
Abstract
Background
The accuracy of internal-transcribed-spacer (ITS) and shotgun metagenomics has not been robustly evaluated, and the effect of diet on the composition and function of the bacterial and fungal gut microbiome in a longitudinal setting has been poorly investigated. Here we compared two approaches to study the fungal community (ITS and shotgun metagenomics), proposed an enrichment protocol to perform a reliable mycobiome analysis using a comprehensive in-house fungal database, and correlated dietary data with both bacterial and fungal communities.
Results
We found that shotgun DNA sequencing after a new enrichment protocol combined with the most comprehensive and novel fungal databases provided a cost-effective approach to perform gut mycobiome profiling at the species level and to integrate bacterial and fungal community analyses in fecal samples. The mycobiome was significantly more variable than the bacterial community at the compositional and functional levels. Notably, we showed that microbial diversity, composition, and functions were associated with habitual diet composition instead of driven by global dietary changes. Our study indicates a potential competitive inter-kingdom interaction between bacteria and fungi for food foraging.
Conclusion
Together, our present work proposes an efficient workflow to study the human gut microbiome integrating robustly fungal, bacterial, and dietary data. These findings will further advance our knowledge of the interaction between gut bacteria and fungi and pave the way for future investigations in human mycobiome.
Video Abstract
Similar content being viewed by others


Introduction
The fungal microbiome, named mycobiome, is believed to play essential roles in human health and disease [1,2,3,4,5,6,7,8,9,10]. However, in comparison with the gut bacterial microbiome, the human gut mycobiome has only been partially investigated (225 results of “human gut mycobiome”, 32,653 results of “human gut microbiome” in PubMed before February 2023). The knowledge gap could reflect the lack of a comprehensive fungal sequence database and bioinformatics pipeline. However, we have recently addressed this issue by creating a gene catalog with over 1.6 million fungal single-copy marker genes and 3 million fungal protein sequences. Additionally, we developed a tool that enables fungal taxonomic and functional profiling from shotgun metagenomic sequence data [16, 35]. To determine the ITS copy numbers using HMM, we created two HMMs respectively for predicting the flanking sequences of the two ends of the whole ITS region. These two HMMs are located separately in the large subunit (LSU) and small subunit (SSU) of the rRNA gene. For creating the HMM of the LSU, a total of 97 sequences in FASTA format were obtained from NCBI (Additional file 1: Data S1) which were then aligned using the “MUSCLE” tool [36] in Stockholm format. This alignment was then used to create the HMM profiles with the hmmbuild function in UNIX [37, 38]. For creating the HMM of the SSU, 100 sequences (Additional file 2: Data S2) were recovered and used through the same process as with the LSU. The resulting HMM profile had a length of 588 bp for LSU and 142 bp for SSU. With the obtained HMM profiles we searched the DNA homologies of the beginning and the ending of the ITS regions in each of the 260 genome assemblies mentioned above, then estimated the number of copies by using the nhmmer function. Then, we applied a filtering step to eliminate those matches that presented an E-value greater than 0.001 [39]. The copy number was then determined using an in-house Python script that evaluated the distance between the start and end of the ITS region. As most ITS lengths were reported in a range of 400 and 800 bp with an average of 550 bp [40, 41], the script accepted a distance between 400 and 800 bp. Moreover, if there was an HMM match for one end of the ITS but not the other, the script determined whether the prospective ITS sequence was at the end of a chromosome/scaffold, in which case the other end of the region could not be found, thus the script counted another copy. Once the copy number was determined for a genome, the complete ITS sequence was extracted by means of the BEDTools tool, which will be used in the map** depth method mentioned below [42]. The resulting file contained three columns for each genome: genome ID, ITS copy number, and ITS sequence. Summary statistics were calculated for the seven species analyzed.
To estimate CNV using the map** depth approach, we calculated the ratio of the ITS depth against the single-copy marker genes’ depth [18]. The ITS sequence for each genome was obtained from the HMM method mentioned above while the single-copy marker genes were obtained from the FunOMIC-T database [45] was used to convert the resulting SAM file into a BAM file. Next, those reads mapped with a q-score inferior to 30 were filtered out and the depth at each base was calculated. The resulting file was then analyzed using R (version 4.0.2) where the map** depth, which was determined as the mean depth of all the bases of each gene, was calculated and normalized by gene lengths. Prior to calculating the mean, the positions at the two ends (which present fewer reads and lead to a bias on the real map** depth) were trimmed by deleting the first and last 50 bp, as done in the report of Lofgren et al. [16]. The copy number was finally estimated as the ratio of the map** depth of the ITS region by the median of the map** depths of all the single-copy marker genes. We used the median in order to avoid a bias of outlier single-copy genes that had a higher-than-usual map** depth. The whole pipeline for this analysis is summarized in Fig. 1a.

Shotgun sequencing provides higher accuracy than ITS sequencing in mycobiome profiling at the species level. a Workflow of the map** depth approach to recover ITS copy numbers. b The distribution of CN-MD (y axis) of the ITS across the strains of the 7 analyzed species (x axis) (n = 260). c The intraspecific CN-MD of S. cerevisiae (x axis) for the 32 strains analyzed (y axis) (n = 32). d In silico comparison at species level between expected abundance (“Expected”) and observed abundance by using the shotgun method (“Shotgun observed”) and the ITS method (“ITS observed”)
To validate the above map** pipeline, we selected 10 S. cerevisiae genome assemblies out of the 260 assemblies downloaded from NCBI. We recovered the ITS sequences from each of these assembled genomes using the CN-HMM method. Then we queried the ITS sequences in chromosome XII of each genome assembly to get the total number of hits, which is used as the reference ITS copy numbers. Then for each of the 10 assemblies, we generated 15 million sequence reads using the InSilicoSeq tool [46]. Those reads were then mapped to their respective ITS and single-copy fungal marker genes’ sequences using the read map** pipeline to identify the copy number estimated by map** depth (CN-MD). The Student t test was done to compare the CN-MD with the references, with a significant threshold p value < 0.05.
