinal microfold cells due to the connection between β-glucans and the membrane phagocytic pattern-recognition receptor Dectin-1 on intestinal microfold cells in intestinal Peyer’s patches. These nanoparticles are engulfed by local macrophages in Peyer’s patches. Taking advantage of the tumor-homing ability of macrophages, which is driven by various chemoattractants secreted by the tumor cells, the macrophage-hitchhiked prodrug enters the circulatory system via the lymphatic system and can transverse the BBB to accumulate in the tumor. Additionally, the overexpressed glutathione can specifically break down the prodrug nanoparticles, resulting in the release of conjugated temozolomide [91].
Considerable research efforts have been directed toward investigating the potential of TAMs as active carriers capable of bypassing the BBB to deliver chemotherapy drugs for the treatment of glioblastoma. However, the impact of exogenous TAM carriers that migrate into tumors in the brain remains uncertain. Li et al. [92] have developed TAMs loaded with Nano-Doxorubicin (DOX), which not only have the ability to infiltrate tumors but also can reprogram the exogenous TAM carriers from a pro-tumor phenotype (M2) to an anti-tumor phenotype (M1), thereby suppressing tumor growth. Specifically, the released Nano-DOX from TAMs induces damage-associated molecular patterns, which in turn promote the recruitment of Nano-DOX-TAMs and TAMs. In addition, the study involving M1 macrophage-carried DOX-loaded PLGA nanoparticles in the U87 glioma model has confirmed the potential efficacy of M1 macrophage carriers, including their tumor-homing properties and their ability to induce cellular apoptosis [93].
In comparison to macrophages, macrophage-derived membranes or exosomes cannot transition into an immunosuppressive TAM phenotype induced by the TME. However, the macrophage-derived exosomes retain the inherent ability to penetrate the BBB [94,95,96,97]. More importantly, exosomes derived from M1 macrophage- can alter the immunosuppressive TME via M2-to-M1 polarization [97]. To enhance BBB penetration, ultrasound exposure has been used [119]. It has been reported that either macrophage-derived exosomes or blood serum-derived exosomes can traverse BBB models and accumulate in glioma cells [97]. Meanwhile, the phospholipid bilayers of exosomes’ membrane ensured the feasibility of constructing tumor cell-targeted exosomes by incubating them with the ligands [98, 99]. Additionally, genetic engineering can confer additional functions to macrophage-derived membranes or exosomes, such as immune checkpoint blockade by overexpression of PD-1 [96].
Microglia
Microglia, the resident macrophages in the CNS, can be distinguished from bone marrow-derived CD45+ macrophages by their CD45 expression [120]. In the glioma microenvironment, microglia can be recruited by gliomas through chemoattractants such as CCL5 [120], CCL2 [121], CX3CL1 [122], and CXCL12 [123], and can penetrate the tumor. These characteristics make microglia suitable candidates for delivering drugs for glioma treatment [100,101,102]. Du et al. [100] developed a liposome-carrying microglia to deliver paclitaxel (PTX) for glioma treatment. The engineered microglia could migrate towards glioma cells across the BBB and penetrate the tumor, as evidenced by stronger fluorescence intensity in the brain of the orthotopic glioma mouse model and the presence of fluorescence in deeper regions of tumor spheroids. Additionally, they observed that the transfer of loaded nanoparticles from the engineered microglia to glioma cells was mediated by the formation of EVs and tunneling nanotubes, overcoming the limitations of traditional nanoparticle delivery systems [100].
The impact of microglia on the development of gliomas is significant and should be considered in the design of microglia-based DDS. Research has shown that the endogenous microglia in the TME can be reprogrammed to promote tumor growth through the transfer of extracellular miR-21 released by glioma cells [124]. However, the role of transmigrated exogenous microglia in glioma development is not well understood. Microglia in the TME plays a central role in brain tumor pathobiology, as they secrete factors such as stress-inducible protein, epidermal growth factor, transforming growth factor-β, and matrix metallopeptidase-2 that can promote tumor growth. Additionally, microglia-induced increased expression of platelet-derived growth factor receptors in tumor cells can accelerate tumor progression [125, 126]. Studies have also demonstrated that depleting microglia can attenuate malignant glioma growth in mice [125, 127]. Therefore, it is essential to disrupt the communication between glioma cells and microglia or use radiation and/or chemotherapy to prevent the promotion of tumor growth and the creation of new functional states with different abilities to promote tumor growth.
Neutrophil
More than 70% of human glioma samples exhibit significant infiltration of neutrophils, which is associated with the grade of the tumor. Neutrophils are capable of permeating the BBB and accessing glioma cells. Furthermore, tumor-associated neutrophils (TANs) have been observed to reside in the vicinity of malignant glioma cells, promoting the recruitment of additional circulating neutrophils. Additionally, the surgical removal of a tumor leads to local brain inflammation, which further facilitates the recruitment of neutrophils. This phenomenon has laid the foundation for the potential application of neutrophil-based DDS in the treatment of brain glioma. Xue et al. [103] creatively reported the use of neutrophils carrying PTX-loaded cationic liposomes (CL) (PTX-CL/NEs) to suppress postoperative glioma recurrence. The highly concentrated inflammatory signals in the brain not only guide the movement of neutrophils into the inflamed brain but also trigger the release of liposomal PTX from the neutrophils, allowing for the delivery of PTX into the remaining invading tumor cells. The results have shown that PTX-CL/NEs present superior inhibitory effects on tumor recurrence in surgically treated glioma mouse models but not in mice with primary gliomas. This indicates that the amplification of inflammatory signals after surgery facilitates the brain tumor targeting and therapeutic efficacy of PTX-CL/NEs. In the following year, Wu et al. [104] clarified the location and behavior of neutrophils after internalizing drug cargoes in the glioma model. In an inflamed mouse glioma model, systemically injected neutrophil carriers can migrate outside the vasculature and move to the inflamed glioma sites along the gradients of molecular guidance signals (chemoattractants or chemokines). Subsequently, the cargoes were unloaded from the neutrophil carriers through neutrophil extracellular trap formulation, but not through exosome secretion in the inflammatory region. Finally, the released cargos were taken up by the glioma cells and performed anticancer efficacy [104]. Thus, it is important for the anticancer efficiency of nanoparticles that the neutrophil carriers accumulate at the inflammatory vascular site and that the drug is accurately unloaded from the neutrophil carriers in the glioma region. In 2021, dual-responsive biohybrid neutrophil microbots were developed. These microbots were equipped with PTX-loaded magnetic nanogels and Escherichia coli membrane, which conferred them with magnetic actuated activity. The magnetically actuated intravascular motion and chemotactic behavior along the gradient of inflammatory factors, combined with the inherent chemotaxis of natural neutrophils, greatly enhance the accumulation of PTX in postoperative glioma [105]. In another research, external ultrasound irradiation can be introduced to release PTX from the nanoparticles in the neutrophil carriers on-demand at glioblastoma sites [106]. Specifically, the nanoparticles can generate reactive oxygen species under external ultrasound irradiation, leading to the instability of the liposomal bilayers and the leakage of PTX from the nanoparticles. This property of neutrophil carrier-internalized nanoparticles resulted in the rapid release of PTX from the carriers under external ultrasound irradiation. Although neutrophil-based DDS is an attractive option for treating glioma, there is an ongoing debate about the role of neutrophil carriers in the TME due to the diverse functions of neutrophils, which can have both pro- and anti-tumor effects. The TANs have been categorized as either antitumorigenic (N1) or protumorigenic (N2) TANs (Fig. 5b). Recently, researchers have utilized neutrophil-derived membranes and exosomes as carriers, which not only preserve the distinct functions of neutrophils such as inflammatory chemotaxis and BBB penetration but also mitigate the potential risk of a switch from N1 to N2 TANs during tumor progression [107, 108]. It is worth noting that the neutrophils and their components applied in DDS for targeting glioma were obtained from the peripheral blood or bone marrow of mice. The different phenotypes of neutrophils used in biomimetic DDS may significantly influence their in vivo behavior.
NK cell
In addition to macrophages and neutrophils, other types of leukocytes, such as NK cells and DC, also play important roles in the immunosurveillance of glioma (Fig. 5c and d). The peripheral NK cells can identify abnormal cells including tumors, without the need for prior exposure to specific antigen. Moreover, peripheral NK cells can eliminate infected and/or malignant cells through the production of cytokines, perforin, and granzyme, as well as by interacting with apoptotic receptors on target cells using CD95-ligand and tumor necrosis factor-α (TNF-α) [128]. Despite their relatively low levels in TAM, NK cells are considered promising candidates for future therapeutic approaches for gliomas due to their unique characteristics [129]. The infiltration of NK cells into TAM involves interactions between integrin on lymphocytes and ICAM-1 or VCAM-1 on endothelial cells, which result in the disruption of TJs and reorganization of the actin cytoskeleton, leading to the formation of intercellular gaps at the BBB [130, 131]. This characteristic can be exploited by using NK cell membrane-coated nanoparticles. Deng et al. [109] investigated the ability of NK@AIEdots, which are NK cell membrane-coated AIE-active polymeric nanoendoskeletons, to cross BBB in vitro and in vivo. The study demonstrated the presence of LFA-1 and VLA-4 on NK@AIEdots. The efficiency of BBB crossing by NK@AIEdots was approximately 8 times higher than that of naked AIEdots in a BBB model in vitro. The inhibiting effect of anti-LFA-1 and anti-VLA-4 antibodies on the BBB crossing efficiency of NK@AIEdots further confirmed the critical role of LFA-1 and VLA-4 proteins in the BBB crossing of NK@AIEdots [109]. Meanwhile, the upregulation of actomyosin stress fiber, which mediates cell motility and contraction, and the downregulation of zonula occludens-1, a TJ-associated protein that maintains endothelial cells morphology and TJ integrity, indicated that NK@AIE dots could serve as TJ modulators to disrupt TJs and reorganize the actin cytoskeleton, thus forming an intercellular “green channel” to facilitate their own crossing of the BBB [109]. Similarly, in nude mice bearing orthotopic glioblastoma U-87 MG, NK@AIEdots showed more pronounced accumulation in the brain and tumor compared to naked NK@AIEdots. The tumor-targeting ability of NK@AIEdots was closely associated with the presence of NKG2D and DNAM-1 on the NK@AIEdots. The recognition and interaction of DNAM-1 and NKG2D with the poliovirus receptor Nectin-2 and major histocompatibility complex class I-related and stress-inducible molecules, which are overexpressed in tumors, resulted in the higher accumulation of NK@AIEdots in U-87 MG glioma cells compared to AIEdots. However, the therapeutic potential of NK cell membranes for treating glioma, particularly due to the expressed CD95-ligand and TNF-α has not been thoroughly investigated, despite the consideration of chimeric antigen receptor NK cell therapy as an effective treatment modality for malignant tumors.
