
Abstract
Background
The fungal component of the human gut microbiome, also known as the mycobiome, plays a vital role in intestinal ecology and human health. However, the overall structure of the gut mycobiome as well as the inter-individual variations in fungal composition remains largely unknown. In this study, we collected a total of 3363 fungal sequencing samples from 16 cohorts across three continents, including 572 newly profiled samples from China.
Results
We identify and characterize four mycobiome enterotypes using ITS profiling of 3363 samples from 16 cohorts. These enterotypes exhibit stability across populations and geographical locations and significant correlation with bacterial enterotypes. Particularly, we notice that fungal enterotypes have a strong age preference, where the enterotype dominated by Candida (i.e., Can_type enterotype) is enriched in the elderly population and confers an increased risk of multiple diseases associated with a compromised intestinal barrier. In addition, bidirectional mediation analysis reveals that the fungi-contributed aerobic respiration pathway associated with the Can_type enterotype might mediate the association between the compromised intestinal barrier and aging.
Conclusions
We show that the human gut mycobiome has stable compositional patterns across individuals and significantly correlates with multiple host factors, such as diseases and host age.
Video Abstract
Similar content being viewed by others


Background
The human gut microbiome, which consists of multi-kingdom microbes of prokaryotes, viruses, protists and fungi, is essential to human health [1]. Current research mainly focuses on the prokaryotic and viral components of the gut ecology [2,3,4]. However, the complicated associations of other types of microorganisms, particularly fungi, with human health remain largely unknown. Although the fungal community, also known as mycobiome, comprises less than 1% of the entire human gut microbiome [5], they have been shown to be involved in disease pathogenesis and to profoundly influence the host immune system [6, 7]. For example, Candida albicans can cause infections in immunocompromised human hosts [8], and alterations of the gut mycobiome composition have been reported in multiple human diseases [9, 10]. While fine-grained fungal taxonomic markers associated with certain phenotypes have been reported [9, 11, 12], the overall structure of the gut mycobiome and the inter-individual variation in fungal composition remain unclear.
Enterotypes, which have been proposed to summarize the human gut microbial characteristics, are effective in stratifying populations and providing a global overview of the inter-individual variations in gut microbial composition [13, 14]. Multiple studies have consistently identified bacterial enterotypes, which are independent of the distribution of the hosts’ age, geography, and gender [13,14,15,16]. Defined based on the prokaryotic compositional patterns, enterotypes could enhance understanding of human health and facilitate intervention [17]. As an integral part of the human gut multi-kingdom microbiome, fungi share microhabitats with the prokaryotic microbiome in the gut through different types of interactions, such as mutualism, commensalism, and competition [18]. Notably, several fungi-bacteria synergistic interactions within the human gut have been reported to be associated with human diseases. For instance, Hoarau et al. [19] found a positive inter-kingdom correlation between Candida tropicalis and two bacterial species, Serratia macesecens and Escherichia coli, in individuals with Crohn’s diseases. The physical interactions among these three species resulted in the formation of robust biofilms, which potentially cause host’s tissue damage and trigger specific immune responses [20]. Hence, the interactions between fungi and bacteria within the human gut play important roles in sha** the ecology of the intestinal microbial community [18, 21]. However, the landscape of the human gut mycobiome and whether fungal enterotype-like structures exist in the human gut are unclear.
In this study, we collected 3,363 fungal sequencing samples from 16 cohorts across Europe, North America, and Asia, including 572 newly sequenced samples from China. Four fungal enterotypes were identified independently of cohorts and geographical regions and were closely correlated with bacterial enterotypes. We noticed strong effects of host phenotypes (including age and diseases) on the fungal enterotypes. Notably, the Candida (Can_type) enterotype, enriched in the elderly population, showed a higher prevalence in patients with multiple diseases, even beyond the age influence, and was associated with a severe compromised intestinal barrier. Furthermore, a Can_type-enriched aerobic respiration pathway mediated the association between the compromised intestinal barrier and aging. Overall, our findings elucidated the highly structured nature of the gut mycobiome and its clinical relevance to human health.
Results
Landscape of human gut mycobiome composition and diversity
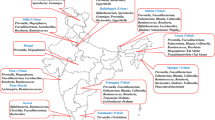
To characterize the human gut fungal diversity and composition, we collected internal transcribed spacer (ITS) sequencing data from 15 published projects (Supplementary Table S1) [12, 22,23,24,25,26,27,28,29,30]. In addition, we recruited 572 Chinese participants (Chinese Gut Mycobiome cohort, or CHGM) aged from 17 to 89 years old and profiled their fecal mycobiome with ITS1 sequencing. In total, 3363 fecal samples with ITS1- (960 samples; hereafter referred to as ‘ITS1-combined’) or ITS2- (2403 samples; hereafter referred to as ‘ITS2-combined’) sequencing data from 16 cohorts covering three continents (Europe, North America, and Asia) were included in our study (Fig. 1a).

Composition and diversity of the human gut mycobiome across studies and geographic sites. a Geographic distribution of study populations and associated fungal enterotypes, where the datasets are sequenced with either ITS1 or ITS2 barcodes. b Genus-level gut mycobiome composition across the three continents (North America, Europe, and Asia). c Cumulative curves of the number of detected genera according to the number of sequenced samples from different study populations. d The distribution of fungal Shannon diversity across study populations. The Venn diagram shows the number of fungal genera detected by ITS1- and ITS2- based amplification. e, The correlation between the Shannon index of bacteria and that of fungi in the Zuo et al. [22] cohort, with shaded region representing 95% confidence intervals of the linear regression
The combined dataset (3363 samples) contained a total of 1,120 genus-level taxonomic groups, where 354 fungi were present in at least 10 samples, and the sequencing depth of most cohorts was sufficient to capture the diversity of the gut mycobiome (Figure S2). With sample rarefaction analysis, we noticed that the number of detected genera in the Germany (Andrea et al. [28]) and Chinese (CHGM) populations dramatically increased with an increasing number of samples, and the number of fungal genera detected in our CHGM cohort far exceeded those of other cohorts (Fig. 1c, Figure S1b). However, the observed number of the fungal genera was still considerably below the estimated saturation level, even when combining all datasets (Figure S1c), suggesting a requirement for a further increase in sample size to characterize the comprehensive gut fungal diversity. At the genus level, Saccharomyces and Candida were the most abundant genera across all samples, followed by Penicillium and Aspergillus (Fig. 1b). These genera are also the most common commensal fungi in other human body sites, including skin, lung, and oral cavity [58]. To better unveil the colonization of fungi in the gut, profiling of active fungal community by ITS cDNA analysis is needed in future work. Secondly, the interactions between the bacteria and fungi were not explored here. The landscape of multi-kingdom interactions can provide insights into the mechanisms underlying the gut mycobiome structure and its association with host physiological conditions. Finally, we explored the functions of gut fungi based on the metagenomics data. However, the metagenomics data is dominated by bacteria, which leads to the underrepresentation of functional profiling of gut mycobiome. Fungi-enriched metagenomics sequencing can be helpful to infer the complete functional profiling of the mycobiome in the future.