In-silico comparison of ITS and shotgun methods
To compare the accuracy of the ITS and shotgun methods in detecting the relative abundance of fungi at strain, species, or genus level in environmental samples, genomes assemblies from 27 strains were used, for which the ITS sequences (by CN-HMM method) and copy numbers of ITS (CN-MD) were extracted. These were used as artificial genomes for creating mock communities. Five different in silico mock fungal communities were generated with randomly selected strains for each species. The strains and their randomly attributed abundances are shown in Additional file 3: Data S3. With the InSilicoSeq tool, we created 15 million reads [50, 51], where various tests were undertaken to determine the optimal filtering value.
A lower map** depth filter would result in the introduction of more off-target species while a higher filtering depth would reduce low-abundant but relevant species [50, 51]. Relative abundance for each map** hit inside each mock population was calculated by dividing the mean depth of each hit by the sum of all mean depths. Then, the relative abundance of each species was retrieved by summing all the hits that corresponded to the relevant species; all other species were marked as off-target. The expected and observed relative abundances were then compared using R (v4.0.2), as described in the Statistical analysis section.
To further evaluate a more diverse fungal community and to mimic a gut microbiome sample, an additional mock community, consisting of the 14 most prevalent fungal species with their relative abundance found in healthy gut controls was created [11] (Additional file 3: Data S3).
Fungal enrichment protocol
Centrifugation was used to separate fungal and bacterial cells based on their different cell sizes. Stoke’s law was used to estimate the centrifugation speed and time, in which D is the particle diameter (cm), η is the fluid viscosity (poise), Rf and Ro are the final and initial radius of rotation respectively (cm), ρp and ρf are the density of the particle and fluid respectively (g/ml), ω is the rotational velocity (radians/sec) and t is the required time for sedimentation from Ro to Rf (sec) (Eq. 1).
Briefly, 15 ml of 1 × PBS solution (Sigma-Aldrich Phosphate Buffered Saline Powder pH 7.4) was added to 500 mg fecal samples together with 10 2 mm glass beads (Merck KGaA glass beads 2 mm) to homogenize the feces into fecal suspension. The suspension was then passed through a 40-micron cell strainer (Clearline® cell strainers 40 µm blue color) to remove large-size undigested particles. We then centrifuged the filtered suspension for 3 min with 201 g using Eppendorf A-4-62. The supernatant was collected in a 50-ml falcon tube (deltalab EUROTUBO® 50 ml conical tubes) and the pellet was resuspended in 15 ml of 1 × PBS. The resuspended solution was centrifuged again for 3 min and 201 g with the same centrifuge to reduce the remaining number of bacterial cells from the fungi-enriched fraction, then the supernatant was collected and combined with the previous supernatant. We then resuspended the pellet with 1 ml of 1 × PBS and centrifuged it for 20 min at 10,000 × g using an Eppendorf Centrifuge 5427R to collect the pellet containing the enriched fungal cell fraction. The combined supernatant was centrifuged parallelly in Fiberlite™ F14-6 × 250LE for 30 min at 10,000 × g, the pellet was collected as the enriched bacterial cell fraction (Additional file 5: Fig. S2).
Collection and processing of habitual diet information
Six volunteers, free of diagnosed diseases, were recruited between August 2021 and October 2021 by disseminating an announcement. During 2 months, each volunteer filled a short Frequency Food Questionnaire (sFFQ) [1a), for which more details are described in the Methods section. Before estimating the ITS copy number, we first validated the map** pipeline. Ten genome assemblies of S. cerevisiae were selected for this validation, and their ITS copy numbers were retrieved from the NCBI nucleotide database to work as the expected ITS copy number. At the same time, we generated 15 million simulated shotgun sequencing reads of the 10 assemblies using the InSilicoSeq tool [46]. The simulated reads were then used as the input of the map** pipeline for calculating the CN-MDs for each of the genomes. At last, the calculated CN-MDs were compared with the reference copy numbers, by applying the Student t-test. The comparison between the two values did not show significant differences (Additional file 7: Data S5, p value = 0.28), which indicates that the pipeline could reliably recover the expected ITS copy numbers from whole genome shotgun sequencing reads.
Next, we applied the map** pipeline to estimate the CN-MDs of the 260 assembled fungal genomes using their shotgun sequencing reads downloaded from NCBI or JGI. The resulting CN-MD ranged from 7 to 170, with an average of 60 (Additional file 8: Data S6). We observed that both the intra- and inter-species variability was high for the ITS copy numbers of the analyzed genomes which cover seven species and three phyla (Fig. 1b). The copy numbers of the ITS region of the 32 collected S. cerevisiae strains were widely distributed, ranging from 15 to 137 (Fig. 1c) and those of the 182 C. albicans strains varied from 11 to 74. The variance between C. albicans and S. cerevisiae was significantly different (p value = 8e−10; Levene’s test). These findings indicate that possible bias could be introduced when profiling the fungal community by using ITS amplicons without normalizing by the actual strain level ITS copy numbers.
Shotgun data are more accurate than ITS data for taxonomic profiling at the species level
To compare the accuracy of the species-level mycobiome profiling generated by ITS sequencing and shotgun sequencing, we created in silico mock communities with different groups of fungal species. A total of 27 artificial fungal genomes with known ITS copy numbers were used to create five in-silico mock communities. We randomly generated relative abundances for the species in each of the five communities (Additional file 3: Data S3). The artificial genomes and their ITS sequences were used to simulate the shotgun sequencing reads and the ITS sequencing reads, respectively. An additional mock community mimicking the gut mycobiome was also created using the 14 most abundant gut fungal species and their observed relative abundance based on a previous study [1d) showed that the two methods differed significantly (p value = 0.005, Student t-test). The ITS method exhibited a mean distance of 0.616 and the shotgun method a mean distance of 0.237 (Additional file 9: Data S7, Additional file 10: Fig. S3), indicating that the shotgun method was able to recover the expected fungal community compositions more reliably at the species level. The same analysis at the strain level was also employed, however, the results revealed that neither shotgun nor ITS sequencing was accurate enough to detect the specific strains.