DC
The use of DC-based immunotherapy has shown promise as a potential treatment for glioblastoma by stimulating T lymphocyte-mediated anti-cancer immunity [132]. Mature DCs, when stimulated by tumor-associated antigens, can promote the proliferation and activation of CD8+ (cytotoxic T cells) and CD4+ T lymphocytes (helper T cells), which are involved with tumor apoptosis [132]. Although DC vaccines have shown promising results in clinical trials, the effectiveness of anti-glioblastoma immunotherapy is hindered by glioblastoma-induced immunosuppression and signaling of tumor-associated antigens. Glioblastoma is characterized by an immunosuppressive TME, with cancerous glioblastoma cells initiating this immunosuppression. Nano-DOX has been found to effectively stimulate damage-associated molecular patterns derived from glioblastoma cells, leading to a shift in the immunosuppressive phenotype of glioblastoma-associated macrophage to an immunostimulatory phenotype [92]. On this basis, Li et al. [110] utilized DC-mediated delivery of Nano-DOX (Nano-DOX-DC) to explore the potential of overcoming the glioblastoma immunosuppressive microenvironment and enhancing the DC-driven anti-glioblastoma immune response. Their findings indicated that the intravenously injected Nano-DOX-DC could migrate to the tumors of mice with orthotopic glioblastoma xenografts. In the presence of Nano-DOX-induced glioblastoma cell damage, the infiltrated DC carriers were effectively activated, resulting in an enhanced mouse lymphocyte-mediated immune response [110]. It is worth noting that DC membranes have been found to retain tumor-associated antigens and T-cell stimulating factors, offering the potential for a cytomembrane vaccine [111]. Additionally, tumor cell membranes contain abundant levels of tumor antigens, including tumor-associated and tumor-specific antigens, which can help decrease immune evasion, break immunological tolerance, and target tumor cells [133]. Inspired by these natural features, Hao et al. [112] developed a hybrid membrane‑coated DTX nanosuspension based on DC membranes and C6 cell membranes (DNS-[C6&DC]m) for multi‑modal anti‑glioma therapy. The delivery of tumor antigens derived from the C6 cell membrane efficiently elicited the antitumor immune responses, leading to a significant increase in the expression of CD8 and CD4 at tumor sites and spleen following in vivo injection of DNS-[C6&DC]m [112]. Furthermore, glioma growth was obviously suppressed by DNS-[C6&DC]m in the C6 glioma-bearing mice, indicating that the combination of drug delivery and antigen delivery resulted in a better chemo-immunotherapeutic effect in gliomas [112].
Ischemic stroke
Ischemic stroke is characterized by necrosis caused by local blood supply obstruction in the brain, with high morbidity, disability, and mortality rates [134]. Current clinical treatments for ischemic stroke primarily involve endovascular thrombectomy and intravenous thrombolytic drug administration [135]. However, the narrow treatment windows and low treatment rates for stroke patients [136] underscore the importance of identifying effective neuroprotective therapy for ischemic stroke. The presence of BBB poses a significant challenge to the delivery of neuroprotective drugs. Recent studies have shown the crucial role of immune cell-mediated immune responses in the pathogenesis of ischemic stroke, including acute intravascular events triggered by blood supply disruption, the inflammatory cascade leading to brain damage, and the subsequent tissue repair phase [137,138,139]. Following ischemia, the adhesion molecule P-selectin is rapidly deployed to the surface membrane of platelet and endothelial cells, while pro-inflammatory cytokines are promptly generated and released, triggered by the upregulated nucleotides (ATP, UTP) from injured cells like neurons [138]. Concurrently, perivascular macrophages and mast cells become activated, leading to the release of proteases and pro-inflammatory cytokines [138]. The increased proteases can facilitate the extravasation of proteins and cells via the paracellular route by downregulating TJ proteins between endothelial cells [139]. Furthermore, the upregulated pro-inflammatory factors can stimulate the release of various chemokines (CCL2, CCL20, and CXCL2) to attract peripheral immune cells to the ischemic region and increase the endothelial expression of ICAM-1 and VCAM-1, thereby facilitating the infiltration of peripheral immune cells into the damaged brain parenchyma. The recruited peripheral immune cells include neutrophil, macrophage/microglia, lymphocyte, and DC (Fig. 6).

The role of infiltrating immune cells in ischemic stroke and their application or non-application in immune cell-based DDS. This includes neutrophil (a), macrophage/microglia (b), T cell (c), Treg cell (d), and NK cell (e). BBB blood–brain barrier, DDS drug delivery systems, NETs neutrophil extracellular traps, NK natural killer. “?” represents no current application in DDS
Neutrophil
Neutrophils are the initial immune cells to enter the damaged area of the brain during acute ischemic stroke [140, 141]. Following the onset of cerebral ischemia, neutrophils gather in the cerebral microvessels and venules within minutes and gradually move to the ischemic brain tissue, guided by increased secretion of chemokines (CXCL1, CXCL2) by astrocytes [142]. The migration of neutrophils peaks after 24 h and persists for over 32 days [143]. This migration behavior has prompted research into utilizing neutrophils for DDS. Additionally, stroke patients exhibit a significant increase in circulating neutrophils due to a systemic immune response. The abundance of neutrophils presents an opportunity for potential drug delivery to the ischemic region to alleviate ischemic injuries. Hou et al. [144] found that cRGD peptide-modified liposomes have a high affinity for neutrophils, as they recognize integrin avb1, which is abundantly expressed on the surface of neutrophils. This recognition efficiently triggers the uptake of cRGD-modified liposomes by neutrophils [144]. Neutrophils effectively deliver cRGD-liposome to the ischemic region owing to the chemotactic nature in a middle cerebral artery occlusion (MCAO) mice model. The delivery efficiency of the cargo is closely linked to the migration efficiency of neutrophils, which depends on secondary inflammatory signals following the onset of ischemia. Similar to the infiltrated neutrophils in the ischemic region, cRGD-liposome significantly accumulated in the ischemic region as early as 3 h and continued to increase up to 24 h after the ischemic insult [144]. The cargo can be transferred from the neutrophils to neuronal cells through transient intercellular connections and the secretion and fusion of exosomes by neutrophils and neuronal cells [145]. The migration process of neutrophils was dominated by integrin β2, macrophage-1 antigen, and LFA-1 on the neutrophil membrane. Efficient delivery of mesoporous Prussian blue nanozyme and resolvin D2, loaded in neutrophil membrane and neutrophil membrane-derived nanovesicles, respectively, further supports the use of neutrophils for delivery into ischemic regions [146, 147]. Interestingly, reduced recruitment of neutrophils was observed in the neutrophil-like cell-membrane-coated mesoporous Prussian blue nanozyme (MPBzyme@NCM) treated tMCAO/stroke mice at day 5 after stroke, as indicated by Western blotting analyses of myeloperoxidase, a marker for neutrophils. This phenomenon may result from the interaction of neutrophil membrane-coated nanoparticles with adhesion molecules on inflamed brain microvascular endothelial cells [146]. In addition, MPBzyme@NCM-treated tMCAO/R model mice exhibited an increased M2 phenotype macrophage/microglia and decreased apoptosis of neurons [146]. However, it is unclear whether these favorable stroke recovery phenomena are associated with neutrophil membranes. Various reports have suggested that the infiltration of neutrophils is linked to BBB breakdown, edema, and brain infarcts [148, 149]. Neutrophil depletion contributes to brain tissue repair after stroke [150, 151].
Macrophage/microglia
It has been documented that the presence of macrophage/microglia (including resident microglia and infiltrated macrophages) in the brain reached its peak approximately 4 days after cerebral ischemia in rats or mice. Resident microglia activation predates and predominates over blood-derived macrophages. Following an ischemic stroke, the release of damage-associated molecular patterns can trigger local immune responses, leading to the activation of glial cells within minutes. The activated microglia primarily exhibit M1-type characteristics, producing and releasing significant amounts of pro-inflammatory factors (IL-1α, IL-1β, IL-6, TNF-α, IFN-γ) [152]. After 1 days, the activated microglia migrated to the infarct core via the annexin-1/casein kinase II pathway [153]. Blood-derived macrophages were recruited into the ischemic brain tissue within 3–4 h after the onset of ischemia. guided by CCL2. The phenotype of infiltrating macrophage/monocytes and activated microglia undergoes dynamic polarized changes [154]. Specifically M1-like cells gradually increased within 14 days after the onset of stroke, while M2-like cells tended to increase in the first 1–2 days and then gradually decreased [152, 154, 155]. Among these, M2-like macrophages promote tissue recovery,d axonal outgrowth, and angiogenesis after ischemic stroke by secreting protective remodeling factors (VEGF, BDNF, progranulin, and transforming growth factor factor-β), anti-inflammatory cytokines, and proteinases [156]. A recent study found that spleen-targeted glabridin-loaded nanoparticles (NPGla-5k) could effectively regulate the polarization of macrophages/monocytes in the spleen into M2-macrophages, accompanied by the infiltration of peripheral macrophages into the ischemic penumbra after tail-vein injection. NPGla-5k treatment can effectively reduce inflammatory damage, protect damaged neurons, and improve nervous system function in MCAO/R mice [157]. Similarly, macrophage-derived exosomes exhibited superior migration capability to cross the BBB and accumulate in the ischemic brain with the loaded drugs [158, 159]. Meanwhile, the microglia and neuronal cells can internalize the drug-loaded migrated exosomes [158]. Surprisingly, lipopolysaccharide (LPS)-induced macrophage exosomes exert therapeutic effects on ischemic stroke by promoting microglia polarization from M1 to M2 [160]. Macrophage-derived membrane-based biomimetic nanoparticles have also made some progress in the treatment of ischemic stroke. These biomimetic nanoparticles were endowed with the natural targeting and migration ability of macrophages to the ischemic brain due to the preservation of most cell membrane surficial proteins, including CD11b, CD44, integrin α4, and integrin β1, as evidenced by the distribution of these biomimetic nanoparticles in the ischemic brain markedly beyond that of the naked nanoparticles [161,162,163,164]. Interestingly, microglia-derived membrane-based biomimetic nanoparticles also demonstrated the ability to cross the BBB, though there were no reports about the migration of microglia from peripheral to CNS [165]. Moreover, the M2 microglia membranes can serve as bioinspired therapeutic agents to repolarize M1 microglia into the M2 phenotype, which may result from the presence of anti-inflammatory proteins on the membrane, such as CD206 [165].
Lymphocyte
Lymphocytes, including T cells, Treg cells, and NK cells, among others, were observed to migrate to the ischemic brain following an ischemic event [166]. While lymphocytes represent a small proportion of the infiltrating immune cells. T cells were found to migrate from subpial and cortical vessels as well as choroid plexus to the infarcted hemisphere on day 1 after ischemia, with this process persisting for an extended period [167]. Peripheral T cells were shown to have a significant impact on preventing hemorrhagic transformation in severe ischemic stroke by interacting with platelets [168]. Infiltrating T cells have been related to promoting the proinflammatory pathway, while another infiltrating lymphocyte, NK cells, have been found to exhibit pathogenic actions in ischemic stroke, including promoting inflammation and neuronal cytotoxicity [169]. In the ischemic cerebral hemisphere, the number of NK cells rapidly increased and peaked at 3 h post-ischemia, and then decreased [169]. Conversely, regulatory lymphocytes, such as Tregs and regulatory B cells (Bregs), have been shown to contribute beneficially to recovery following ischemic stroke [170]. Treg cells were observed to accumulate in the ischemic lesion at 15 days and persist at 30 days post-stroke [171]. Attenuation of Treg cells function resulted in aggravated tissue loss and impaired neurological function in MCAO mice. In contrast, enhancing the number or functions of Treg cells was found to be conducive to recovery [172]. These results support the potential of Treg cell therapy. Up to now, there have been no reported studies on Treg cell-based DDS in the treatment of ischemic stroke. The application of immune cell-based DDS in ischemic stroke is summarized in Table 3 [144,145,146,147, 157,158,159,160,161,162,163,164,165].
Neurodegenerative diseases
AD and PD are the most prevalent neurodegenerative diseases, with AD being more common among the elderly, affecting around 10% of individuals over 65. AD is characterized by accumulated amyloid-β (Aβ) plaques, neurofibrillary tangles, and neuronal loss, leading to progressive cognitive deterioration. Accumulating evidence indicates that the amyloid cascade hypothesis and tau protein, although well-established, may not be the sole causative factors in AD. The presence of inflammatory markers in AD patients suggested an association between immune cells and pathological processes underlying AD [173,174,175]. Microglia, as innate immune cells, play a complex role in AD pathogenesis through various activation pathways [176]. They display diverse phenotypes and engage in multifaceted interactions with Aβ and tau species, as well as neuronal circuits [176]. Microglia recruited to the sites of Aβ plaque deposition can phagocytose Aβ, thereby facilitating its elimination [177]. Moreover, soluble hyperphosphorylated tau protein can induce degeneration of microglia, impairing their immunosurveillance function and promoting the formation of neurofibrillary tangles [178]. Typically, these neurofibrillary tangles are internalized by microglia [179]. Although AD brains generally have fewer peripheral immune cell infiltrations compared to glioma or other neuroinflammatory diseases like MS [180], evidence indicates that under Aβ stimulation, immune cells such as neutrophils, monocytes, and T cells can infiltrate the brain (Fig. 7) [181].