Conclusions
In this study, we characterized the human gut fungal community structures with a broad spectrum of ITS sequencing samples from 16 cohorts across 11 countries worldwide, including 572 newly ITS-profiled and metagenomically sequenced samples from China. We confirmed the existence of four fungal enterotypes that varied in taxonomic and functional compositions. These enterotypes showed close associations with both age and diseases, with the Candida-dominated enterotype being particularly enriched in the elderly population and associated with multiple human diseases accompanied by a compromised intestinal barrier. Bidirectional mediation analysis further revealed that the Can_type-associated fungi-contributed aerobic respiration pathway could mediate the association between aging and the compromised intestinal barrier. These findings reveal both the biological and clinical significance of fungal enterotypes and offer a new perspective on host-microbe interactions.
Materials and methods
Data collection
We downloaded ITS sequencing data of fecal samples from public databases including National Center for Biotechnology Information (NCBI) sequence read archive (SRA) and China National GeneBank database (CNGBdb). Samples with read number fewer than 10,000 were discarded. Due to the instability and large difference in the human gut mycobiome of infants, we excluded samples from infants. Metadata including demographics (e.g., age, gender, BMI, country) and human disease phenotypes were also retrieved from corresponding publications or databases. As a result, we collected a total of 2791 public samples from 11 countries covering multiple human disease phenotypes including clostridium difficile infection (CDI), alcohol use disorder (AUD), coronavirus disease 2019 (COVID-19), type 2 diabetes (T2D), irritable bowel syndrome (IBS), alcoholic hepatitis (ALHP), Crohn’s disease (CD), and melanoma. The details for each project including the number of samples, country, associated disease phenotype and used amplicon targets were listed in Supplementary Table S1.
We additionally collected human fecal samples from newly recruited 572 Chinese volunteers (CHGM cohort) with age ranging from 18 to 89 years old, where the fecal mycobiome was profiled with ITS1 amplification. Of these samples, 74 were collected from subjects with Alzheimer’s disease (AD) enrolled in Shanghai Sixth People’s Hospital, whereas others were obtained from healthy volunteers recruited in Wuhan, Shanghai, and Zhengzhou. Subjects who take antibiotics, antifungals or probiotics up to 1 month prior to sample collection were excluded from this study. The study protocol was approved by the Human Ethics Committee of the School of Life Science of Fudan University (No. BE1940) and the Ethics Committee of the Tongji Medical College of Huazhong University of Science. All subjects provided informed consent before participation and were asked to complete questionnaires. In total, the combined dataset consisted of 3363 samples from 16 cohorts and covered 11 countries from three continents, including Europe (615 samples), North America (344 samples) and Asia (2404 samples); among which, the fungal compositions of six and nine cohorts were determined by ITS1- (960 samples) and ITS2- (2403 samples) sequencing.
DNA extraction from fecal samples
After sample collection, the fecal samples from the CHGM cohort were immediately stored on dry ice and transported to a refrigerator at – 80 ℃ within 5 h. Total DNA was extracted from fecal samples using semi-automated DNeasy PowerSoil HTP 96 Kit (Qiagen, 12,955–4) according to manufacturer’s instructions. The purified DNAs were quality-checked by 1% agarose gel, and DNA concentration and purity were determined with NanoDrop 2000 UV–vis spectrophotometer (Thermo Scientific, Wilmingtom, USA).
ITS sequencing and procession
The mycobiome of CHGM cohort was profiled by the sequencing of Internal Transcribed Spacer (ITS), and the ITS1 hypervariable region was amplified with primer pairs ITS1F (5′-CTTGGTCATTTAGAGGAAGTAA-3′) and ITS2R (5′-GCTGCGTTCTTCATCGATGC-3′) [59] by an BI GeneAmp® 9700 PCR thermocycler (ABI, CA, USA). The PCR amplification was conducted as follows: initial denaturation at 95 ℃ for 3 min, followed by 27 cycles of denaturing at 95 ℃ for 30 s, annealing at 55 ℃ for 30 s, elongation at 72 ℃ for 45 s and a final extension at 72 ℃ for 10 min. The PCR mixtures (20 μL total value) contained 4 μL of 5 × FastPfu buffer, 2 μL of 2.5 mM dNTPs, 0.8 μL of each primer (5 μM concentration), 0.4 μL of FastPfu DNA Polymerase and 10 ng of template DNA. The PCR products were extracted from 2% agarose gel and purified using the AxyPrep DNA Gel Extraction Kit (Axygen Biosciences, Union City, CA, USA) according to manufacturer’s instructions, and further quantified using Quantus™ Fluorometer (Promega, USA). Purified amplicons were pooled and paired-end sequenced on Illumina MiSeq PE300 platform (Illumina, San Diego, USA) according to the standard protocols by Majorbio Bio-Pharm Technology Co. Ltd. (Shanghai, China).