A fungal enrichment protocol effectively concentrates fungal cells in human fecal samples
As demonstrated by previous studies [11, 26], the proportion of fungal sequences obtained upon shotgun sequencing of DNA prepared from human fecal samples consists of less than 0.08% of the total sequences, which limits the accuracy of the recovered fungal community composition results if the sequencing depth is not high enough. However, the cost of deep shotgun sequencing is still not easily affordable by all researchers. We thus proposed an enrichment protocol based on a series of centrifugations to separate fungal and bacterial cells prior to the regular DNA extraction method.
To evaluate the practical efficiency of this enrichment protocol, we collected fecal samples from six healthy volunteers that included three females and three males. Each of the volunteers donated their fecal samples weekly during an 8-week span, making up a batch of 48 fecal samples. Then, for each of the 48 samples, two aliquots of 500 mg were kept, from which one aliquot underwent the enrichment protocol to be separated into a fungal enriched partition and a bacterial enriched partition, while the other aliquot did not pass any further operation and was used as the unenriched control. Finally, a total of 143 partitions (one of the volunteers did not provide enough feces for two aliquots) of fecal samples were sent for shotgun sequencing using the Illumina Novaseq 6000 platform (Fig. 2a). The sequencing provided an average of 6.4 Gb, and 21.5 million pair reads which are comparable with other studies using shotgun sequencing [5, 72, 73]. Next, we annotated the bacterial and fungal communities for all 143 samples using HUMANn [57] and FunOMIC pipelines [11]. A total of 411 bacterial species and 208 KEGG pathways were found in the bacterial community, and 91 fungal species and 154 KEGG pathways were found in the fungal community. To assess whether the sequencing depth was sufficient to recover the majority of both fungal and bacterial richness, we selected eight samples that had the highest number of Gb to perform rarefaction curves. Each sample was subsampled and annotated with a gradient of sequencing depths. With the cutoff of the 6.4 Gb, around 80% of fungal taxonomic richness, more than 70% of fungal functional richness, 100% of bacterial richness, and almost 100% of bacterial functional richness were recovered, showing that our shotgun sequencing run was able to capture most of the microbiome information (Fig. 2b, Additional file 11: Fig. S4). The plateau was reached at 7.5 Gb for fungal taxonomy, 15 Gb for fungal functions, and 6.7 Gb for bacterial functions. Together, these results showed that a sequencing depth of 15 Gb would allow the capture of the entire bacterial and fungal communities.

Study design and quality of shotgun sequencing. a Design and workflow of this study. b Rarefaction curves of the shotgun sequencing, x-axis represents the depth of sequencing, y-axis represents the percentage of richness. The rows of the panel are different microbial communities, and the columns of the panel represent the taxonomic or functional level richness. The black solid line is the average percentage of the richness of the 8 samples at the specific depth of subsampling. The red vertical solid lines represent the average sequencing depth 6.4 Gb, the blue horizontal dotted lines represent the threshold of 80% richness
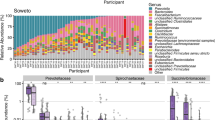
Then, we mapped each of the 143 samples to fungal and bacterial databases [11, 57] to calculate the enrichment efficiencies and meanwhile get their fungal and bacterial, taxonomic, and functional profiles. Notably, the fungal profiling was annotated with the unpublished updated version of the FunOMIC database that contains 2 million single-copy marker genes and 21 million protein sequences extracted from more than 3000 fungal species. In total, we have detected fungi in 96 samples out of 143 (67%). We observed that by applying the enrichment protocol, the proportion of samples that have fungi detected increased from 58.3 (28 out of 48) to 95.8% (46 out of 48). We then calculated the ratio of the reads that were mapped to the fungal database against the reads that were mapped to the bacterial database for all 143 samples. Then, we used this ratio in the fungal partition and divided by this ratio into their corresponding control partitions to estimate the extent to which the fungal sequences have been enriched. The ratio increased on average 18.47 times (ranging from 0.07 to 235) after applying the enrichment protocol, and the fungal alpha diversity in fungal enriched partitions was found significantly higher than in both bacterial (q = 4.3e−5 Shannon index, q = 2e−7 Chao1 index) and control partitions (q = 5.2e−5 Shannon index, q = 3.7e−6 Chao1 index) (Fig. 3a, b).

Enrichment efficiency in fungi. a The genus-level taxa bar plot of the fungal community compositions in bacterial, control, and fungal partitions. b Boxplots of the species-level fungal community alpha diversities (observed species, Shannon, and Chao1 indices) in control, bacterial, and fungal partitions (n = 96), ordered by their mean from smallest to largest (left to right)
Similar but less significant results were found in the bacterial partitions (Additional file 12: Fig. S5a, b). Genus-level taxa bar plot of the fungal and bacterial communities grouped by different time points can also be found in Additional file 13: Fig. S6. Since bacterial reads were still present in the fungal partitions, they were merged with those in their corresponding bacterial partitions. After applying a paired Wilcoxon test between the bacterial alpha diversities before and after merging, we found the Chao1 index after merging had a trend of being higher than that of before merging (p = 0.06, Additional file 12: Fig. S5c). This result was not observed for the fungal alpha diversity. For the purpose of capturing more information, in the subsequent analysis, the merged bacterial microbiome profiling was used to represent the bacterial community, and the fungal microbiome profiling in fungal partitions was used to represent the fungal community in each sample.