The role of infiltrating immune cells in neurodegenerative diseases and their application or non-application in immune cell-based DDS, including AD (a), MS (b), and PD (c). AD Alzheimer’s disease, Aβ amyloid-β, BBB blood–brain barrier, CCL2 c–c motif chemokine ligand 2, DDS drug delivery systems, MS multiple sclerosis, PD Parkinson’s disease, ROS reactive oxygen species, TLR2 toll-like receptor 2. “?” represents no current application in DDS
AD
In the brains of AD transgenic mice, there is a notable accumulation of monocytes specifically around Aβ plaques. Unlike in other neurodegenerative diseases, the monocytes that infiltrate the AD brain have a positive impact on the disease’s progression by limiting Aβ plaques [182]. This unique ability of monocytes to enter the brain and aid in Aβ elimination has led to the exploration of monocyte or its differentiated cells (including macrophages and microglia)-based delivery systems for proteins, miRNA, and drugs. Böttger et al. [183] utilized Bioporter™ to facilitate the recombination of NGF-loaded primary rat monocytes. The results revealed that over 30% of NGF-loaded monocytes were able to adhere to the monolayer of rat brain capillary endothelial cells (BCEC) and efficiently traverse a simplified artificial BCEC monolayer. Additionally, the loaded NGF can be released by migrating monocytes into the basolateral medium, thereby providing neuroprotection for cholinergic neurons against degeneration [183]. Furthermore, the migration ability of monocytes loaded with proteins, miRNA, and drugs, was further demonstrated. Moreover, monocyte-derived membranes, exosomes, and EVs have been found to serve as effective transport vehicles across the BBB in AD model mice [184,185,186]. The mechanism of exosome-loaded drugs’ migration was investigated, revealing that the specific interaction between monocyte-derived exosomes’ LFA-1 and endothelial ICAM-1 mediates their migration across the BBB. Although there was no obvious evidence that the monocyte-derived exosomes specifically accumulate around specific brain cells, they have been shown to fuse with astrocytes and deliver drugs to specific brain regions [186].
To improve the delivery efficiency of exosomes- or membrane-based DDS for targeting neurons, microglia, or organelles such as lysosome and mitochondria, various chemicals, proteins, and antibodies like rabies virus glycoprotein (RVG29), mannose, or triphenylphosphine cation (TPP) can be modified on the surface of the exosomes. Mitochondrial dysfunction is a contributing factor to the production and abnormal aggregation of Aβ, thus leading to neuron apoptosis. Han et al. [187] used RVG29 and TPP-modified macrophage membrane-coated solid lipid nanoparticles (SLNs) to deliver Genistein (GS) (RVG/TPP-MASLNs-GS), a natural flavonoid that effectively inhibits neuronal apoptosis induced by Aβ in vitro, to abnormal mitochondria in the neuron. The attachment of RVG29 and TPP on the macrophage membrane did not affect the macrophage membrane’s ability to evade the reticuloendothelial system, as evidenced by similar pharmacokinetic curves and parameters between MASLNs-GS and RVG/TPP-MASLNs-GS groups [187]. Additionally, RVG29 modification reinforced the macrophage membrane’s innate ability to cross the BBB, a crucial requirement for RVG/TPP-MASLNs-GS to target neuronal mitochondria [187]. In this research, MASLNs presented poor BBB permeability in an in vitro BBB model and in vivo imaging, likely due to the absence of chemokines. Conversely, the significant signal was observed in the lower neuron of the BBB model in the RVG-MASLNs and RVG/TPP-MASLNs groups, consistent with in vivo imaging results. Notably, a large number of RVG/TPP-MASLNs were internalized by neurons rather than astrocytes in the bEnd.3/HT22 or bEnd.3/astrocytes co-culture BBB models, as RVG29 exhibits higher selectivity for neurons [188, 189]. Furthermore, RVG/TPP-MASLNs effectively inhibit mitochondrial reactive oxygen species in Aβ-treated HT22 neuronal cells, facilitated by TPP-mediated mitochondria targeting [190].
Interestingly, exosomes can attach to Aβ because of the high presence of GM1 ganglioside in their membranes. This is known to cause conformational changes in Aβ, leading to the formation of non-toxic amyloid fibrils and promoting the absorption of Aβ [191,192,193,194]. Additionally, microglia with impaired lysosomal function are associated with abnormal endocytosis and insufficient clearance of Aβ through lysosome-mediated degradation. Building on this, Hao et al. [185] developed mannose-conjugated macrophage-derived exosomes (MExo) to promote the targeted delivery of gemfibrozil (Gem) and restore the lysosomal activity of microglia in clearing Aβ aggregation. MExo-Gem was found to reduce the formation of amyloid fibrils by binding to Aβ and delivering Aβ precisely to microglia via the interaction between the mannose modified on exosomes and mannose receptors expressed in microglia [195]. This was demonstrated by a decrease in Thioflavin T fluorescence intensity and an increase in the concentration of Aβ in microglia in the MExo-treated group [185]. The accumulated Gem effectively restored lysosomal activity by promoting the nuclear translocation of transcription factor EB and activating peroxisome proliferator-activated receptor α. The activated lysosomes in microglia contributed to accelerating lysosome-mediated clearance of the accumulated Aβ in microglia, as demonstrated in vivo and in vitro. Finally, MExo-Gem significantly reduced neuronal injury and improved learning and memory ability, as evidenced by the highest target quadrant occupancy and crossing numbers in Aβ-induced AD mice [185]. In contrast to monocyte-derived components (exosomes, EVs, or membranes), an excess of monocytes may have uncontrolled effects in the brain as carriers of the BBB, and it is unclear whether an increase in macrophages/microglia in the brain leads to beneficial or harmful effects in the context of nerve injury.
In contrast to macrophages, the infiltrating neutrophils and T lymphocytes play an important role in the development of AD [196, 197]. Activated circulating neutrophils cause damage to BBB and neurotoxicity in AD by producing inflammatory mediators such as myeloperoxidase and reactive oxygen species, and by releasing neutrophil extracellular traps (NETs) [198, 199]. In AD models, reducing the number of neutrophils or inhibiting their movement through blocking LFA-1 or integrins α4 can decrease AD-like neuropathology and improve cognitive function [200,201,202]. Infiltrating T cells were classified as CD4+ T cells and CD8+ T cells, with CD8+ T cells infiltrating the CNS and causing harmful effects [196]. The effects of infiltrating CD4+ T cells varied depending on their phenotypes. Specifically, Th1 and Th17 cell types may have negative effects on nearby neurons or glial cells by producing pro-inflammatory cytokines, while Th2 or Treg cells may suppress neuroinflammation in AD by secreting anti-inflammatory cytokines or through cell–cell contact [194]. Additionally, the increased expression of CCR on T cells and their corresponding ligands in the AD brain further facilitates the recruitment of peripheral immune cells into the CNS [196]. The application of immune cell-based DDS in AD is outlined in Table 4 [59, 183,184,185,186,187].
PD
PD is characterized by the significant loss of dopaminergic neurons in the substantia nigra and the aggregation of α-synuclein in Lewy bodies [203]. Additionally, there is evidence of inflammation and activation of microglia, as represented in postmortem tissue studies of PD patients and an increase in cytokines in the brain and CSF [204]. The activated microglia and astroglia released pro-inflammatory cytokines, leading to endothelium-derived inflammatory responses and the expression of adhesion molecules [203], which in turn recruit circulating blood cells into the brain microvasculature. Furthermore, there is an innate immune response in the periphery triggered by the antigenicity of peripheral α-synuclein. Post-mortem brain samples of PD patients have shown the presence of infiltrating immune cells, including T cells, NK cells, and monocytes/macrophages, in the substantia nigra [205, 206]. The interaction between immune cells in the periphery and the brain not only influences the overall immune response in PD but also provides a potential avenue for delivering proteins, mRNA, and drugs into the brain [207,208,209,210,211,212,213,214,215,216,217,218,219].
Batrakova et al. [40] and other researchers utilized a bone-marrow-derived macrophage system to deliver catalase to brain regions affected by PD [207,208,209]. They encapsulated catalase/PEI-PEG complexes into the macrophages to protect catalase from degradation and ensure its sustained release [40, 207]. The loaded macrophages were observed to move along microvessels, adhere to endothelial walls, and cross the BBB into the parenchyma during brain inflammation, [208]. Compared to free nanozyme, more nanozyme was transferred from the loaded macrophage to various target cells, including endothelial cells, neurons, and astrocytes, leading to decreased reactive oxygen species, reduced neuroinflammation, and protection against degeneration. The enhanced transfer of nanozyme from macrophages to target cells involved the fusion of cell membranes, the formation of macrophage bridging conduits, and the release of exosomes containing nanozyme [209]. In this study, macrophages loaded with catalase were utilized as depots, while subsequent investigations subtly advanced the development of macrophage carriers as production facilities. Genetically modified macrophages were constructed by transfection of pDNA encoding catalase. The transfected gene enables these macrophages to effectively reach the brain and prolong the secretion of catalase in the brain of PD mice [210]. Interestingly, the exosomes from catalase-transfected macrophages efficiently transferred their contents (DNA, mRNA, transcription factors molecules, and the encoded protein) to neighboring neurons [210] leading to sustained catalase expression and contributing to anti-inflammatory and neuroprotective effects in murine models of neuroinflammation and PD.
In addition to the efficient delivery of catalase and catalase pDNA into the brain of PD model mice via macrophages, these macrophages when transfected with glial cell line-derived neurotrophic factor (GDNF) were recruited to substantia nigra during neurodegeneration. This recruitment significantly improved neurodegeneration and neuroinflammation in 6-hydroxidophamine (6-OHDA)-intoxicated mice and transgenic Parkin Q311X(A) mice [211,212,213,214,215,216]. The accumulated GDNF-transfected macrophages were able to differentiate into the regenerative M2 phenotype. Furthermore, the GDNF formed by these macrophages could transfer to target neurons, facilitated by the targeted ability of macrophage-derived exosomes containing GDNF. Similarly, bone marrow-derived microglia transfected with neurturin could also cross the BBB and were recruited in large numbers to sites of neurodegeneration, where they become activated microglia capable of secreting trophic factors [217]. It has been noted that the targeted ability of exosomes or EVs is related to the cell source. A previous study has shown that the brain accumulation levels of mEVs in a transgenic mouse model of PD are significantly higher than nEVs and aEVs [80]. Research using mEVs as the carriers of catalase for PD treatment has further demonstrated the targeted ability of these mEVs to reach inflamed brain tissues [218]. The application of immune cell-based DDS in PD is summarized in Table 5 [40, 207,208,209,210,211,212,213,214,215,216,217,218,219].