The raw ITS reads were first demultiplexed, quality-filtered by fastp version 0.20.0 [60] and merged by FLASH version 1.2.7 [61] with the following criteria: (i) the 300 bp reads were truncated at any site with an average quality score < 20 over a 50-bp sliding window, and the truncated reads shorter than 50 bp were discarded; (ii) only overlap** sequences longer than 10 bp were assembled according to their overlapped sequence, and the maximum mismatch ratio of overlap region is 0.2. QIIME2 (version 2019.7) was used for the downstream analysis [62]. The quality-filtered ITS reads were then denoised and clustered into amplicon sequence variants (ASVs) using DADA2 [63], and chimeric sequences were identified and removed. Then the Naïve Bayes classifier trained on the UNITE reference database [64] was used for taxonomy assignment of individual ASVs. \(\alpha\)- and \(\beta\)-diversity analysis was conducted on samples at the sampling depth of 10,000 by utilizing the R packages “vegan” (version 2.5–7) [65] and “phyloseq” (version 1.34.0) [66]. \(\alpha\)-diversity was estimated by the Shannon index (evenness and richness of community within a sample), Simpson index (a qualitive measure of community diversity that accounts for both the number and the abundance of features), Faith’s phylogenetic diversity (or Faith’s PD; a qualitative measure of community diversity that incorporates both the phylogenetic relationship and abundance of the observed features) and richness (observed number of features). The fungal genera presented in less than 10 samples were excluded from downstream analysis.
Metagenomics sequencing and processing
The fecal bacterial microbiome of CHGM cohort was profiled by whole-genome shotgun sequencing with Illumina HiSeq 2000 platform (Novogen, Bei**g, China). DNA libraries were prepared as described previously [67]. The raw sequencing reads were quality-filtered using fastp version 0.20.0, followed by the use of Bowtie2 [68] to remove host-derived reads by map** to the human reference genome (hg38). Quantitative profiling of the taxonomic composition of the microbial communities was performed via MetaPhlAn2 [69]. Profiling of microbial pathways was performed with HUMAnN2 v2.8.1 [70] by map** reads to Uniref90 [71] and MetaCyc [72] reference databases. Both the abundance output of MetaPhlAn2 and HUMAnN2 were normalized into the relative abundance. We extracted the metabolic pathways of gut fungi for downstream analysis. The metabolic pathways or bacterial species presented in less than 10 samples were excluded from downstream analysis. To estimate the percentage of human DNA contents (HDCs) within CHGM cohort, we aligned the clean reads to the human reference genome with bowtie2, and the HDCs was calculated as the percentage of mapped reads to the total number of clean reads.
16S rRNA sequencing data processing
The 16S rRNA sequencing data available for four cohorts including Lemoinne et al. [27], Vitali et al. [73], Prochazkova et al. [30], and Zuo et al. [22] were downloaded from NCBI SRA. Raw 16S reads were quality filtered, clustered into ASVs and taxonomic annotated using QIIME2 (version 2019.7) as described above. The taxonomies of ASVs were annotated by using the SILVA database [74]. \(\alpha\)- and \(\beta\)-diversity analysis was conducted on samples at the sampling depth of 25,000. The bacterial genera presented in less than 10 samples were excluded from consideration.
Fungal enterotype clustering
The fecal samples of ITS1 and ITS2 amplification were separately clustered into fungal enterotypes by using a partitioning around medoid (PAM) clustering method [75] as those previously described for bacterial enterotype discovery [13, 14]. Briefly, the samples were grouped into clusters with partitioning around medoid (PAM) based on the between-sample Bray–Curtis distance calculated at genus-level, where three other widely used distance matrices including Jaccard, Kulcxynski, and Jensen-Shannon distance (JSD) were also considered to validate the robustness of fungal enterotypes (Figure S4a). The optimal number of clusters was determined by the silhouette index. The driver genera of each enterotype was determined as the genus with the highest relative abundance in the enterotype.
We further validated the structural similarity of fungal enterotypes obtained separately from ITS1 and ITS2-combined fungal datasets. Specifically, we performed cross-dataset validation between ITS1 and ITS2 datasets with one dataset used for training a LASSO logistic regression model [76] to predict the fungal enterotype in the other dataset. To determine whether the fungal enterotypes can reflect the overall community structure and not only the difference of the driver genera, we further removed driver genera, Candida, Saccharomyces, Aspergillus, Saccharomyces sp and Ascomycota sp from the data and re-performed cross-validation as described above.
Gut aging index
We calculated the gut aging index (GAI) by using the relative abundance of 21 age-associated gut fungal genera. Subjects with diseases or age below 18 years old were excluded from this analysis. To identify age-associated fungi, we adopted a multivariate linear regression analysis on 531 healthy subjects with age ranging from 18 to 90 years from four cohorts (i.e., the CHGM cohort, Gao et al. [23], Limon et al. [12], and Zuo et al. [22]) to examine the associations between age and the relative abundance of fungal genera with the adjustment of gender and cohort. The fungal genera associated with a p values < 0.05 in the linear regression test were considered as “age-associated”. We grouped these age-associated gut fungal genera into two sets \({M}_{P}\) and \({M}_{N}\), where \({M}_{P}\) was the set of fungal genera positively associated with age and vice versa for \({M}_{N}\). We then coupled these two fungal genera sets with a computational procedure (see below) to define a gut aging index (GAI) for a mycobiome sample. The GAI of sample i is defined as.
where \({R}_{{M}_{P,i}}\) denotes the richness of \({M}_{P}\) (or the number of present fungal genera of \({M}_{P}\) in sample i) in sample i, \(|{M}_{P}|\) is the size of set \({M}_{P}\) (or the overall number of fungal genera in \({M}_{P}\)), \({x}_{j,i}\) denotes the relative abundance of fungi j in sample i and the same for \({R}_{{M}_{N,i}}\) and \(|{M}_{N}|\). The calculation of GAI considered both the richness and the relative abundance of age-associated gut fungal genera to quantify the balance between \({M}_{P}\) and \({M}_{N}\). Due to the difference between the set sizes of \({M}_{P}\) and \({M}_{N}\), we calculated the proportion of the present fungi of these two sets for each sample (\(\frac{{R}_{{M}_{P}}}{|{M}_{P}|}\) and \(\frac{{R}_{{M}_{N}}}{|{M}_{N}|}\)) instead of the richness \({R}_{{M}_{P}}\) and \({R}_{{M}_{N}}\). As such, a higher GAI or GAI > 0 indicates that a more age-positive related fungal profile rather than an age-negative related fungal profile in one sample, and thus suggests a higher intestinal aging degree.