Keystone bacterial and fungal species in the human gut
To determine the keystone species in the human gut microbiome, we constructed networks based on the SparCC correlation matrix and the corresponding BH-adjusted P-values matrix. In each network, the nodes represent the microbial species that were included in this network, and the edges connecting the nodes represent the significant inter-kingdom correlations (FDR < 0.001). This network captured 625 associations among 199 microbial species which includes 111 bacterial species, 87 fungal species, and 1 Archaea species (Fig. 4a). Among the 625 associations 349 were positive and 276 negative associations. This network consisted of only one large connected group (199 out of 199 microbial species (100%)). The global network had an average node degree (number of edges adjacent to the node) of 6.28 (7.66 for bacteria and 4.5 for fungi), and it perfectly followed a scale-free degree distribution (power law) (Fig. 4b), indicating that most nodes had low-degree values, and only a few nodes had the highest degree values, which are often called “hubs”, and are thought to serve specific purposes in the networks. In this network, “hubs” are microbial species that have a much higher number of correlations among all the species, indicating that they are more active within the gut microbiome context.

Inter-kingdom network and keystone species. a Network of the SparCC correlation between the fungal and bacterial taxonomic composition at the species level (FDR < 0.01). Each node shows a unique microbial species, each edge showing the SparCC correlation between the two nodes linked. The edges connecting the nodes represent significant correlations (FDR < 0.001). The color of the nodes represents the kingdom of the specific species and highlights the keystone species. The size of the nodes represents the node degrees for each node. The color of the edges represents the symbol of the correlation, and the width of the edges represents the intensity of the correlation. b Degree distribution of the network following a scale-free distribution. c Candida albicans, one of the fungal species that has a high node degree and the highest betweenness centrality
Two fungal species Eremothecium sinecaudum and Candida albicans, were found to have the highest betweenness centrality (the number of shortest paths going through a node) (945), and high node degree (11) among all the fungal species in this network (Fig. 4c), suggesting a critical role in the gut microbial community. Among them, C. albicans is a known fungal pathogen [74], was found to form in the gut microbiome seven cross-domain associations with Ruminococcus gnavus, Firmicutes bacterium CAG 110, Streptococcus salivarius, Holdemanella biformis, Eubacterium sp CAG 274, Proteobacteria bacterium CAG 139, and Alistipes inops. From the species analysis (using betweenness centrality and node degree), we identified one fungal species and 13 bacterial species as potential gut keystone species (Table 1) as they were the species that appeared in both the list of the top 20 highest node degree and the top 20 highest betweenness centrality (top 20).
Short-term dynamics of the human gut microbiome
To determine the intra- and inter-individual variability of the volunteers’ gut microbiome, we measured the pairwise dissimilarities using the Bray-Curtis dissimilarity values between longitudinal samples donated by the same volunteer and between samples donated by different volunteers for both fungal and bacterial microbiomes. The results revealed that both bacterial and fungal communities exhibited higher inter-individual than intra-individual dissimilarities (Fig. 5a), while this difference was significantly more pronounced in the bacterial community. We then compared the dissimilarity between fungal and bacterial communities; the variabilities in the fungal community were significantly higher than in the bacterial community (Fig. 5b).

Dynamics of the human gut microbiome. a Intra- and inter-individual beta diversity (Bray-Curtis) in fungal and bacterial communities at taxonomic and functional levels. b Comparison of the beta diversities between fungal and bacterial communities intra- and inter-individually at taxonomic and functional levels. c Dynamics of fungal and bacterial communities at taxonomic and functional levels. The x-axis represents different time points, the y-axis represents Bray-Curtis values
Then, to investigate the stability of the gut microbiome over time, we considered the first time point as a baseline, and for each of the individuals, we measured the Bray-Curtis dissimilarities of other time points against the baseline. In both taxonomy and function, despite the high degree of short-term longitudinal change in both communities, we found that the fungal community displayed increased dynamics as compared to the bacterial community (Fig. 5c). Notably, the mean Bray-Curtis values calculated from data of the six individuals were significantly higher for the fungal microbiome than bacterial microbiome (Wilcoxon, p < 0.01 for both taxonomy and functions). To determine whether dietary changes drove the dynamics of the gut microbiome, we correlated the pairwise Bray-Curtis dissimilarity of the microbiome (fungal and bacterial, taxonomy and function) with the pairwise Bray-Curtis dissimilarity of dietary data (nutrient macromolecules and food groups). However, no significant correlations were found, which may indicate the absence of an effect of the diet on the microbiome composition and function at the global level but does not exclude the effect of specific food groups or nutrients.
Microbial diversity and composition are associated with habitual diet
We then assessed the correlation between habitual diet (nutrients and food groups) and the alpha diversity of the human gut microbiome to get a broad view of how habitual diet could modulate microbial communities. Interestingly, using the Spearman correlation coefficient, we detected 21 significant associations (FDR < 0.05) with fungal taxa while no significant associations were found with bacterial taxa (Fig. 6a). Furthermore, 31 significant associations were found with fungal functions, and five significant associations with bacterial functions (Fig. 6b, c). The overlapped associations found with fungal taxa and fungal function were all consistent, whereas the overlapped associations detected with fungal function and bacterial function were all opposite, indicating that fungal and bacterial communities are likely to act competitively for some dietary products (Fig. 6d).

Microbial taxonomic and functional alpha diversities are associated with habitual diet. a Significant (FDR < 0.05) Spearman correlations found between the fungal taxonomic alpha diversity and diet categories. The x-axis is the value of the correlation coefficient, the y-axis is the name of the diet categories. b Significant (FDR < 0.05) Spearman correlations found between the fungal functional alpha diversity and diet categories. The x-axis is the value of the correlation coefficient, the y-axis is the name of the diet categories. c Significant (FDR < 0.05) Spearman correlations were found between the bacterial functional alpha diversity and diet categories. The x-axis is the value of the correlation coefficient, the y-axis is the name of the diet categories. d Network of the fungal functional alpha diversity, bacterial functional alpha diversity, and diet categories detected to be significantly (FDR < 0.01) correlated with them. The r values are labeled for the overlapped diet categories. The edges are the Spearman correlation coefficients between the two nodes linked. The color of the edges represents the symbol of the correlation. The size of the nodes represents the node degree values
Next, we calculated the Spearman correlation coefficients between a habitual diet, specific gut microbiome components, and functional pathways. We found 23 fungal species were significantly correlated with one or more dietary categories. Among them, the strongest correlations were Lactarius pseudohatsudake with biscuits (rho = −0.32, FDR = 0.027), Penicillium lancoscoeruleum with fish (rho = 0.31, FDR = 0.027), Candida albicans with iron (rho = −0.29, FDR = 0.038) (Fig. 7a). More significant correlations were detected in the bacterial community. At a broad level, we found three apparent groups of species clustered to a group of foods mainly classified as related to more animal-based foods (fish, sauces, sausages, processed food, dairy products) and two others related to less animal-based foods (fruit, vegetables) (Fig. 7b). Similar but less obvious grou**s were also found when correlating habitual diet with microbial functions (Additional file 15: Fig. S8; Additional file 16: Fig. S9b).