MS
MS is a chronic autoimmune, inflammatory, and neurodegenerative disease that affects more than 2.5 million people worldwide. The disease is considered to be triggered by the activation of CNS-reactive T cells in the periphery, which are then arrested by endothelial cells in the CNS and subsequently migrate across the BBB into the CNS [197, 220]. This process involves the expression of chemokines, integrin, and selectin by inflammatory endothelial cells, leading to the recruitment and migration of immune cells [220]. The resulting immune dysregulation in the CNS is the primary cause of MS, leading to demyelination, axonal damage, and neurodegeneration [221]. Similar to AD, the pathogenesis of MS involves the activation of microglia and astrocytes, resulting in the release of pro-inflammatory cytokines and chemokines, which in turn recruit more immune cells from the periphery to the CNS [220]. The main therapeutic objective for MS is to suppress the immune response in the CNS. There are three treatment strategies approved by the Food and Drug Administration (FDA) for MS patients: regulating the immune state to induce tolerance, blocking T cell trafficking to the CNS, and inhibiting T cell division and proliferation [222]. However, these non-specific immunosuppressive treatments may pose serious risks in the medium to long term. As a result, numerous nanocarriers have been designed to target peripheral immune cells, such as macrophages, DC, and B cells, to deplete monocytes, induce specific antigen tolerance, or deliver corticosteroids. Additionally, immune cells have been utilized as “Trojan horse”-mediated DDS to efficiently migrate into the CNS parenchyma, overcoming systemic treatment dispersion, and reducing the number of B cells in spinal cord infiltrates [223]. One specific example is the development of a T cell-mediated DDS, where iron-oxide nanoparticles (NBR) conjugated with the monoclonal antibody anti-CD20 (NBR-anti-CD20) were loaded in MOG35-55 antigen-specific T cells. Anti-CD20 is the only FDA-approved disease-modifying therapy for primary progressive MS and depletes B cells [224]. This approach has shown promise in depleting B cells and preserving neurons and the axonal state in experimental autoimmune encephalomyelitis mice. [223].
Immune cell-based DDS has also been applied in epilepsy and depression, both of which involve neuroinflammation [225, 226]. The application of immune cell-based DDS in MS, depression, and epilepsy is further detailed in Table 6 [223, 225, 226].
Conclusion and future perspectives
The interaction between immune cells and the CNS during disease provides a potential chance for delivering drugs to the brain. Substantial data have demonstrated the potential of immune cell-based DDS, which involves using immune cells to transport nanoparticles into the brain. These strategies include immune cells carrying nanoparticles (hitchhiking nanoparticles, immune trojan horses, immune cells with backpacks), nanoparticles coated with the immune cell membrane, and exosomes derived from immune cells loaded with nanoparticles. These methods take advantage of the natural migratory ability of immune cells across the BBB due to receptors on their membranes. However, it's important to consider the role of immune cells in CNS diseases when using these strategies. The effectiveness and properties of these DDS depend on the specific immune cells chosen. Here are the factors to consider in the design process of immune cell-based DDS. (1) The number and timing of immune cells infiltrating are crucial factors determining the efficiency of immune cell-based DDS delivery. (2) The migrated mechanism of immune cells is mainly mediated by adhesion molecules on the endothelial cells, such as VCAM, ICAM, and P-selectin, as well as their integrin ligands on immune cells like LFA, Mac, and selection. Additionally, other upregulated adhesion molecules on endothelial cells can also serve as ligands for immune cell-based nanoparticles to improve CNS barrier penetration. Based on this premise, targeting multiple adhesion molecules through genetic engineering modification can enhance the penetration of immune cell-based DDS into the BBB. Interestingly, chemokine receptors can strengthen the interactions between integrin and cell adhesion molecules by mediating integrin clustering and conformational changes during the migration of immune cells. When designing immune cell-based nanoparticles, exogenous modification or construction with upregulation of chemokine receptors can be employed. (3) The function of immune cells is vital in the pathogenesis of CNS diseases. It is crucial to understand the role of cell-mediated immunity to consider leveraging the immunotherapeutic potential of immune cells beyond just their migratory abilities. (4) Safety. The infiltration of exogenous immune cell-based nanoparticles into the CNS, especially immune cell carriers, can potentially preserve the functional immune response of specific immune cells. Simultaneously, targeted integration of immune cell-based nanoparticles with classical cell adhesion molecules may effectively inhibit CNS infiltration by immune cells. These actions have the potential to interfere with the host immune response or result in serious side effects.
This review highlights the potential of using immune cell-based nanoparticles for treating CNS diseases. While the findings are promising, they are primarily based on laboratory studies. The translation of these nanoparticles into clinical use faces significant challenges. (1) Standard procedures are crucial for quality controls, including cell culture, cell membrane and exosome extraction, and nanoparticles package. Precise and reproducible procedures are necessary to maintain the functions of these nanoparticles, such as immune evasion, targeting, and immunomodulatory effects. (2) Strict control over storage conditions (solvent type, particle concentration, freeze-drying method, temperature, and reconstituted condition) is essential to preserve protein activity. (3) Ensuring the immune safety of these nanoparticles, particularly immune cell carriers are vital, and their effects on different immune cell subsets should be thoroughly investigated to minimize potential side effects. Overall, the migration of immune cells to CNS presents an opportunity for drug delivery using immune cell-based nanoparticles and other strategies.
Availability of data and materials
Not applicable.
Abbreviations
- Aβ:
-
Amyloid-β
- AD:
-
Alzheimer’s disease
- BBB:
-
Blood–brain barrier
- Bregs:
-
Regulatory B cells
- CNS:
-
Central nervous system
- CSF:
-
Cerebrospinal fluid
- CXCL12:
-
C-X-C motif chemokine ligand 12
- CXCR4:
-
Chemokine (C-X-C motif) receptor 4
- C6-Luc:
-
C6 glioma cells that luciferase reporter-gene labeled
- DCs:
-
Dendritic cells
- DDS:
-
Drug delivery systems
- DOX:
-
Doxorubicin
- EVs:
-
Extracellular vesicles
- GDNF:
-
Glial cell line-derived neurotrophic factor
- ICAM-1:
-
Intracellular adhesion molecule-1
- ICAM-2:
-
Intracellular adhesion molecule-2
- IL-1β:
-
Interleukin-1β
- LFA-1:
-
Lymphocyte function-associated antigen-1
- MCAO:
-
Middle cerebral artery occlusion
- MS:
-
Multiple sclerosis
- NETs:
-
Neutrophil extracellular traps
- NK:
-
Natural killer
- PD:
-
Parkinson’s disease
- PSGL-1:
-
P-selectin glycoprotein ligand-1
- PTX:
-
Paclitaxel
- TAMs:
-
Tumor-associated macrophages
- TANs:
-
Tumor-associated neutrophils
- TJs:
-
Tight junctions
- TNF-α:
-
Tumor necrosis factor-α
- TME:
-
Tumor microenvironment
- TPP:
-
Triphenylphosphine cation
- Tregs:
-
Regulatory T cells
- VCAM-1:
-
Vascular cell adhesion molecule-1
- VLA-4:
-
Very late antigen-4
References
Szeto JYY, Lewis SJG. Current treatment options for Alzheimer’s disease and Parkinson’s disease dementia. Curr Neuropharmacol. 2016;14(4):326–38.
Katan M, Luft A. Global burden of stroke. Semin Neurol. 2018;38(2):208–11.
Pardridge WM. Blood–brain barrier delivery. Drug Discov Today. 2007;12(1–2):54–61.
Zhang W, Mehta A, Tong Z, Esser L, Voelcker NH. Development of polymeric nanoparticles for blood–brain barrier transfer—strategies and challenges. Adv Sci. 2021;8(10):2003937.
Terstappen GC, Meyer AH, Robert DB, Zhang W. Strategies for delivering therapeutics across the blood–brain barrier. Nat Rev Drug Discov. 2021;20(5):362–83.
Mastorakos P, McGavern D. The anatomy and immunology of vasculature in the central nervous system. Sci Immunol. 2019;4(37):eaav0492.
Prinz M, Priller J. The role of peripheral immune cells in the CNS in steady state and disease. Nat Neurosci. 2017;20(2):136–44.
Buckley MW, McGavern DB. Immune dynamics in the CNS and its barriers during homeostasis and disease. Immunol Rev. 2022;306(1):58–75.
Ransohoff RM, Brown MA. Innate immunity in the central nervous system. J Clin Investig. 2012;122(4):1164–71.
Marchetti L, Engelhardt B. Immune cell trafficking across the blood–brain barrier in the absence and presence of neuroinflammation. Vasc Biol. 2020;2(1):H1–18.
Engelhardt B, Ransohoff RM. The ins and outs of T-lymphocyte trafficking to the CNS: anatomical sites and molecular mechanisms. Trends Immunol. 2005;26(9):485–95.
Ransohoff RM. Chemokines and chemokine receptors: standing at the crossroads of immunobiology and neurobiology. Immunity. 2009;31(5):711–21.
Lopes Pinheiro MA, Kooij G, Mizee MR, Kamermans A, Enzmann G, Lyck R, et al. Immune cell trafficking across the barriers of the central nervous system in multiple sclerosis and stroke. Biochim Biophys Acta. 2016;1862(3):461–71.
Nourshargh S, Alon R. Leukocyte migration into inflamed tissues. Immunity. 2014;41(5):694–707.
Kliche S, Worbs T, Wang X, Degen J, Patzak I, Meineke B, et al. CCR7-mediated LFA-1 functions in T cells are regulated by 2 independent ADAP/SKAP55 modules. Blood. 2012;119(3):777–85.
Wang LL, Janes ME, Kumbhojkar N, Kapate N, Clegg JR, Prakash S, et al. Cell therapies in the clinic. Bioeng Transl Med. 2021;6(2):e10214.
Yu H, Yang Z, Li F, Xu L, Sun Y. Cell-mediated targeting drugs delivery systems. Drug Deliv. 2020;27(1):1425–37.
Charabati M, Rabanel JM, Ramassamy C, Prat A. Overcoming the brain barriers: from immune cells to nanoparticles. Trends Pharmacol Sci. 2020;41(1):42–54.
Champion JA, Mitragotri S. Role of target geometry in phagocytosis. Proc Natl Acad Sci U S A. 2006;103(13):4930–4.
Doshi N, Swiston AJ, Gilbert JB, Alcaraz ML, Cohen RE, Rubner MF, et al. Cell-based drug delivery devices using phagocytosis-resistant backpacks. Adv Mater. 2011;23(12):H105–9.
Polak R, Lim RM, Beppu MM, Pitombo RN, Cohen RE, Rubner MF. Liposome-loaded cell backpacks. Adv Healthc Mater. 2015;4(18):2832–41.
Klyachko NL, Polak R, Haney MJ, Zhao Y, Gomes Neto RJ, Hill MC, et al. Macrophages with cellular backpacks for targeted drug delivery to the brain. Biomaterials. 2017;140:79–87.
Shields CW, Evans MA, Wang LL, Baugh N, Iyer S, Wu D, et al. Cellular backpacks for macrophage immunotherapy. Sci Adv. 2020;6(18):eaaz6579.
Prakash S, Kumbhojkar N, Clegg JR, Mitragotri S. Cell-bound nanoparticles for tissue targeting and immunotherapy: engineering of the particle-membrane interface. Curr Opin Colloid Interface. 2021;52:101408.
Swiston AJ, Cheng C, Um SH, Irvine DJ, Cohen RE, Rubner MF. Surface functionalization of living cells with multilayer patches. Nano Lett. 2008;8(12):4446–53.
Liu T, Gao C, Gu D, Tang H. Cell-based carrier for targeted hitchhiking delivery. Drug Deliv Transl Res. 2022;12(11):2634–48.
Anselmo AC, Mitragotri S. Cell-mediated delivery of nanoparticles: taking advantage of circulatory cells to target nanoparticles. J Control Release. 2014;190:531–41.
Udofa E, Zhao Z. In situ cellular hitchhiking of nanoparticles for drug delivery. Adv Drug Deliv Rev. 2023;204:115143.
Chambers E, Mitragotri S. Prolonged circulation of large polymeric nanoparticles by non-covalent adsorption on erythrocytes. J Control Release. 2004;100(1):111–9.