Statistical analysis
All statistical analysis were conducted using R version 4.0.5 within RStudio and all figures were visualized by using “ggplot2” package version 3.3.5 [77]. The quantification of the variance explained by factors (e.g., continent, amplicon target) was calculated using the permutational multivariate analysis of variance (PERMANOVA, permutations = 999, distance = “bray”) as implemented by the “adonis” function in the R package “vegan”. Correlation between the \(\alpha\)-diversity and chronological age was assessed with Spearman’s correlation. Comparisons of enterotype characteristics (e.g., diversity), host phenotypes (e.g., BMI, age, gender, disease) and health related index (e.g., HDCs, GAI, and GMHI) across fungal enterotypes were performed using Fisher’s exact test or chi-square test for categorical variables and Wilcoxon rank-sum tests for continuous variables. The pathways enriched in each enterotype were determined by using a Wilcoxon-rank-sum test, where the other three enterotypes were combined into a single group. The bi-directional mediation analysis was performed using the “mediate” function within the R package “mediation” (version 4.5.0) [78] with 1000 bootstrap sampling times to infer the causal role of the aging in contributing to the compromised intestinal barrier through the fungi-contributed aerobic respiration pathway. For analysis regarding multiple comparisons, the Benjamini–Hochberg false discovery rate (adjusted p) [79] was employed to correct for multiple testing. The results with adjusted p < 0.05 were considered significant without statement specially.
Availability of data and materials
The raw data of the ITS1 sequencing and metagenomic sequencing of the CHGM cohort are available in the China National Center for Bioinformation (CNCB) under accession number PRJCA010668). Other datasets used for analysis are available from the corresponding author and can be downloaded from NCBI database directly (see Supplementary Table S1). All custom code used in this work is available at the following GitHub repository: https://github.com/ZhaoXM-Lab/Mycobiome_cohort_analysis.
References
Barrera-Vazquez OS, Gomez-Verjan JC. The unexplored world of human virome, mycobiome, and archaeome in aging. J Gerontol A Biol Sci Med Sci. 2020;75:1834–7.
Gregory AC, Zablocki O, Zayed AA, Howell A, Bolduc B, Sullivan MB. The gut virome database reveals age-dependent patterns of virome diversity in the human gut. Cell Host Microbe. 2020;28(724–740): e728.
Camarillo-Guerrero LF, Almeida A, Rangel-Pineros G, Finn RD, Lawley TD. Massive expansion of human gut bacteriophage diversity. Cell. 2021;184(1098–1109): e1099.
Pasolli E, Asnicar F, Manara S, Zolfo M, Karcher N, Armanini F, Beghini F, Manghi P, Tett A, Ghensi P, et al. Extensive unexplored human microbiome diversity revealed by over 150,000 genomes from metagenomes spanning age, geography, and lifestyle. Cell. 2019;176(649–662): e620.
Huffnagle GB, Noverr MC. The emerging world of the fungal microbiome. Trends Microbiol. 2013;21:334–41.
Underhill DM, Iliev ID. The mycobiota: interactions between commensal fungi and the host immune system. Nat Rev Immunol. 2014;14:405–16.
Zhai B, Ola M, Rolling T, Tosini NL, Joshowitz S, Littmann ER, Amoretti LA, Fontana E, Wright RJ, Miranda E, et al. High-resolution mycobiota analysis reveals dynamic intestinal translocation preceding invasive candidiasis. Nat Med. 2020;26:59–64.
Dai Q, Zhang FL, Feng T. Sesquiterpenoids specially produced by fungi: structures, biological activities, chemical and biosynthesis (2015–2020). J Fungi (Basel). 2021;7(12):1026.
Das A, O’Herlihy E, Shanahan F, O’Toole PW, Jeffery IB. The fecal mycobiome in patients with Irritable Bowel Syndrome. Sci Rep. 2021;11:124.
Frau A, Ijaz UZ, Slater R, Jonkers D, Penders J, Campbell BJ, Kenny JG, Hall N, Lenzi L, Burkitt MD, et al. Inter-kingdom relationships in Crohn’s disease explored using a multi-omics approach. Gut Microbes. 2021;13:1930871.
Ling Z, Zhu M, Liu X, Shao L, Cheng Y, Yan X, Jiang R, Wu S. Fecal fungal dysbiosis in chinese patients with Alzheimer’s disease. Front Cell Dev Biol. 2020;8: 631460.
Limon JJ, Tang J, Li D, Wolf AJ, Michelsen KS, Funari V, Gargus M, Nguyen C, Sharma P, Maymi VI, et al. Malassezia is associated with crohn’s disease and exacerbates colitis in mouse models. Cell Host Microbe. 2019;25(377–388): e376.
Arumugam M, Raes J, Pelletier E, Le Paslier D, Yamada T, Mende DR, Fernandes GR, Tap J, Bruls T, Batto JM, et al. Enterotypes of the human gut microbiome. Nature. 2011;473:174–80.
Costea PI, Hildebrand F, Arumugam M, Backhed F, Blaser MJ, Bushman FD, de Vos WM, Ehrlich SD, Fraser CM, Hattori M, et al. Enterotypes in the landscape of gut microbial community composition. Nat Microbiol. 2018;3:8–16.
Liang C, Tseng HC, Chen HM, Wang WC, Chiu CM, Chang JY, Lu KY, Weng SL, Chang TH, Chang CH, et al. Diversity and enterotype in gut bacterial community of adults in Taiwan. BMC Genomics. 2017;18:932.
Mobeen F, Sharma V, Tulika P. Enterotype variations of the healthy human gut microbiome in different geographical regions. Bioinformation. 2018;14:560–73.