Microbial taxonomic compositions are associated with habitual diet. a Heatmap of all the detected significant correlations between fungal taxonomic compositions and diet categories. b Heatmap of all the detected significant correlations between bacterial taxonomic compositions and diet categories. The asterisk indicates that the correlation index for the corresponding species metadata pair is significant. For better visualization, this plot with higher resolutions can be found in Additional file 14: Fig. S7
Discussion
In this study, we took advantage of our recent implementation of the most comprehensive fungal databases that contain 2 million single-copy marker genes and 21 million protein sequences extracted from more than 3000 fungal species [73, 76], indicating that the fungal compositions in human gut shift rapidly instead of level off to a stable status such as the bacterial community. Since the habitual diet was found to have an influence on the composition of the fungal microbiome in both human and mice models [77,78,79,80,81], we sought the relationship between the dynamics of habitual diet and the dynamics of the gut microbiome. Although the microbiome changes were not driven by global dietary changes, we showed that microbial diversity, composition, and functions were associated with habitual diet composition. We have found that bacterial alpha diversity and fungal alpha diversity were oppositely correlated with three diet categories, sweets, protein, and iron. The level of iron in the habitual diet was found to negatively correlate with the fungal functional alpha diversity. To the best of our knowledge, this is the first demonstration of the effect of iron on the fungal functional alpha diversity, though some studies have discussed that high iron levels promote the growth of specific fungal species [82]. Our study indicates a potential competitive inter-kingdom interaction between bacteria and fungi for food foraging.
The data that have been generated for six healthy individuals sampled over a 2-month period provide a new vision of the link between the diet and the composition of the bacterial and fungal microbiomes. Altogether, our present work proposes an efficient workflow to study the human gut microbiome integrating robustly fungal, bacterial, and dietary data.
Conclusion
The data that have been generated for six healthy individuals sampled over a 2-month period provide a new vision of the link between the diet and the composition of the bacterial and fungal microbiomes. Together, our present work proposes an efficient workflow to study the human gut microbiome integrating robustly fungal, bacterial, and dietary data. With this workflow, our findings demonstrated the interkingdom association between intestinal bacteria and fungi at taxonomic and functional levels and their correlation with diet.
Availability of data and materials
Shotgun metagenomic sequencing raw data (short-read archives, SRA) are available via NCBI Project Number PRJNA925700, which includes the sequences from the 143 fecal samples described in the Methods section. The in-house scripts for performing bioinformatics analyses in this work can be found on GitHub at https://github.com/ManichanhLab/LongitudinalMycobiomeWithDiet.
Abbreviations
- ITS:
-
Internal transcribed spacer
- HMM:
-
Hidden Markov model
- CN:
-
Copy number
- MD:
-
Map** depth
- SCMG:
-
Single copy marker gene
- SD:
-
Standard deviation
- Gbp:
-
Giga base pair
- FDR:
-
False discovery rate
- LSU:
-
Large subunit
- SSU:
-
Small subunit
- ICC:
-
Intraclass correlation coefficient
- sFFQ:
-
Short food frequency questionnaire
- RMT:
-
Random matrix theory
References
Auchtung TA, Stewart CJ, Smith DP, Triplett EW, Agardh D, Hagopian WA, et al. Temporal changes in gastrointestinal fungi and the risk of autoimmunity during early childhood: the TEDDY study. Nat Commun. 2022;13(1):3151. https://doi.org/10.1038/s41467-022-30686-w.
El Mouzan M, Wang F, Al Mofarreh M, Menon R, Al Barrag A, Korolev KS, et al. Fungal microbiota profile in newly diagnosed treatment-naive children with Crohn’s disease. J Crohns Colitis. 2017;11(5):586–92. https://doi.org/10.1093/ecco-jcc/jjw197.
Gao R, Kong C, Li H, Huang L, Qu X, Qin N, et al. Dysbiosis signature of mycobiota in colon polyp and colorectal cancer. Eur J Clin Microbiol Infect Dis. 2017;36(12):2457–68. https://doi.org/10.1007/s10096-017-3085-6.
Gupta S, Hjelmso MH, Lehtimaki J, Li X, Mortensen MS, Russel J, et al. Environmental sha** of the bacterial and fungal community in infant bed dust and correlations with the airway microbiota. Microbiome. 2020;8(1):115. https://doi.org/10.1186/s40168-020-00895-w.
Liu NN, Jiao N, Tan JC, Wang Z, Wu D, Wang AJ, et al. Multi-kingdom microbiota analyses identify bacterial-fungal interactions and biomarkers of colorectal cancer across cohorts. Nat Microbiol. 2022;7(2):238–50. https://doi.org/10.1038/s41564-021-01030-7.
Nagpal R, Neth BJ, Wang S, Mishra SP, Craft S, Yadav H. Gut mycobiome and its interaction with diet, gut bacteria and Alzheimer’s disease markers in subjects with mild cognitive impairment: a pilot study. EBioMedicine. 2020;59:102950. https://doi.org/10.1016/j.ebiom.2020.102950.
Qiu X, Ma J, Jiao C, Mao X, Zhao X, Lu M, et al. Alterations in the mucosa-associated fungal microbiota in patients with ulcerative colitis. Oncotarget. 2017;8(64):107577–88. https://doi.org/10.18632/oncotarget.22534.