Chambers E, Mitragotri S. Long circulating nanoparticles via adhesion on red blood cells: mechanism and extended circulation. Exp Biol Med (Maywood). 2007;232(7):958–66.
Anselmo AC, Gupta V, Zern BJ, Pan D, Zakrewsky M, Muzykantov V, et al. Delivering nanoparticles to lungs while avoiding liver and spleen through adsorption on red blood cells. ACS Nano. 2013;7(12):11129–37.
Hu Q, Sun W, Wang J, Ruan H, Zhang X, Ye Y, et al. Conjugation of haematopoietic stem cells and platelets decorated with anti-PD-1 antibodies augments anti-leukaemia efficacy. Nat Biomed Eng. 2018;2(11):831–40.
Tang L, Zheng Y, Melo MB, Mabardi L, Castaño AP, **e YQ, et al. Enhancing T cell therapy through TCR-signaling-responsive nanoparticle drug delivery. Nat Biotechnol. 2018;36(8):707–16.
Scott MD, Murad KL, Koumpouras F, Talbot M, Eaton JW. Chemical camouflage of antigenic determinants: stealth erythrocytes. Proc Natl Acad Sci U S A. 1997;94(14):7566–71.
Rossi NA, Constantinescu I, Kainthan RK, Brooks DE, Scott MD, Kizhakkedathu JN. Red blood cell membrane grafting of multi-functional hyperbranched polyglycerols. Biomaterials. 2010;31(14):4167–78.
Clafshenkel WP, Murata H, Andersen J, Creeger Y, Koepsel RR, Russell AJ. The effect of covalently-attached ATRP-synthesized polymers on membrane stability and cytoprotection in human erythrocytes. PLoS One. 2016;11(6):e0157641.
Ayer M, Klok HA. Cell-mediated delivery of synthetic nano- and microparticles. J Control Release. 2017;259:92–104.
Xu L, Zolotarskaya OY, Yeudall WA, Yang H. Click hybridization of immune cells and polyamidoamine dendrimers. Adv Healthc Mater. 2014;3:1430–8.
Choi MR, Stanton-Maxey KJ, Stanley JK, Levin CS, Bardhan R, Akin D, et al. A cellular Trojan Horse for delivery of therapeutic nanoparticles into tumors. Nano Lett. 2007;7(12):3759–65.
Batrakova EV, Li S, Reynolds AD, Mosley RL, Bronich TK, Kabanov AV, et al. A macrophage-nanozyme delivery system for Parkinson’s disease. Bioconjug Chem. 2007;18(5):1498–506.
Wroblewska A, Szczygiel A, Szermer-Olearnik B, Pajtasz-Piasecka E. Macrophages as promising carriers for nanoparticle delivery in anticancer therapy. Int J Nanomed. 2023;18:4521–39.
Chugh V, Vijaya Krishna K, Pandit A. Cell membrane-coated mimics: a methodological approach for fabrication, characterization for therapeutic applications, and challenges for clinical translation. ACS Nano. 2021;15(11):17080–123.
Liu W, Ruan M, Wang Y, Song R, Ji X, Xu J, et al. Light-triggered biomimetic nanoerythrocyte for tumor-targeted lung metastatic combination therapy of malignant melanoma. Small. 2018;14(38):e1801754.
Dehaini D, Wei X, Fang RH, Masson S, Angsantikul P, Luk BT, et al. Erythrocyte-platelet hybrid membrane coating for enhanced nanoparticle functionalization. Adv Mater. 2017. https://doi.org/10.1002/adma.201606209.
Hu CM, Fang RH, Wang KC, Luk BT, Thamphiwatana S, Dehaini D, et al. Nanoparticle biointerfacing by platelet membrane cloaking. Nature. 2015;526(7571):118–21.
Ren N, Liang N, Dong M, Feng Z, Meng L, Sun C, et al. Stem cell membrane-encapsulated zeolitic imidazolate framework-8: a targeted nano-platform for osteogenic differentiation. Small. 2022;18(26):e2202485.
Parodi A, Quattrocchi N, van de Ven AL, Chiappini C, Evangelopoulos M, Martinez JO, et al. Synthetic nanoparticles functionalized with biomimetic leukocyte membranes possess cell-like functions. Nat Nanotechnol. 2013;8(1):61–8.
Xuan M, Shao J, Dai L, He Q, Li J. Macrophage cell membrane camouflaged mesoporous silica nanocapsules for in vivo cancer therapy. Adv Healthc Mater. 2015;4(11):1645–52.
Kang T, Zhu Q, Wei D, Feng J, Yao J, Jiang T, et al. Nanoparticles coated with neutrophil membranes can effectively treat cancer metastasis. ACS Nano. 2017;11(2):1397–411.
Fang RH, Hu CM, Luk BT, Gao W, Copp JA, Tai Y, et al. Cancer cell membrane-coated nanoparticles for anticancer vaccination and drug delivery. Nano Lett. 2014;14(4):2181–8.
Gao W, Fang RH, Thamphiwatana S, Luk BT, Li J, Angsantikul P, et al. Modulating antibacterial immunity via bacterial membrane-coated nanoparticles. Nano Lett. 2015;15(2):1403–9.
Chen Z, Hu Q, Gu Z. Leveraging engineering of cells for drug delivery. Acc Chem Res. 2018;51(3):668–77.
Oroojalian F, Beygi M, Baradaran B, Mokhtarzadeh A, Shahbazi MA. Immune cell membrane-coated biomimetic nanoparticles for targeted cancer therapy. Small. 2021;17(12):e2006484.
Rao L, Cai B, Bu LL, Liao QQ, Guo SS, Zhao XZ, et al. Microfluidic electroporation-facilitated synthesis of erythrocyte membrane-coated magnetic nanoparticles for enhanced imaging-guided cancer therapy. ACS Nano. 2017;11(4):3496–505.
Chai Z, Hu X, Wei X, Zhan C, Lu L, Jiang K, et al. A facile approach to functionalizing cell membrane-coated nanoparticles with neurotoxin-derived peptide for brain-targeted drug delivery. J Control Release. 2017;264:102–11.
Kang T, Jiang M, Jiang D, Feng X, Yao J, Song Q, et al. Enhancing glioblastoma-specific penetration by functionalization of nanoparticles with an iron-mimic peptide targeting transferrin/transferrin receptor complex. Mol Pharm. 2015;12(8):2947–61.
Chen H, Sha H, Zhang L, Qian H, Chen F, Ding N, et al. Lipid insertion enables targeted functionalization of paclitaxel-loaded erythrocyte membrane nanosystem by tumor-penetrating bispecific recombinant protein. Int J Nanomed. 2018;13:5347–59.
Yang Y, Hong Y, Nam GH, Chung JH, Koh E, Kim IS. Virus-mimetic fusogenic exosomes for direct delivery of integral membrane proteins to target cell membranes. Adv Mater. 2017. https://doi.org/10.1002/adma.201605604.
Alvarez-Erviti L, Seow Y, Yin H, Betts C, Lakhal S, Wood MJA. Delivery of siRNA to the mouse brain by systemic injection of targeted exosomes. Nat Biotechnol. 2011;29(4):341–5.
Rufino-Ramos D, Albuquerque PR, Carmona V, Perfeito R, Nobre RJ, Pereira de Almeida L. Extracellular vesicles: novel promising delivery systems for therapy of brain diseases. J Control Release. 2017;262:247–58.
György B, Szabó TG, Pásztói M, Pál Z, Misják P, Aradi B, et al. Membrane vesicles, current state-of-the-art: emerging role of extracellular vesicles. Cell Mol Life Sci. 2011;68(16):2667–88.
Abels ER, Breakefield XO. Introduction to extracellular vesicles: biogenesis, RNA cargo selection, content, release, and uptake. Cell Mol Neurobiol. 2016;36(3):301–12.
Akers JC, Gonda D, Kim R, Carter BS, Chen CC. Biogenesis of extracellular vesicles (EV): exosomes, microvesicles, retrovirus-like vesicles, and apoptotic bodies. J Neurooncol. 2013;113(1):1–11.
Wang J, Chen D, Ho EA. Challenges in the development and establishment of exosome-based drug delivery systems. J Control Release. 2021;329:894–906.
Mathivanan S, Ji H, Simpson RJ. Exosomes: extracellular organelles important in intercellular communication. J Proteom. 2010;73(10):1907–20.
Théry C, Witwer KW, Aikawa E, Alcaraz MJ, Anderson JD, Andriantsitohaina R, et al. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): a position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. J Extracell Vesicles. 2018;7(1):1535750.
Bashyal S, Thapa C, Lee S. Recent progresses in exosome-based systems for targeted drug delivery to the brain. J Control Release. 2022;348:723–44.
Li P, Kaslan M, Lee SH, Yao J, Gao Z. Progress in exosome isolation techniques. Theranostics. 2017;7(3):789–804.
An M, Wu J, Zhu J, Lubman DM. Comparison of an optimized ultracentrifugation method versus size-exclusion chromatography for isolation of exosomes from human serum. J Proteome Res. 2018;17(10):3599–605.
Kowal J, Arras G, Colombo M, Jouve M, Morath JP, Primdal-Bengtson B, et al. Proteomic comparison defines novel markers to characterize heterogeneous populations of extracellular vesicle subtypes. Proc Natl Acad Sci U S A. 2016;113(8):E968–77.
Heinemann ML, Ilmer M, Silva LP, Hawke DH, Recio A, Vorontsova MA, et al. Benchtop isolation and characterization of functional exosomes by sequential filtration. J Chromatogr A. 2014;1371:125–35.
Manandhar S, Kothandan VK, Oh J, Yoo SH, Hwang J, Hwang SR. A pharmaceutical investigation into exosomes. J Pharm Investig. 2018;48(6):617–26.
Xu Y, Shen L, Li F, Yang J, Wan X, Ouyang M. microRNA-16-5p-containing exosomes derived from bone marrow-derived mesenchymal stem cells inhibit proliferation, migration, and invasion, while promoting apoptosis of colorectal cancer cells by downregulating ITGA2. J Cell Physiol. 2019;234(11):21380–94.
Yang D, Zhang W, Zhang H, Zhang F, Chen L, Ma L, et al. Progress, opportunity, and perspective on exosome isolation—efforts for efficient exosome-based theranostics. Theranostics. 2020;10(8):3684–707.
Batrakova EV, Kim MS. Development and regulation of exosome-based therapy products. Wiley Interdiscip Rev Nanomed Nanobiotechnol. 2016;8(5):744–57.
Vader P, Mol EA, Pasterkamp G, Schiffelers RM. Extracellular vesicles for drug delivery. Adv Drug Deliv Rev. 2016;106(Pt A):148–56.
** XM, **a SJ, Lu R. Drug loading techniques for exosome-based drug delivery systems. Pharmazie. 2021;76(2):61–7.
Kim JK, Youn YJ, Lee YB, Kim SH, Song DK, Shin M, et al. Extracellular vesicles from dHL-60 cells as delivery vehicles for diverse therapeutics. Sci Rep. 2021;11(1):8289.
Long Q, Upadhya D, Hattiangady B, Kim DK, An SY, Shuai B, et al. Intranasal MSC-derived A1-exosomes ease inflammation, and prevent abnormal neurogenesis and memory dysfunction after status epilepticus. Proc Natl Acad Sci U S A. 2017;114(17):E3536–45.
Haney MJ, Zhao Y, Fallon JK, Yue W, Li SM, Lentz EE, et al. Extracellular vesicles as drug delivery system for treatment of neurodegenerative disorders: optimization of the cell source. Adv Nanobiomed Res. 2021;1(12):2100064.