Di Pierro F. A possible perspective about the compositional models, evolution, and clinical meaning of human enterotypes. Microorganisms. 2021;9(11):2341.
Richard ML, Sokol H. The gut mycobiota: insights into analysis, environmental interactions and role in gastrointestinal diseases. Nat Rev Gastroenterol Hepatol. 2019;16:331–45.
Hoarau G, Mukherjee PK, Gower-Rousseau C, Hager C, Chandra J, Retuerto MA, Neut C, Vermeire S, Clemente J, Colombel JF, et al. Bacteriome and mycobiome interactions underscore microbial dysbiosis in familial Crohn's disease. mBio. 2016;7(5):e01250–16.
Cavalcanti YW, Morse DJ, da Silva WJ, Del-Bel-Cury AA, Wei X, Wilson M, Milward P, Lewis M, Bradshaw D, Williams DW. Virulence and pathogenicity of Candida albicans is enhanced in biofilms containing oral bacteria. Biofouling. 2015;31:27–38.
Sokol H, Leducq V, Aschard H, Pham HP, Jegou S, Landman C, Cohen D, Liguori G, Bourrier A, Nion-Larmurier I, et al. Fungal microbiota dysbiosis in IBD. Gut. 2017;66:1039-1048.
Zuo T, Wong SH, Cheung CP, Lam K, Lui R, Cheung K, Zhang F, Tang W, Ching JYL, Wu JCY, et al. Gut fungal dysbiosis correlates with reduced efficacy of fecal microbiota transplantation in Clostridium difficile infection. Nat Commun. 2018;9:3663.
Gao B, Zhang X, Schnabl B. Fungi-bacteria correlation in alcoholic hepatitis patients. Toxins (Basel). 2021;13(2):143.
Nash AK, Auchtung TA, Wong MC, Smith DP, Gesell JR, Ross MC, Stewart CJ, Metcalf GA, Muzny DM, Gibbs RA, et al. The gut mycobiome of the Human Microbiome Project healthy cohort. Microbiome. 2017;5:153.
Lv L, Gu S, Jiang H, Yan R, Chen Y, Chen Y, Luo R, Huang C, Lu H, Zheng B, et al. Gut mycobiota alterations in patients with COVID-19 and H1N1 infections and their associations with clinical features. Commun Biol. 2021;4:480.
Jayasudha R, Das T, Kalyana Chakravarthy S, Sai Prashanthi G, Bhargava A, Tyagi M, Rani PK, Pappuru RR, Shivaji S. Gut mycobiomes are altered in people with type 2 Diabetes Mellitus and Diabetic Retinopathy. PLoS ONE. 2020;15: e0243077.
Lemoinne S, Kemgang A, Ben Belkacem K, Straube M, Jegou S, Corpechot C, Saint-Antoine IBDN, Chazouilleres O, Housset C, Sokol H. Fungi participate in the dysbiosis of gut microbiota in patients with primary sclerosing cholangitis. Gut. 2020;69:92–102.
Marfil-Sanchez A, Zhang L, Alonso-Pernas P, Mirhakkak M, Mueller M, Seelbinder B, Ni Y, Santhanam R, Busch A, Beemelmanns C, et al. An integrative understanding of the large metabolic shifts induced by antibiotics in critical illness. Gut Microbes. 2021;13:1993598.
Demir M, Lang S, Hartmann P, Duan Y, Martin A, Miyamoto Y, Bondareva M, Zhang X, Wang Y, Kasper P, et al. The fecal mycobiome in non-alcoholic fatty liver disease. J Hepatol. 2022;76:788–99.
Prochazkova P, Roubalova R, Dvorak J, Kreisinger J, Hill M, Tlaskalova-Hogenova H, Tomasova P, Pelantova H, Cermakova M, Kuzma M, et al. The intestinal microbiota and metabolites in patients with anorexia nervosa. Gut Microbes. 2021;13:1–25.
Liu H, Liang Z, Cao N, Yi X, Tan X, Liu Z, Wang F, Yang Y, Li C, **ang Z, et al. Airway bacterial and fungal microbiome in chronic obstructive pulmonary disease. Med Microecol. 2021;7(2021):100035.
Leung MH, Chan KC, Lee PK. Skin fungal community and its correlation with bacterial community of urban Chinese individuals. Microbiome. 2016;4:46.
Suzuki TA, Worobey M. Geographical variation of human gut microbial composition. Biol Lett. 2014;10:20131037.
Getzke F, Thiergart T, Hacquard S. Contribution of bacterial-fungal balance to plant and animal health. Curr Opin Microbiol. 2019;49:66–72.
Bellemain E, Carlsen T, Brochmann C, Coissac E, Taberlet P, Kauserud H. ITS as an environmental DNA barcode for fungi: an in silico approach reveals potential PCR biases. BMC Microbiol. 2010;10:189.
Cheng M, Ning K. Stereotypes about enterotype: the old and new ideas. Genomics Proteomics Bioinformatics. 2019;17:4–12.
Gupta VK, Kim M, Bakshi U, Cunningham KY, Davis JM 3rd, Lazaridis KN, Nelson H, Chia N, Sung J. A predictive index for health status using species-level gut microbiome profiling. Nat Commun. 2020;11:4635.
Kim BS, Choi CW, Shin H, ** SP, Bae JS, Han M, Seo EY, Chun J, Chung JH. Comparison of the gut microbiota of centenarians in longevity villages of South Korea with those of other age groups. J Microbiol Biotechnol. 2019;29:429–40.
Badal VD, Vaccariello ED, Murray ER, Yu KE, Knight R, Jeste DV, Nguyen TT. The gut microbiome, aging, and longevity: a systematic review. Nutrients. 2020;12(12):3759.
Jiang P, Lai S, Wu S, Zhao XM, Chen WH. Host DNA contents in fecal metagenomics as a biomarker for intestinal diseases and effective treatment. BMC Genomics. 2020;21:348.
Leite G, Pimentel M, Barlow GM, Chang C, Hosseini A, Wang JJ, Parodi G, Sedighi R, Rezaie A, Mathur R. Age and the aging process significantly alter the small bowel microbiome. Cell Rep. 2021;36(13):109765.