Sokol H, Leducq V, Aschard H, Pham HP, Jegou S, Landman C, et al. Fungal microbiota dysbiosis in IBD. Gut. 2017;66(6):1039–48. https://doi.org/10.1136/gutjnl-2015-310746.
Ventin-Holmberg R, Saqib S, Korpela K, Nikkonen A, Peltola V, Salonen A, et al. The effect of antibiotics on the infant gut fungal microbiota. J Fungi (Basel). 2022;8(4):328. https://doi.org/10.3390/jof8040328.
Zuo T, Zhan H, Zhang F, Liu Q, Tso EYK, Lui GCY, et al. Alterations in fecal fungal microbiome of patients with COVID-19 during time of hospitalization until discharge. Gastroenterology. 2020;159(4):1302–10.e5. https://doi.org/10.1053/j.gastro.2020.06.048.
**e Z, Manichanh C. FunOMIC: pipeline with built-in fungal taxonomic and functional databases for human mycobiome profiling. Comput Struct Biotechnol J. 2022;20:3685–94. https://doi.org/10.1016/j.csbj.2022.07.010.
Bellemain E, Carlsen T, Brochmann C, Coissac E, Taberlet P, Kauserud H. ITS as an environmental DNA barcode for fungi: an in silico approach reveals potential PCR biases. BMC Microbiol. 2010;10:189. https://doi.org/10.1186/1471-2180-10-189.
Longo AV, Rodriguez D, da Silva LD, Toledo LF, Mendoza Almeralla C, Burrowes PA, et al. ITS1 copy number varies among Batrachochytrium dendrobatidis strains: implications for qPCR estimates of infection intensity from field-collected amphibian skin swabs. PLoS One. 2013;8(3):e59499. https://doi.org/10.1371/journal.pone.0059499.
Herrera ML, Vallor AC, Gelfond JA, Patterson TF, Wickes BL. Strain-dependent variation in 18S ribosomal DNA Copy numbers in Aspergillus fumigatus. J Clin Microbiol. 2009;47(5):1325–32. https://doi.org/10.1128/JCM.02073-08.
Lavrinienko A, Jernfors T, Koskimaki JJ, Pirttila AM, Watts PC. Does intraspecific variation in rDNA copy number affect analysis of microbial communities? Trends Microbiol. 2021;29(1):19–27. https://doi.org/10.1016/j.tim.2020.05.019.
Lofgren LA, Uehling JK, Branco S, Bruns TD, Martin F, Kennedy PG. Genome-based estimates of fungal rDNA copy number variation across phylogenetic scales and ecological lifestyles. Mol Ecol. 2019;28(4):721–30. https://doi.org/10.1111/mec.14995.
Saka K, Takahashi A, Sasaki M, Kobayashi T. More than 10% of yeast genes are related to genome stability and influence cellular senescence via rDNA maintenance. Nucleic Acids Res. 2016;44(9):4211–21. https://doi.org/10.1093/nar/gkw110.
Sharma D, Denmat SH, Matzke NJ, Hannan K, Hannan RD, O’Sullivan JM, et al. A new method for determining ribosomal DNA copy number shows differences between Saccharomyces cerevisiae populations. Genomics. 2022;114(4):110430. https://doi.org/10.1016/j.ygeno.2022.110430.
Ranjan R, Rani A, Metwally A, McGee HS, Perkins DL. Analysis of the microbiome: advantages of whole genome shotgun versus 16S amplicon sequencing. Biochem Biophys Res Commun. 2016;469(4):967–77. https://doi.org/10.1016/j.bbrc.2015.12.083.
Durazzi F, Sala C, Castellani G, Manfreda G, Remondini D, De Cesare A. Comparison between 16S rRNA and shotgun sequencing data for the taxonomic characterization of the gut microbiota. Sci Rep. 2021;11(1):3030. https://doi.org/10.1038/s41598-021-82726-y.
Ellis-Pegler RB, Crabtree C, Lambert HP. The faecal flora of children in the United Kingdom. J Hyg (Lond). 1975;75(1):135–42. https://doi.org/10.1017/s002217240004715x.
Krawczyk A, Salamon D, Kowalska-Duplaga K, Bogiel T, Gosiewski T. Association of fungi and archaea of the gut microbiota with Crohn’s disease in pediatric patients-pilot study. Pathogens. 2021;10(9):1119. https://doi.org/10.3390/pathogens10091119.
Simon GL, Gorbach SL. Intestinal flora in health and disease. Gastroenterology. 1984;86(1):174–93.
Sender R, Fuchs S, Milo R. Revised estimates for the number of human and bacteria cells in the body. PLoS Biol. 2016;14(8):e1002533. https://doi.org/10.1371/journal.pbio.1002533.
Heintz-Buschart A, May P, Laczny CC, Lebrun LA, Bellora C, Krishna A, et al. Erratum: Integrated multi-omics of the human gut microbiome in a case study of familial type 1 diabetes. Nat Microbiol. 2016;2:16227. https://doi.org/10.1038/nmicrobiol.2016.227.
Qin J, Li R, Raes J, Arumugam M, Burgdorf KS, Manichanh C, et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature. 2010;464(7285):59–65. https://doi.org/10.1038/nature08821.
Adhikari M, Gurung SK, Kim HS, Bazie S, Lee HG, Lee HB, et al. Three new records of ascomycetes isolates from field soils in Korea. Mycobiology. 2017;45(4):327–37. https://doi.org/10.5941/MYCO.2017.45.4.327.
Fernandes KE, Carter DA. Cellular plasticity of pathogenic fungi during infection. PLoS Pathog. 2020;16(6):e1008571. https://doi.org/10.1371/journal.ppat.1008571.
Walker K, Skelton H, Smith K. Cutaneous lesions showing giant yeast forms of Blastomyces dermatitidis. J Cutan Pathol. 2002;29(10):616–8. https://doi.org/10.1034/j.1600-0560.2002.291009.x.