Yuan D, Zhao Y, Banks WA, Bullock KM, Haney M, Batrakova E, et al. Macrophage exosomes as natural nanocarriers for protein delivery to inflamed brain. Biomaterials. 2017;142:1–12.
Fossati G, Ricevuti G, Edwards SW, Walker C, Dalton A, Rossi ML. Neutrophil infiltration into human gliomas. Acta Neuropathol. 1999;98(4):349–54.
Chen H, Li M, Guo Y, Zhong Y, He Z, Xu Y, et al. Immune response in glioma’s microenvironment. Innov Surg Sci. 2021;5(3–4):20190001.
Gieryng A, Pszczolkowska D, Walentynowicz KA, Rajan WD, Kaminska B. Immune microenvironment of gliomas. Lab Investig. 2017;97(5):498–518.
Basheer AS, Abas F, Othman I, Naidu R. Role of inflammatory mediators, macrophages, and neutrophils in glioma maintenance and progression: mechanistic understanding and potential therapeutic applications. Cancers. 2021;13(16):4226.
Ibarra LE, Beaugé L, Arias-Ramos N, Rivarola VA, Chesta CA, López-Larrubia P, et al. Trojan horse monocyte-mediated delivery of conjugated polymer nanoparticles for improved photodynamic therapy of glioblastoma. Nanomedicine. 2020;15(17):1687–707.
Wang C, Li K, Li T, Chen Z, Wen Y, Liu X, et al. Monocyte-mediated chemotherapy drug delivery in glioblastoma. Nanomedicine. 2018;13(2):157–78.
Wang S, Shen H, Mao Q, Tao Q, Yuan G, Zeng L, et al. Macrophage-mediated porous magnetic nanoparticles for multimodal imaging and postoperative photothermal therapy of gliomas. ACS Appl Mater Interfaces. 2021;13(48):56825–37.
Pang L, Qin J, Han L, Zhao W, Liang J, **e Z, et al. Exploiting macrophages as targeted carrier to guide nanoparticles into glioma. Oncotarget. 2016;7(24):37081–91.
Shin D, Christie C, Ju D, Nair RK, Molina S, Berg K, et al. Photochemical internalization enhanced macrophage delivered chemotherapy. Photodiagnosis Photodyn Ther. 2018;21:156–62.
Miao YB, Chen KH, Chen CT, Mi FL, Lin YJ, Chang Y, et al. A noninvasive gut-to-brain oral drug delivery system for treating brain tumors. Adv Mater. 2021;33(34):e2100701.
Li TF, Li K, Wang C, Liu X, Wen Y, Xu YH, et al. Harnessing the cross-talk between tumor cells and tumor-associated macrophages with a nano-drug for modulation of glioblastoma immune microenvironment. J Control Release. 2017;268:128–46.
Pang L, Zhu Y, Qin J, Zhao W, Wang J. Primary M1 macrophages as multifunctional carrier combined with PLGA nanoparticle delivering anticancer drug for efficient glioma therapy. Drug Deliv. 2018;25(1):1922–31.
**ao T, He M, Xu F, Fan Y, Jia B, Shen M, et al. Macrophage membrane-camouflaged responsive polymer nanogels enable magnetic resonance imaging-guided chemotherapy/chemodynamic therapy of orthotopic glioma. ACS Nano. 2021;15(12):20377–90.
Lai J, Deng G, Sun Z, Peng X, Li J, Gong P, et al. Scaffolds biomimicking macrophages for a glioblastoma NIR-Ib imaging guided photothermal therapeutic strategy by crossing blood-brain barrier. Biomaterials. 2019;211:48–56.
Yin T, Fan Q, Hu F, Ma X, Yin Y, Wang B, et al. Engineered macrophage-membrane-coated nanoparticles with enhanced PD-1 expression induce immunomodulation for a synergistic and targeted antiglioblastoma activity. Nano Lett. 2022;22(16):6606–14.
Wang X, Ding H, Li Z, Peng Y, Tan H, Wang C, et al. Exploration and functionalization of M1-macrophage extracellular vesicles for effective accumulation in glioblastoma and strong synergistic therapeutic effects. Signal Transduct Target Ther. 2022;7(1):74.
Jia G, Han Y, An Y, Ding Y, He C, Wang X, et al. NRP-1 targeted and cargo-loaded exosomes facilitate simultaneous imaging and therapy of glioma in vitro and in vivo. Biomaterials. 2018;178:302–16.
Shan S, Chen J, Sun Y, Wang Y, **a B, Tan H, et al. Functionalized macrophage exosomes with panobinostat and PPM1D-siRNA for diffuse intrinsic pontine gliomas therapy. Adv Sci. 2022;9(21):e2200353.
Du Y, Yang Z, Sun Q, Lin M, Wang R, Peng Y, et al. Engineered microglia potentiate the action of drugs against glioma through extracellular vesicles and tunneling nanotubes. Adv Healthc Mater. 2021;10(9):e2002200.
Qiao S, Cheng Y, Liu M, Ji Q, Zhang B, Mei Q, et al. Chemoattractants driven and microglia based biomimetic nanoparticle treating TMZ-resistant glioblastoma multiforme. J Control Release. 2021;336:54–70.
Liu B, Ji Q, Cheng Y, Liu M, Zhang B, Mei Q, et al. Biomimetic GBM-targeted drug delivery system boosting ferroptosis for immunotherapy of orthotopic drug-resistant GBM. J Nanobiotechnology. 2022;20(1):161.
Xue J, Zhao Z, Zhang L, Xue L, Shen S, Wen Y, et al. Neutrophil-mediated anticancer drug delivery for suppression of postoperative malignant glioma recurrence. Nat Nanotechnol. 2017;12(7):692–700.
Wu M, Zhang H, Tie C, Yan C, Deng Z, Wan Q, et al. MR imaging tracking of inflammation-activatable engineered neutrophils for targeted therapy of surgically treated glioma. Nat Commun. 2018;9(1):4777.
Zhang H, Li Z, Gao C, Fan X, Pang Y, Li T, et al. Dual-responsive biohybrid neutrobots for active target delivery. Sci Robot. 2021;6(52):eaaz9519.
Li Y, Teng X, Wang Y, Yang C, Yan X, Li J. Neutrophil delivered hollow titania covered persistent luminescent nanosensitizer for ultrosound augmented chemo/immuno glioblastoma therapy. Adv Sci. 2021;8(17):e2004381.
Wang J, Tang W, Yang M, Yin Y, Li H, Hu F, et al. Inflammatory tumor microenvironment responsive neutrophil exosomes-based drug delivery system for targeted glioma therapy. Biomaterials. 2021;273:120784.
Yin Y, Tang W, Ma XY, Tang L, Zhang Y, Yang M, et al. Biomimetic neutrophil and macrophage dual membrane-coated nanoplatform with orchestrated tumor-microenvironment responsive capability promotes therapeutic efficacy against glioma. Chem Eng J. 2022;433(3):133848.
Deng G, Peng X, Sun Z, Zheng W, Yu J, Du L, et al. Natural-killer-cell-inspired nanorobots with aggregation-induced emission characteristics for near-infrared-II fluorescence-guided glioma theranostics. ACS Nano. 2020;14(9):11452–62.
Li TF, Li K, Zhang Q, Wang C, Yue Y, Chen Z, et al. Dendritic cell-mediated delivery of doxorubicin-polyglycerol-nanodiamond composites elicits enhanced anti-cancer immune response in glioblastoma. Biomaterials. 2018;181:35–52.
Ma X, Kuang L, Yin Y, Tang L, Zhang Y, Fan Q, et al. Tumor-antigen activated dendritic cell membrane-coated biomimetic nanoparticles with orchestrating immune responses promote therapeutic efficacy against glioma. ACS Nano. 2023;17(3):2341–55.
Hao W, Cui Y, Fan Y, Chen M, Yang G, Wang Y, et al. Hybrid membrane-coated nanosuspensions for multi-modal anti-glioma therapy via drug and antigen delivery. J Nanobiotechnol. 2021;19(1):378.
Hambardzumyan D, Gutmann DH, Kettenmann H. The role of microglia and macrophages in glioma maintenance and progression. Nat Neurosci. 2016;19(1):20–7.
Hickey WF. Migration of hematogenous cells through the blood-brain barrier and the initiation of CNS inflammation. Brain Pathol. 1991;1(2):97–105.
Oude Engberink RD, Blezer EL, Hoff EI, van der Pol SM, van der Toorn A, Dijkhuizen RM, et al. MRI of monocyte infiltration in an animal model of neuroinflammation using SPIO-labeled monocytes or free USPIO. J Cereb Blood Flow Metab. 2008;28(4):841–51.
Valable S, Barbier EL, Bernaudin M, Roussel S, Segebarth C, Petit E, et al. In vivo MRI tracking of exogenous monocytes/macrophages targeting brain tumors in a rat model of glioma. Neuroimage. 2008;40(2):973–83.
Madsen SJ, Christie C, Hong SJ, Trinidad A, Peng Q, Uzal FA, et al. Nanoparticle-loaded macrophage-mediated photothermal therapy: potential for glioma treatment. Lasers Med Sci. 2015;30(4):1357–65.
Christie C, Madsen SJ, Peng Q, Hirschberg H. Photothermal therapy employing gold nanoparticle-loaded macrophages as delivery vehicles: comparing the efficiency of nanoshells versus nanorods. J Environ Pathol Toxicol Oncol. 2017;36(3):229–35.
Bai L, Liu Y, Guo K, Zhang K, Liu Q, Wang P, et al. Ultrasound facilitates naturally equipped exosomes derived from macrophages and blood serum for orthotopic glioma treatment. ACS Appl Mater Interfaces. 2019;11(16):14576–87.
Brandenburg S, Blank A, Bungert AD, Vajkoczy P. Distinction of microglia and macrophages in glioblastoma: Close relatives, different tasks?. Int J Mol Sci. 2020;22(1):194.
Vakilian A, Khorramdelazad H, Heidari P, Sheikh Rezaei Z, Hassanshahi G. CCL2/CCR2 signaling pathway in glioblastoma multiforme. Neurochem Int. 2017;103:1–7.
Held-Feindt J, Hattermann K, Müerköster SS, Wedderkopp H, Knerlich-Lukoschus F, Ungefroren H, et al. CX3CR1 promotes recruitment of human glioma-infiltrating microglia/macrophages (GIMs). Exp Cell Res. 2010;316(9):1553–66.
Woerner BM, Warrington NM, Kung AL, Perry A, Rubin JB. Widespread CXCR4 activation in astrocytomas revealed by phospho-CXCR4-specific antibodies. Cancer Res. 2005;65(24):11392–9.
Abels ER, Maas SLN, Nieland L, Wei Z, Cheah PS, Tai E, et al. Glioblastoma-associated microglia reprogramming is mediated by functional transfer of extracellular miR-21. Cell Rep. 2019;28(12):3105-3119.e7.
Gutmann DH, Kettenmann H. Microglia/brain macrophages as central drivers of brain tumor pathobiology. Neuron. 2019;104(3):442–9.
Wei J, Chen P, Gupta P, Ott M, Zamler D, Kassab C, et al. Immune biology of glioma-associated macrophages and microglia: functional and therapeutic implications. Neuro Oncol. 2020;22(2):180–94.
Coniglio SJ, Eugenin E, Dobrenis K, Stanley ER, West BL, Symons MH, et al. Microglial stimulation of glioblastoma invasion involves epidermal growth factor receptor (EGFR) and colony stimulating factor 1 receptor (CSF-1R) signaling. Mol Med. 2012;18(1):519–27.
Domingues P, González-Tablas M, Otero Á, Pascual D, Miranda D, Ruiz L, et al. Tumor infiltrating immune cells in gliomas and meningiomas. Brain Behav Immun. 2016;53:1–15.