Keesing F, Belden LK, Daszak P, Dobson A, Harvell CD, Holt RD, Hudson P, Jolles A, Jones KE, Mitchell CE, et al. Impacts of biodiversity on the emergence and transmission of infectious diseases. Nature. 2010;468:647–52.
Ghosh TS, Shanahan F, O’Toole PW. The gut microbiome as a modulator of healthy ageing. Nat Rev Gastroenterol Hepatol. 2022;19(9):565–84.
Shiao SL, Kershaw KM, Limon JJ, You S, Yoon J, Ko EY, Guarnerio J, Potdar AA, McGovern DPB, Bose S, et al. Commensal bacteria and fungi differentially regulate tumor responses to radiation therapy. Cancer Cell. 2021;39(1202–1213): e1206.
Mims TS, Abdallah QA, Stewart JD, Watts SP, White CT, Rousselle TV, Gosain A, Bajwa A, Han JC, Willis KA, Pierre JF. The gut mycobiome of healthy mice is shaped by the environment and correlates with metabolic outcomes in response to diet. Commun Biol. 2021;4:281.
Dong TS, Gupta A. Influence of Early Life, Diet, and the Environment on the Microbiome. Clin Gastroenterol Hepatol. 2019;17:231–42.
van Leeuwen PT, van der Peet JM, Bikker FJ, Hoogenkamp MA, Paiva AMO, Kostidis S, Mayboroda OA, Smits WK, Krom BP. Interspecies Interactions between Clostridium difficile and Candida albicans. Msphere. 2016;1(6):e00187–16.
Morales DK, Hogan DA. Candida albicans interactions with bacteria in the context of human health and disease. PLoS Pathog. 2010;6: e1000886.
Sardi JC, Duque C, Mariano FS, Peixoto IT, Hofling JF, Goncalves RB. Candida spp. in periodontal disease: a brief review. J Oral Sci. 2010;52:177–185.
Elghandour MMY, Tan ZL, Abu Hafsa SH, Adegbeye MJ, Greiner R, Ugbogu EA, Cedillo Monroy J, Salem AZM. Saccharomyces cerevisiae as a probiotic feed additive to non and pseudo-ruminant feeding: a review. J Appl Microbiol. 2020;128:658–74.
Ponomarova O, Gabrielli N, Sevin DC, Mulleder M, Zimgibl K, Bulyha K, Andrejev S, Kafkia E, Typas A, Sauer U, et al. Yeast Creates a Niche for Symbiotic Lactic Acid Bacteria through Nitrogen Overflow. Cell Syst. 2017;5(4):345–57.
Ren CC, Dokter-Fokkens J, Lozano SF, Zhang QX, de Haan BJ, Zhang H, Faas MM, de Vos P. Lactic acid bacteria may impact intestinal barrier function by modulating goblet cells. Mol Nutr Food Res. 2018;62(6):e1700572.
Seiwert N, Heylmann D, Hasselwander S, Fahrer J. Mechanism of colorectal carcinogenesis triggered by heme iron from red meat. Biochim Biophys Acta Rev Cancer. 2020;1873: 188334.
Sasso A, Latella G. Role of heme iron in the association between red meat consumption and colorectal cancer. Nutr Cancer. 2018;70:1173–83.
Campbell EL, Colgan SP. Control and dysregulation of redox signalling in the gastrointestinal tract. Nat Rev Gastroenterol Hepatol. 2019;16:106–20.
Turnbaugh PJ, Ley RE, Mahowald MA, Magrini V, Mardis ER, Gordon JI. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature. 2006;444:1027–31.
Auchtung TA, Fofanova TY, Stewart CJ, Nash AK, Wong MC, Gesell JR, Auchtung JM, Ajami NJ, Petrosino JF. Investigating colonization of the healthy adult gastrointestinal tract by fungi. mSphere. 2018;3(2):e00092–18.
Shuai M, Fu Y, Zhong HL, Gou W, Jiang Z, Liang Y, Miao Z, Xu JJ, Huynh T, Wahlqvist ML, et al. Map** the human gut mycobiome in middle-aged and elderly adults: multiomics insights and implications for host metabolic health. Gut. 2022;71(9):1812–20.
White T BT, Lee S, Taylor J. Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics. PCI protocols: a guide to methods and applications. 1990;18(1):315–22.
Chen S, Zhou Y, Chen Y, Gu J. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics. 2018;34:i884–90.
Magoc T, Salzberg SL. FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinformatics. 2011;27:2957–63.
Bolyen E, Rideout JR, Dillon MR, Bokulich NA, Abnet CC, Al-Ghalith GA, Alexander H, Alm EJ, Arumugam M, Asnicar F, et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat Biotechnol. 2019;37:852–7.
Callahan BJ, McMurdie PJ, Rosen MJ, Han AW, Johnson AJ, Holmes SP: DADA2. High-resolution sample inference from Illumina amplicon data. Nat Methods. 2016;13:581–583.
Nilsson RH, Larsson KH, Taylor AFS, Bengtsson-Palme J, Jeppesen TS, Schigel D, Kennedy P, Picard K, Glockner FO, Tedersoo L, et al. The UNITE database for molecular identification of fungi: handling dark taxa and parallel taxonomic classifications. Nucleic Acids Res. 2019;47:D259–64.
Dixon P. VEGAN, a package of R functions for community ecology. Journal of vegetation science. 2003;14(6):927–30.
McMurdie PJ, Holmes S. phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PLoS ONE. 2013;8: e61217.
Zeller G, Tap J, Voigt AY, Sunagawa S, Kultima JR, Costea PI, Amiot A, Bohm J, Brunetti F, Habermann N, et al. Potential of fecal microbiota for early-stage detection of colorectal cancer. Mol Syst Biol. 2014;10:766.
Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9:357–9.
Truong DT, Franzosa EA, Tickle TL, Scholz M, Weingart G, Pasolli E, Tett A, Huttenhower C, Segata N. MetaPhlAn2 for enhanced metagenomic taxonomic profiling. Nat Methods. 2015;12:902–3.