Wang L, Lin X. Morphogenesis in fungal pathogenicity: shape, size, and surface. PLoS Pathog. 2012;8(12):e1003027. https://doi.org/10.1371/journal.ppat.1003027.
Osiro D, Filho RB, Assis OB, Jorge LA, Colnago LA. Measuring bacterial cells size with AFM. Braz J Microbiol. 2012;43(1):341–7. https://doi.org/10.1590/S1517-838220120001000040.
Yanez F, Soler Z, Oliero M, **e Z, Oyarzun I, Serrano-Gomez G, et al. Integrating dietary data into microbiome studies: a step forward for nutri-metaomics. Nutrients. 2021;13(9):2978. https://doi.org/10.3390/nu13092978.
Wheeler DL, Chappey C, Lash AE, Leipe DD, Madden TL, Schuler GD, et al. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2000;28(1):10–4. https://doi.org/10.1093/nar/28.1.10.
Grigoriev IV, Nikitin R, Haridas S, Kuo A, Ohm R, Otillar R, et al. MycoCosm portal: gearing up for 1000 fungal genomes. Nucleic Acids Res. 2014;42(Database issue):D699–704. https://doi.org/10.1093/nar/gkt1183.
Shen Y, Gu Y, Pe’er I. A hidden Markov model for copy number variant prediction from whole genome resequencing data. BMC Bioinformatics. 2011;12 Suppl 6(Suppl 6):S4. https://doi.org/10.1186/1471-2105-12-S6-S4.
Madeira F, Park YM, Lee J, Buso N, Gur T, Madhusoodanan N, et al. The EMBL-EBI search and sequence analysis tools APIs in 2019. Nucleic Acids Res. 2019;47(W1):W636–41. https://doi.org/10.1093/nar/gkz268.
Baum LE, Petrie T. Statistical inference for probabilistic functions of finite state Markov chains. Ann Math Stat. 1966;37(6):1554–63.
Wheeler TJ, Eddy SR. nhmmer: DNA homology search with profile HMMs. Bioinformatics. 2013;29(19):2487–9. https://doi.org/10.1093/bioinformatics/btt403.
Alam I, Hubbard SJ, Oliver SG, Rattray M. A kingdom-specific protein domain HMM library for improved annotation of fungal genomes. BMC Genomics. 2007;8:97. https://doi.org/10.1186/1471-2164-8-97.
Nilsson RH, Tedersoo L, Ryberg M, Kristiansson E, Hartmann M, Unterseher M, et al. A Comprehensive, automatically updated fungal ITS sequence dataset for reference-based chimera control in environmental sequencing efforts. Microbes Environ. 2015;30(2):145–50. https://doi.org/10.1264/jsme2.ME14121.
Schoch CL, Robbertse B, Robert V, Vu D, Cardinali G, Irinyi L, et al. Finding needles in haystacks: linking scientific names, reference specimens and molecular data for Fungi. Database (Oxford). 2014;2014:bau061. https://doi.org/10.1093/database/bau061.
Quinlan AR, Hall IM. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26(6):841–2. https://doi.org/10.1093/bioinformatics/btq033.
Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30(15):2114–20. https://doi.org/10.1093/bioinformatics/btu170.
Langmead B, Wilks C, Antonescu V, Charles R. Scaling read aligners to hundreds of threads on general-purpose processors. Bioinformatics. 2019;35(3):421–32. https://doi.org/10.1093/bioinformatics/bty648.
Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25(16):2078–9. https://doi.org/10.1093/bioinformatics/btp352.
Gourle H, Karlsson-Lindsjo O, Hayer J, Bongcam-Rudloff E. Simulating Illumina metagenomic data with InSilicoSeq. Bioinformatics. 2019;35(3):521–2. https://doi.org/10.1093/bioinformatics/bty630.
Liu J, Wang X, **e H, Zhong Q, **a Y. Analysis and evaluation of different sequencing depths from 5 to 20 million reads in shotgun metagenomic sequencing, with optimal minimum depth being recommended. Genome. 2022;65(9):491–504. https://doi.org/10.1139/gen-2021-0120.
Nilsson RH, Larsson KH, Taylor AFS, Bengtsson-Palme J, Jeppesen TS, Schigel D, et al. The UNITE database for molecular identification of fungi: handling dark taxa and parallel taxonomic classifications. Nucleic Acids Res. 2019;47(D1):D259–64. https://doi.org/10.1093/nar/gky1022.
O’Leary NA, Wright MW, Brister JR, Ciufo S, Haddad D, McVeigh R, et al. Reference sequence (RefSeq) database at NCBI: current status, taxonomic expansion, and functional annotation. Nucleic Acids Res. 2016;44(D1):D733–45. https://doi.org/10.1093/nar/gkv1189.
Patel ZH, Kottyan LC, Lazaro S, Williams MS, Ledbetter DH, Tromp H, et al. The struggle to find reliable results in exome sequencing data: filtering out Mendelian errors. Front Genet. 2014;5:16. https://doi.org/10.3389/fgene.2014.00016.
Song K, Li L, Zhang G. Coverage recommendation for genoty** analysis of highly heterologous species using next-generation sequencing technology. Sci Rep. 2016;6:35736. https://doi.org/10.1038/srep35736.
Shrout PE, Fleiss JL. Intraclass correlations: uses in assessing rater reliability. Psychol Bull. 1979;86(2):420.
Koo TK, Li MY. A guideline of selecting and reporting intraclass correlation coefficients for reliability research. J Chiropr Med. 2016;15(2):155–63.
Serrano-Gomez G, Mayorga L, Oyarzun I, Roca J, Borruel N, Casellas F, et al. Dysbiosis and relapse-related microbiome in inflammatory bowel disease: a shotgun metagenomic approach. Comput Struct Biotechnol J. 2021;19:6481–9. https://doi.org/10.1016/j.csbj.2021.11.037.