Rudek LS, Zimmermann K, Galla M, Meyer J, Kuehle J, Stamopoulou A, et al. Generation of an NFκB-driven alpharetroviral “all-in-one” vector construct as a potent tool for CAR NK cell therapy. Front Immunol. 2021;12:751138.
Takeda A, Sasaki N, Miyasaka M. The molecular cues regulating immune cell trafficking. Proc Jpn Acad Ser B Phys Biol Sci. 2017;93(4):183–95.
Daniel AE, van Buul JD. Endothelial junction regulation: a prerequisite for leukocytes crossing the vessel wall. J Innate Immun. 2013;5(4):324–35.
Sokratous G, Polyzoidis S, Ashkan K. Immune infiltration of tumor microenvironment following immunotherapy for glioblastoma multiforme. Hum Vaccin Immunother. 2017;13(11):2575–82.
Kroll AV, Fang RH, Jiang Y, Zhou J, Wei X, Yu CL, et al. Nanoparticulate delivery of cancer cell membrane elicits multiantigenic antitumor immunity. Adv Mater. 2017. https://doi.org/10.1002/adma.201703969.
Prabhakaran S, Ruff I, Bernstein RA. Acute stroke intervention: a systematic review. JAMA. 2015;313(14):1451–62.
Powers WJ, Rabinstein AA, Ackerson T, Adeoye OM, Bambakidis NC, Becker K, et al. Guidelines for the early management of patients with acute ischemic stroke: 2019 update to the 2018 guidelines for the early management of acute ischemic stroke: a guideline for healthcare professionals from the American Heart Association/American Stroke Association. Stroke. 2019;50(12):e344–418.
Leigh R, Jen SS, Hillis AE, Krakauer JW, Barker PB. Pretreatment blood–brain barrier damage and post-treatment intracranial hemorrhage in patients receiving intravenous tissue-type plasminogen activator. Stroke. 2014;45(7):2030–5.
Jayaraj RL, Azimullah S, Beiram R, Jalal FY, Rosenberg GA. Neuroinflammation: friend and foe for ischemic stroke. J Neuroinflammation. 2019;16(1):142.
Iadecola C, Anrather J. The immunology of stroke: from mechanisms to translation. Nat Med. 2011;17(7):796–808.
Mattila OS, Strbian D, Saksi J, Pikkarainen TO, Rantanen V, Tatlisumak T, et al. Cerebral mast cells mediate blood-brain barrier disruption in acute experimental ischemic stroke through perivascular gelatinase activation. Stroke. 2011;42(12):3600–5.
Ruhnau J, Schulze J, Dressel A, Vogelgesang A. Thrombosis, neuroinflammation, and poststroke infection: the multifaceted role of neutrophils in stroke. J Immunol Res. 2017;2017:5140679.
Ao LY, Yan YY, Zhou L, Li CY, Li WT, Fang WR, et al. Immune cells after ischemic stroke onset: roles, migration, and target intervention. J Mol Neurosci. 2018;66(3):342–55.
Gelderblom M, Weymar A, Bernreuther C, Velden J, Arunachalam P, Steinbach K, et al. Neutralization of the IL-17 axis diminishes neutrophil invasion and protects from ischemic stroke. Blood. 2012;120(18):3793–802.
Weston RM, Jones NM, Jarrott B, Callaway JK. Inflammatory cell infiltration after endothelin-1-induced cerebral ischemia: histochemical and myeloperoxidase correlation with temporal changes in brain injury. J Cereb Blood Flow Metab. 2007;27(1):100–14.
Hou J, Yang X, Li S, Cheng Z. Accessing neuroinflammation sites: monocyte/neutrophil-mediated drug delivery for cerebral ischemia. Sci Adv. 2019;5(7):eaau8301.
Zhang C, Ling CL, Pang L, Wang Q, Liu JX, Wang BS, et al. Direct macromolecular drug delivery to cerebral ischemia area using neutrophil-mediated nanoparticles. Theranostics. 2017;7(13):3260–75.
Feng L, Dou C, **a Y, Li B, Zhao M, Yu P, et al. Neutrophil-like cell-membrane-coated nanozyme therapy for ischemic brain damage and long-term neurological functional recovery. ACS Nano. 2021;15(2):2263–80.
Dong X, Gao J, Zhang CY, Hayworth C, Frank M, Wang Z. Neutrophil membrane-derived nanovesicles alleviate inflammation to protect mouse brain injury from ischemic stroke. ACS Nano. 2019;13(2):1272–83.
Jickling GC, Liu D, Ander BP, Stamova B, Zhan X, Sharp FR. Targeting neutrophils in ischemic stroke: translational insights from experimental studies. J Cereb Blood Flow Metab. 2015;35(6):888–901.
Hermann DM, Kleinschnitz C, Gunzer M. Implications of polymorphonuclear neutrophils for ischemic stroke and intracerebral hemorrhage: predictive value, pathophysiological consequences and utility as therapeutic target. J Neuroimmunol. 2018;321:138–43.
Kang L, Yu H, Yang X, Zhu Y, Bai X, Wang R, et al. Neutrophil extracellular traps released by neutrophils impair revascularization and vascular remodeling after stroke. Nat Commun. 2020;11(1):2488.
Zhang CY, Dong X, Gao J, Lin W, Liu Z, Wang Z. Nanoparticle-induced neutrophil apoptosis increases survival in sepsis and alleviates neurological damage in stroke. Sci Adv. 2019;5(11):eaax7964.
Jiang CT, Wu WF, Deng YH, Ge JW. Modulators of microglia activation and polarization in ischemic stroke (Review). Mol Med Rep. 2020;21(5):2006–18.
Liu S, Gao Y, Yu X, Zhao B, Liu L, Zhao Y, et al. Annexin-1 mediates microglial activation and migration via the CK2 pathway during oxygen-glucose deprivation/reperfusion. Int J Mol Sci. 2016;17(10):1770.
Kanazawa M, Ninomiya I, Hatakeyama M, Takahashi T, Shimohata T. Microglia and monocytes/macrophages polarization reveal novel therapeutic mechanism against stroke. Int J Mol Sci. 2017;18(10):2135.
Huang Y, Chen S, Luo Y, Han Z. Crosstalk between inflammation and the BBB in stroke. Curr Neuropharmacol. 2020;18(12):1227–36.
Amantea D, Bagetta G. Drug repurposing for immune modulation in acute ischemic stroke. Curr Opin Pharmacol. 2016;26:124–30.
Li S, Wang Y, Wu M, Younis MH, Olson AP, Barnhart TE, et al. Spleen-targeted glabridin-loaded nanoparticles regulate polarization of monocyte/macrophage (Mo/Mφ) for the treatment of cerebral ischemia-reperfusion injury. Adv Mater. 2022;34(39):e2204976.
Li F, Zhao L, Shi Y, Liang J. Edaravone-loaded macrophage-derived exosomes enhance neuroprotection in the rat permanent middle cerebral artery occlusion model of stroke. Mol Pharm. 2020;17(9):3192–201.
He R, Jiang Y, Shi Y, Liang J, Zhao L. Curcumin-laden exosomes target ischemic brain tissue and alleviate cerebral ischemia-reperfusion injury by inhibiting ROS-mediated mitochondrial apoptosis. Mater Sci Eng C Mater Biol Appl. 2020;117:111314.
Zheng Y, He R, Wang P, Shi Y, Zhao L, Liang J. Exosomes from LPS-stimulated macrophages induce neuroprotection and functional improvement after ischemic stroke by modulating microglial polarization. Biomater Sci. 2019;7(5):2037–49.
Li C, Zhao Z, Luo Y, Ning T, Liu P, Chen Q, et al. Macrophage-disguised manganese dioxide nanoparticles for neuroprotection by reducing oxidative stress and modulating inflammatory microenvironment in acute ischemic stroke. Adv Sci. 2021;8(20):e2101526.
Su Y, Guo C, Chen Q, Guo H, Wang J, Mu K, et al. Construction of bionanoparticles based on Angelica polysaccharides for the treatment of stroke. Nanomedicine. 2022;44:102570.
Su Y, Guo C, Chen Q, Guo H, Wang J, Kaihang M, et al. Novel multifunctional bionanoparticles modified with sialic acid for stroke treatment. Int J Biol Macromol. 2022;214:278–89.
Long Y, **ang Y, Liu S, Zhang Y, Wan J, Ci Z, et al. Macrophage membrane modified baicalin liposomes improve brain targeting for alleviating cerebral ischemia reperfusion injury. Nanomedicine. 2022;43:102547.
Duan R, Sun K, Fang F, Wang N, He R, Gao Y, et al. An ischemia-homing bioengineered nano-scavenger for specifically alleviating multiple pathogeneses in ischemic stroke. J Nanobiotechnol. 2022;20(1):397.
Qiu YM, Zhang CL, Chen AQ, Wang HL, Zhou YF, Li YN, et al. Immune cells in the BBB disruption after acute ischemic stroke: Targets for immune therapy? Front Immunol. 2021;12:678744.
Gelderblom M, Leypoldt F, Steinbach K, Behrens D, Choe CU, Siler DA, et al. Temporal and spatial dynamics of cerebral immune cell accumulation in stroke. Stroke. 2009;40(5):1849–57.
Salas-Perdomo A, Miró-Mur F, Urra X, Justicia C, Gallizioli M, Zhao Y, et al. T cells prevent hemorrhagic transformation in ischemic stroke by P-selectin binding. Arterioscler Thromb Vasc Biol. 2018;38(8):1761–71.
Gan Y, Liu Q, Wu W, Yin JX, Bai XF, Shen R, et al. Ischemic neurons recruit natural killer cells that accelerate brain infarction. Proc Natl Acad Sci U S A. 2014;111(7):2704–9.
Planas AM. Role of immune cells migrating to the ischemic brain. Stroke. 2018;49(9):2261–7.
Stubbe T, Ebner F, Richter D, Engel O, Klehmet J, Royl G, et al. Regulatory T cells accumulate and proliferate in the ischemic hemisphere for up to 30 days after MCAO. J Cereb Blood Flow Metab. 2013;33(1):37–47.
Liesz A, Suri-Payer E, Veltkamp C, Doerr H, Sommer C, Rivest S, et al. Regulatory T cells are key cerebroprotective immunomodulators in acute experimental stroke. Nat Med. 2009;15(2):192–9.
Wyss-Coray T. Inflammation in Alzheimer disease: Driving force, bystander or beneficial response?. Nat Med. 2006;12(9):1005–15.
Schwartz M, Kipnis J, Rivest S, Prat A. How do immune cells support and shape the brain in health, disease, and aging? J Neurosci. 2013;33(45):17587–96.
Krstic D, Knuesel I. Deciphering the mechanism underlying late-onset Alzheimer disease. Nat Rev Neurol. 2013;9(1):25–34.
Leng F, Edison P. Neuroinflammation and microglial activation in Alzheimer disease: Where do we go from here?. Nat Rev Neurol. 2021;17(3):157–72.
Zhao Y, Wu X, Li X, Jiang LL, Gui X, Liu Y, et al. TREM2 is a receptor for beta-amyloid that mediates microglial function. Neuron. 2018;97(5):1023-1031.e7.
Sanchez-Mejias E, Navarro V, Jimenez S, Sanchez-Mico M, Sanchez-Varo R, Nuñez-Diaz C, et al. Soluble phospho-tau from Alzheimer’s disease hippocampus drives microglial degeneration. Acta Neuropathol. 2016;132(6):897–916.
Bolós M, Llorens-Martín M, Jurado-Arjona J, Hernández F, Rábano A, Avila J. Direct evidence of internalization of Tau by microglia in vitro and in vivo. J Alzheimers Dis. 2016;50(1):77–87.
Rezai-Zadeh K, Gate D, Town T. CNS infiltration of peripheral immune cells: D-Day for neurodegenerative disease?. J Neuroimmune Pharmacol. 2009;4(4):462–75.