Franzosa EA, McIver LJ, Rahnavard G, Thompson LR, Schirmer M, Weingart G, Lipson KS, Knight R, Caporaso JG, Segata N, Huttenhower C. Species-level functional profiling of metagenomes and metatranscriptomes. Nat Methods. 2018;15:962–8.
Suzek BE, Huang H, McGarvey P, Mazumder R, Wu CH. UniRef: comprehensive and non-redundant UniProt reference clusters. Bioinformatics. 2007;23:1282–8.
Caspi R, Billington R, Keseler IM, Kothari A, Krummenacker M, Midford PE, Ong WK, Paley S, Subhraveti P, Karp PD. The MetaCyc database of metabolic pathways and enzymes - a 2019 update. Nucleic Acids Res. 2020;48:D445–53.
Vitali F, Colucci R, Di Paola M, Pindo M, De Filippo C, Moretti S, Cavalieri D. Early melanoma invasivity correlates with gut fungal and bacterial profiles. Br J Dermatol. 2022;186:106–16.
Quast C, Pruesse E, Yilmaz P, Gerken J, Schweer T, Yarza P, Peplies J, Glockner FO. The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 2013;41:D590-596.
Blashfield RK. Finding Groups in Data - an Introduction to Cluster-Analysis - Kaufman, L, Rousseeuw Pj. J Classif. 1991;8:277–9.
Tibshirani R. Regression shrinkage and selection via the Lasso. J R Stat Soc Ser B Methodol. 1996;58:267–88.
Villanueva RAM, Chen ZJ. ggplot2: elegant graphics for data analysis, 2nd edition. Measurement-Interdisciplinary Res Perspect. 2019;17:160–167.
Dustin Tingley TY, Kentaro Hirose, Luke Keele, Kosuke Imai: “mediation”. R package for Causal Mediation Analysis. J Stat Softw. 2014;59:1–38.
Benjamini YHY. Controlling the false discovery rate: a practical and powerful approach to multiple tesing. J R Stat Soc Series B Stat Methodol. 1995;57:289–300.
Acknowledgements
Not applicable.
Funding
This work was partly supported by the National Key Research and Development Program of China (Nos. 2020YFA0712403), National Natural Science Foundation of China (NSFC) (Nos. T2225015 and 61932008), and Shanghai Municipal Science and Technology Major Project (No. 2018SHZDZX01 to X.M.Z, No. 2019SHZDZX02 to Y.Y).
Author information
Authors and Affiliations
Contributions
XMZ, YZ, WHC and PB conceived the study and supervised the project. YY, YNP, SCL, JGQ and BHJ managed the sampling and did most of the experiments; SYL, XMZ, YZ, WHC and PB designed the method and performed analysis. S.L wrote the first draft of the manuscript. All authors contributed to the revision of manuscript prior to submission and all authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
This study was approved by the Human Ethics Committee of the School of Life Sciences of Fudan University (No. BE1940) and the Ethics Committee of the Tongji Medical College of Huazhong University of Science and Technology (No. S1241).
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1:
Figure S1. Composition and diversity of the human gut mycobiome across studies and geographic sites. a, The distribution of four highly abundant fungal taxa across three continents. b, The distribution of the number of total reads per sample across study populations. c, Cumulative curves of the number of detected genera according to the number of sequenced samples for different amplicon targets. d, The distribution of fungal Shannon diversity across study continents. e, Comparison of the Shannon diversity (left) and Bray-Curtis pairwise dissimilarities (right) of bacteriome (16S) and mycobiome (ITS) at genus level. f, The correlation between fungal Bray-Curtis distance (FBCD) and bacterial Bray-Curtis distance (BBCD), where the Bray-Curtis distance is calculated between two samples. The shaded gray region represents 95% confidence intervals of the linear regression. In boxplots, boxes span from the first to the third quantiles and black horizontal lines represent the median, with whiskers extending 1.5 times the interquartile range (IQR). p values of two-sided Mann-Whitney U-test are shown. Figure S2. Cumulative curves of the number of detected genera (fungal richness) according to the number of sequenced reads from different study populations. Figure S3. Principal Coordinate Analysis (PCoA) plot of fungal community composition based on Bray-Curtis dissimilarity index. Each point represents a sample and is colored by their dataset (a), continent (b) and Phenotype (c). Figure S4. Composition of the human mycobiome across continents with the removal of the dataset of Limon (2019). a, Genus-level gut mycobiome composition across the three continents (North America, Europe, and Asia). b, The distribution of four highly abundant fungal taxa across three continents. c, The composition of fungal enterotypes across continents in ITS1- and ITS2-combined datasets, respectively. To avoid the bias introduced by the dataset of Limon (2019) from North America (the dataset is missing the Sacc_type enterotype), we re-examined the mycobiome composition across different continents after removing the dataset of the Limon (2019). Figure S5. The robustness of fungal enterotype clustering. a, The optimal clustering number calculated within each distance-matrix determined by Silhouette score. b, The optimal cluster number under varying sampling sizes as determined by Silhouette score for ITS- and ITS2-sequencing datasets, respectively. c, The effect of removing samples from the datasets on the overall clustering behavior for ITS1- and ITS2-sequencing datasets, respectively. We repeated 100 times with different random samples removed in each iteration. The re-clustering results indicated that the enterotypes generally clustered stably with various sample size and less than 10% of samples were wrongly categorized even when half of the samples were removed. d, Hierarchical clustering on the combined ITS1 and ITS2 datasets. e, Clustering results of fungal enterotypes on ITS1 and ITS2- combined datasets visualized by Principal coordinate analysis based on Family and Order levels. The adjusted rand index (ARI) values measuring the similarity between the enterotype clustering results at the family- or order-level against that at the genus-level are shown. e, The optimal clustering number determined by Silhouette score at different taxonomic levels. Partitioning around medoid (PAM) clustering was employed based on the between-sample Bray Curtis distance. Figure S6. Characteristics of fungal enterotypes. a, Abundance of the main contributors of each fungal enterotype within ITS1- (top) and ITS2-combined datasets (bottom). b, The distribution of fungal diversity across fungal enterotypes within ITS1- (top) and ITS2-combined datasets (bottom) as measured by common alpha-diversity indices. c, The distribution of functional richness across fungal enterotypes within the CHGM cohort. In boxplots, boxes span from the first to the third quantiles and black horizontal lines represent the median, with whiskers extending 1.5 times the interquartile range (IQR). p values of two-sided Mann-Whitney U-test are shown. Figure S7. The distribution of the unclassified Ascomycota phylum (Ascomycota.sp) and the unclassified Saccharomycetales order (Saccharomycetales.sp) in ITS1 and ITS2 sequencing datasets. In boxplots, boxes span from the first to the third quantiles and black horizontal lines represent the median, with whiskers extending 1.5 times the interquartile range (IQR). p values of two-sided Mann-Whitney U-test are shown. Figure S8. Robust classification of fungal enterotypes across datasets. a-c, The 5-fold cross-validation results of four-enterotype classifier on ITS1-sequencing dataset (a), ITS2-sequencing dataset (b) and ITS1- and ITS2-combined datasets (c), separately. d-e, The cross-dataset validation performance of four-enterotype classifier between ITS1 and ITS2-sequencing datasets. “Without drivers” refers to excluding the driver genera Candida, Saccharomyces, Aspergillus, Saccharomycetales.sp, and Ascomycota.sp when training the classifiers. “Average” refers to the micro-averaging ROC curve. Figure S9. Bacterial enterotype clustering results for the CHGM metagenomics dataset. a-b, The optimal clustering number calculated within each distance-matrix determined by Silhouette score (a) and CH-index (b). c, Abundance of the main contributors of each bacterial enterotype within each bacterial enterotype. Figure S10. The inter-kingdom interactions between bacterial and fungal communities. a, The correlations between fungal enterotypes and bacterial enterotypes in cohorts with paired 16S sequencing data. The color reflects the O/E ratio (the ratio of observed count to expected count), and asterisks represent the statistical significance of Fisher’s exact test for each pair of comparison: *p < 0.05, **p < 0.01. b, The distribution of Bacteroides and Prevotella across four fungal enterotypes. In boxplots, boxes span from the first to the third quantiles and black horizontal lines represent the median, with whiskers extending 1.5 times the interquartile range (IQR). p values of two-sided Mann-Whitney U-test are shown. Figure S11. The impacts of host phenotypes on the human gut mycobiome. a, Enterotype clustering results on randomly down-sampled datasets (sampled by age group) with 50 repetitions. The left panel shows the optimal cluster number calculated within each distance-matric using Silhouette score for each, and the inner panel of which shows the adjusted rand score (ARI) compared to the original enterotype clusters for each repetition. The right panel shows the distribution of age across re-clustered enterotypes on down-sampled datasets. b, The relationship between Shannon diversity index and age for each fungal enterotype with shaded region representing 95% confidence intervals of the linear regression. c, The correlation between gut aging index (GAI) and Eastern Cooperative Oncology Group (ECOG) score with shaded region representing 95% confidence intervals of the linear regression. d, The distribution of GAI across bacterial enterotypes (E1_bac, E2_bac, E3_bac and E4_bac). e, The distribution of gut aging index (GAI) between non-healthy (Case) and healthy (Control) subjects. f, The distribution of BMI values across fungal enterotypes. g, The distribution of Shannon diversity of the human gut mycobiome between non-healthy (Case) and healthy (Control) subjects. h, The distribution of bacterial Shannon diversity between non-healthy (AD) and healthy (Control) subjects in the CHGM cohort from China. AUD: alcohol use disorder; T2D: type 2 diabetes; CDI: clostridium difficile infection; IBS: irritable bowel syndrome; COVID-19: coronavirus disease 2019; AD: Alzheimer’s disease. In boxplots, boxes span from the first to the third quantiles and black horizontal lines represent the median, with whiskers extending 1.5 times the interquartile range (IQR). p values of two-sided Mann-Whitney U-test are shown. Figure S12. Enterotype clustering results on healthy individuals a., Clustering results of fungal enterotypes on ITS1- and ITS2-sequencing healthy datasets and visualized by principal coordinate analysis (PCoA). b., The optimal cluster number under varying sampling sizes as determined by Silhouette score for ITS1- and ITS2-sequencing healthy datasets, respectively. c., The effect of removing samples from the healthy datasets on the overall clustering behavior for ITS1- and ITS2-sequencing healthy datasets. We repeated 100 times with different random samples removed in each iteration. The re-clustering results indicated that the enterotypes generally clustered stably with various sample size and less than 10% of samples were wrongly categorized even when half of the samples were removed. Supplementary Table S1. Statistic summary of study populations. Supplementary Table S2. The effect size of different factors on fungal community using permutational MANOVA. Supplementary Table S3. Associations between gut fungal and bacterial alpha diversity indices, where Spearman’s coefficient and corresponded p-values were shown in table. Results of Vitali et al (2021) are not shown given its small sample size to calculate correlation. Supplementary Table S4. The effect size of metadata variables in human gut mycobiome variation within each cohort measured by “envfit” function within the R package “vegan” (*p < 0.05, **p < 0.01, ***p < 0.001). Supplementary Table S5. 21 age-associated fungal genera (p-value < 0.05). The p-values of association between these genera and fungal enterotype are also shown. The p-values are determined by the multiple linear regression with adjusted for gender and study. Supplementary Table S6. 31 fungal enterotype-associated metabolic pathways. The p-values are determined by the Wilcoxon-Rank Sum test, and the Benjamini-Hochberg false discovery rate (adjusted p) is employed to correct for multiple testing. Log(FC) denotes the log-transformed fold change of the pathway within respective fungal enterotype relative to other three enterotypes. Supplementary note. The robustness of fungal enterotype clustering results.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Lai, S., Yan, Y., Pu, Y. et al. Enterotypes of the human gut mycobiome. Microbiome 11, 179 (2023). https://doi.org/10.1186/s40168-023-01586-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40168-023-01586-y