Hohsfield LA, Humpel C. Migration of blood cells to β-amyloid plaques in Alzheimer’s disease. Exp Gerontol. 2015;65:8–15.
Town T, Laouar Y, Pittenger C, Mori T, Szekely CA, Tan J, et al. Blocking TGF-beta-Smad2/3 innate immune signaling mitigates Alzheimer-like pathology. Nat Med. 2008;14(6):681–7.
Böttger D, Ullrich C, Humpel C. Monocytes deliver bioactive nerve growth factor through a brain capillary endothelial cell-monolayer in vitro and counteract degeneration of cholinergic neurons. Brain Res. 2010;1312:108–19.
Wang H, Sui H, Zheng Y, Jiang Y, Shi Y, Liang J, et al. Curcumin-primed exosomes potently ameliorate cognitive function in AD mice by inhibiting hyperphosphorylation of the Tau protein through the AKT/GSK-3β pathway. Nanoscale. 2019;11(15):7481–96.
Hao Y, Su C, Liu X, Sui H, Shi Y, Zhao L. Bioengineered microglia-targeted exosomes facilitate Aβ clearance via enhancing activity of microglial lysosome for promoting cognitive recovery in Alzheimer’s disease. Biomater Adv. 2022;136: 212770.
Huo Q, Shi Y, Qi Y, Huang L, Sui H, Zhao L. Biomimetic silibinin-loaded macrophage-derived exosomes induce dual inhibition of Aβ aggregation and astrocyte activation to alleviate cognitive impairment in a model of Alzheimer’s disease. Mater Sci Eng C Mater Biol Appl. 2021;129:112365.
Han Y, Gao C, Wang H, Sun J, Liang M, Feng Y, et al. Macrophage membrane-coated nanocarriers co-modified by RVG29 and TPP improve brain neuronal mitochondria-targeting and therapeutic efficacy in Alzheimer’s disease mice. Bioact Mater. 2020;6(2):529–42.
Park TE, Singh B, Li H, Lee JY, Kang SK, Choi YJ, et al. Enhanced BBB permeability of osmotically active poly(mannitol-co-PEI) modified with rabies virus glycoprotein via selective stimulation of caveolar endocytosis for RNAi therapeutics in Alzheimer’s disease. Biomaterials. 2015;38:61–71.
Dos Santos Rodrigues B, Arora S, Kanekiyo T, Singh J. Efficient neuronal targeting and transfection using RVG and transferrin-conjugated liposomes. Brain Res. 2020;1734:146738.
Marrache S, Dhar S. Engineering of blended nanoparticle platform for delivery of mitochondria-acting therapeutics. Proc Natl Acad Sci U S A. 2012;109(40):16288–93.
Sapoń K, Maziarz D, Janas T, Sikorski AF, Janas T. Cholera toxin subunit B for sensitive and rapid determination of exosomes by gel filtration. Membranes. 2020;10(8):172.
He X, Guan F, Lei L. Structure and function of glycosphingolipids on small extracellular vesicles. Glycoconj J. 2022;39(2):197–205.
Yanagisawa K. GM1 ganglioside and the seeding of amyloid in Alzheimer’s disease: endogenous seed for Alzheimer amyloid. Neuroscientist. 2005;11(3):250–60.
Fernández-Pérez EJ, Sepúlveda FJ, Peoples R, Aguayo LG. Role of membrane GM1 on early neuronal membrane actions of Aβ during onset of Alzheimer’s disease. Biochim Biophys Acta Mol Basis Dis. 2017;1863(12):3105–16.
Zimmer H, Riese S, Régnier-Vigouroux A. Functional characterization of mannose receptor expressed by immunocompetent mouse microglia. Glia. 2003;42(1):89–100.
McManus RM, Mills KH, Lynch MA. T cells-protective or pathogenic in Alzheimer’s disease?. J Neuroimmune Pharmacol. 2015;10(4):547–60.
Rossi B, Santos-Lima B, Terrabuio E, Zenaro E, Constantin G. Common peripheral immunity mechanisms in multiple sclerosis and Alzheimer’s disease. Front Immunol. 2021;12:639369.
Pietronigro EC, Della Bianca V, Zenaro E, Constantin G. NETosis in Alzheimer’s disease. Front Immunol. 2017;8:211.
Manda-Handzlik A, Demkow U. The brain entangled: the contribution of neutrophil extracellular traps to the diseases of the central nervous system. Cells. 2019;8(12):1477.
Zenaro E, Pietronigro E, Della Bianca V, Piacentino G, Marongiu L, Budui S, et al. Neutrophils promote Alzheimer’s disease-like pathology and cognitive decline via LFA-1 integrin. Nat Med. 2015;21(8):880–6.
Gabbita SP, Johnson MF, Kobritz N, Eslami P, Poteshkina A, Varadarajan S, et al. Oral TNFα modulation alters neutrophil infiltration, improves cognition and diminishes Tau and amyloid pathology in the 3xTgAD mouse model. PLoS One. 2015;10(10):e0137305.
Pietronigro E, Zenaro E, Bianca VD, Dusi S, Terrabuio E, Iannoto G, et al. Blockade of α4 integrins reduces leukocyte-endothelial interactions in cerebral vessels and improves memory in a mouse model of Alzheimer’s disease. Sci Rep. 2019;9(1):12055.
Zhu B, Yin D, Zhao H, Zhang L. The immunology of Parkinson’s disease. Semin Immunopathol. 2022;44(5):659–72.
Öberg M, Fabrik I, Fabrikova D, Zehetner N, Härtlova A. The role of innate immunity and inflammation in Parkinson´s disease. Scand J Immunol. 2021;93(5):e13022.
Pajares M, Rojo AI, Manda G, Boscá L, Cuadrado A. Inflammation in Parkinson’s disease: mechanisms and therapeutic implications. Cells. 2020;9(7):1687.
Harms AS, Ferreira SA, Romero-Ramos M. Periphery and brain, innate and adaptive immunity in Parkinson’s disease. Acta Neuropathol. 2021;141(4):527–45.
Brynskikh AM, Zhao Y, Mosley RL, Li S, Boska MD, Klyachko NL, et al. Macrophage delivery of therapeutic nanozymes in a murine model of Parkinson’s disease. Nanomedicine. 2010;5(3):379–96.
Zhao Y, Haney MJ, Mahajan V, Reiner BC, Dunaevsky A, Mosley RL, et al. Active targeted macrophage-mediated delivery of catalase to affected brain regions in models of Parkinson’s disease. J Nanomed Nanotechnol. 2011;S4:003.
Haney MJ, Zhao Y, Li S, Higginbotham SM, Booth SL, Han HY, et al. Cell-mediated transfer of catalase nanoparticles from macrophages to brain endothelial, glial and neuronal cells. Nanomedicine. 2011;6(7):1215–30.
Haney MJ, Zhao YL, Harrison EB, Mahajan V, Ahmed S, He Z, et al. Specific transfection of inflamed brain by macrophages: a new therapeutic strategy for neurodegenerative diseases. PLoS One. 2013;8(4):e61852.
Biju K, Zhou Q, Li G, Imam SZ, Roberts JL, Morgan WW, et al. Macrophage-mediated GDNF delivery protects against dopaminergic neurodegeneration: a therapeutic strategy for Parkinson’s disease. Mol Ther. 2010;18(8):1536–44.
Chen C, Li X, Ge G, Liu J, Biju KC, Laing SD, et al. GDNF-expressing macrophages mitigate loss of dopamine neurons and improve Parkinsonian symptoms in MitoPark mice. Sci Rep. 2018;8(1):5460.
Chen C, Guderyon MJ, Li Y, Ge G, Bhattacharjee A, Ballard C, et al. Non-toxic HSC transplantation-based macrophage/microglia-mediated GDNF delivery for Parkinson’s disease. Mol Ther Methods Clin Dev. 2019;17:83–98.
Zhao Y, Haney MJ, Gupta R, Bohnsack JP, He Z, Kabanov AV, et al. GDNF-transfected macrophages produce potent neuroprotective effects in Parkinson’s disease mouse model. PLoS One. 2014;9(9):e106867.
Zhao Y, Haney MJ, ** YS, Uvarov O, Vinod N, Lee YZ, et al. GDNF-expressing macrophages restore motor functions at a severe late-stage, and produce long-term neuroprotective effects at an early-stage of Parkinson’s disease in transgenic Parkin Q311X(A) mice. J Control Release. 2019;315:139–49.
Zhao Y, Haney MJ, Fallon JK, Rodriguez M, Swain CJ, Arzt CJ, et al. Using extracellular vesicles released by GDNF-transfected macrophages for therapy of Parkinson disease. Cells. 2022;11(12):1933.
Biju KC, Santacruz RA, Chen C, Zhou Q, Yao J, Rohrabaugh SL, et al. Bone marrow-derived microglia-based neurturin delivery protects against dopaminergic neurodegeneration in a mouse model of Parkinson’s disease. Neurosci Lett. 2013;535:24–9.
Haney MJ, Klyachko NL, Zhao Y, Gupta R, Plotnikova EG, He Z, et al. Exosomes as drug delivery vehicles for Parkinson’s disease therapy. J Control Release. 2015;207:18–30.
Shen LM, Li MC, Wei WJ, Guan X, Liu J. In vitro neuroprotective effects of macrophage membrane-derived curcumin-loaded carriers against 1-methyl-4-phenylpyridinium-induced neuronal damage. ACS Omega. 2021;6(47):32133–41.
Rodríguez Murúa S, Farez MF, Quintana FJ. The immune response in multiple sclerosis. Annu Rev Pathol. 2022;17:121–39.
Wang J, Wang J, Wang J, Yang B, Weng Q, He Q. Targeting microglia and macrophages: a potential treatment strategy for multiple sclerosis. Front Pharmacol. 2019;10:286.
Nally FK, de Santi C, McCoy CE. Nanomodulation of macrophages in multiple sclerosis. Cells. 2019;8(6):543.
Carnasciali A, Amoriello R, Bonechi E, Mazzoni A, Ravagli C, Doumett S, et al. T cell delivery of nanoparticles-bound anti-CD20 monoclonal antibody: successful B cell depletion in the spinal cord during experimental autoimmune encephalomyelitis. J Neuroimmune Pharmacol. 2021;16(2):376–89.
Montalban X, Hauser SL, Kappos L, Arnold DL, Bar-Or A, Comi G, et al. Ocrelizumab versus placebo in primary progressive multiple sclerosis. N Engl J Med. 2017;376(3):209–20.
Qin J, Yang X, Zhang RX, Luo YX, Li JL, Hou J, et al. Monocyte mediated brain targeting delivery of macromolecular drug for the therapy of depression. Nanomedicine. 2015;11(2):391–400.
Han H, Eyal S, Portnoy E, Mann A, Shmuel M, Benifla M, et al. Monocytes as carriers of magnetic nanoparticles for tracking inflammation in the epileptic rat brain. Curr Drug Deliv. 2019;16(7):637–44.
Acknowledgements
Not applicable.
Funding
This work was supported by the National Natural Science Foundation of China (82204634, 82174047, 81622051), the Zhejiang Provincial Natural Science Foundation of China (LQ22H280010), and the Foundation of Zhejiang Chinese Medical University (2021ZR03).
Author information
Authors and Affiliations
Contributions
SZ collected the data and wrote the manuscript. RL helped in data collection. ZC, XW, ASD, and XF revised the manuscript. ASD, XF conceived and supervised the study. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Zhang, SS., Li, RQ., Chen, Z. et al. Immune cells: potential carriers or agents for drug delivery to the central nervous system. Military Med Res 11, 19 (2024). https://doi.org/10.1186/s40779-024-00521-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40779-024-00521-y