Pereira MB, Wallroth M, Jonsson V, Kristiansson E. Comparison of normalization methods for the analysis of metagenomic gene abundance data. BMC Genomics. 2018;19(1):274. https://doi.org/10.1186/s12864-018-4637-6.
Robinson MD, Oshlack A. A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. 2010;11(3):R25. https://doi.org/10.1186/gb-2010-11-3-r25.
Franzosa EA, McIver LJ, Rahnavard G, Thompson LR, Schirmer M, Weingart G, et al. Species-level functional profiling of metagenomes and metatranscriptomes. Nat Methods. 2018;15(11):962–8. https://doi.org/10.1038/s41592-018-0176-y.
Caspi R, Billington R, Ferrer L, Foerster H, Fulcher CA, Keseler IM, et al. The MetaCyc database of metabolic pathways and enzymes and the BioCyc collection of pathway/genome databases. Nucleic Acids Res. 2016;44(D1):D471–80. https://doi.org/10.1093/nar/gkv1164.
Kanehisa M, Sato Y, Kawashima M, Furumichi M, Tanabe M. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 2016;44(D1):D457–62. https://doi.org/10.1093/nar/gkv1070.
Friedman J, Alm EJ. Inferring correlation networks from genomic survey data. PLoS Comput Biol. 2012;8(9):e1002687. https://doi.org/10.1371/journal.pcbi.1002687.
Feizi S, Marbach D, Medard M, Kellis M. Network deconvolution as a general method to distinguish direct dependencies in networks. Nat Biotechnol. 2013;31(8):726–33. https://doi.org/10.1038/nbt.2635.
Ma B, Wang H, Dsouza M, Lou J, He Y, Dai Z, et al. Geographic patterns of co-occurrence network topological features for soil microbiota at continental scale in eastern China. ISME J. 2016;10(8):1891–901. https://doi.org/10.1038/ismej.2015.261.
Luo F, Zhong J, Yang Y, Scheuermann RH, Zhou J. Application of random matrix theory to biological networks. Phys Lett A. 2006;357(6):420–3.
Csardi G, Nepusz T. The igraph software package for complex network research. Int J Complex Syst. 2006;1695(5):1–9.
Lozupone C, Lladser ME, Knights D, Stombaugh J, Knight R. UniFrac: an effective distance metric for microbial community comparison. ISME J. 2011;5(2):169–72. https://doi.org/10.1038/ismej.2010.133.
McMurdie PJ, Holmes S. phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PLoS One. 2013;8(4):e61217. https://doi.org/10.1371/journal.pone.0061217.
William S. The probable error of a mean. Biometrika. 1908;6(1):1–25.
Shapiro SS, Wilk MB. An analysis of variance test for normality (complete samples). Biometrika. 1965;52(3/4):591–611.
Freire-Beneitez V, Price RJ, Tarrant D, Berman J, Buscaino A. Candida albicans repetitive elements display epigenetic diversity and plasticity. Sci Rep. 2016;6:22989. https://doi.org/10.1038/srep22989.
Pendrak ML, Roberts DD. Ribosomal RNA processing in Candida albicans. RNA. 2011;17(12):2235–48. https://doi.org/10.1261/rna.028050.111.
Rustchenko EP, Curran TM, Sherman F. Variations in the number of ribosomal DNA units in morphological mutants and normal strains of Candida albicans and in normal strains of Saccharomyces cerevisiae. J Bacteriol. 1993;175(22):7189–99. https://doi.org/10.1128/jb.175.22.7189-7199.1993.
Asnicar F, Berry SE, Valdes AM, Nguyen LH, Piccinno G, Drew DA, et al. Microbiome connections with host metabolism and habitual diet from 1,098 deeply phenotyped individuals. Nat Med. 2021;27(2):321–32. https://doi.org/10.1038/s41591-020-01183-8.
Nash AK, Auchtung TA, Wong MC, Smith DP, Gesell JR, Ross MC, et al. The gut mycobiome of the Human Microbiome Project healthy cohort. Microbiome. 2017;5(1):153. https://doi.org/10.1186/s40168-017-0373-4.
d’Enfert C, Kaune AK, Alaban LR, Chakraborty S, Cole N, Delavy M, et al. The impact of the Fungus-Host-Microbiota interplay upon Candida albicans infections: current knowledge and new perspectives. FEMS Microbiol Rev. 2021;45(3):fuaa060. https://doi.org/10.1093/femsre/fuaa060.
Schoch CL, Seifert KA, Huhndorf S, Robert V, Spouge JL, Levesque CA, et al. Nuclear ribosomal internal transcribed spacer (ITS) region as a universal DNA barcode marker for Fungi. Proc Natl Acad Sci U S A. 2012;109(16):6241–6. https://doi.org/10.1073/pnas.1117018109.
Amenyogbe N, Adu-Gyasi D, Enuameh Y, Asante KP, Konadu DG, Kaali S, et al. Bacterial and fungal gut community dynamics over the first 5 years of life in predominantly rural communities in Ghana. Front Microbiol. 2021;12:664407. https://doi.org/10.3389/fmicb.2021.664407.
Auchtung TA, Fofanova TY, Stewart CJ, Nash AK, Wong MC, Gesell JR, et al. Investigating colonization of the healthy adult gastrointestinal tract by fungi. mSphere. 2018;3(2):e00092–18. https://doi.org/10.1128/mSphere.00092-18.
David LA, Maurice CF, Carmody RN, Gootenberg DB, Button JE, Wolfe BE, et al. Diet rapidly and reproducibly alters the human gut microbiome. Nature. 2014;505(7484):559–63. https://doi.org/10.1038/nature12820.
Hallen-Adams HE, Kachman SD, Kim J, Legge RM, Martínez I. Fungi inhabiting the healthy human gastrointestinal tract: a diverse and dynamic community. Fungal Ecol. 2015;15:9–17.
Mims TS, Abdallah QA, Stewart JD, Watts SP, White CT, Rousselle TV, et al. The gut mycobiome of healthy mice is shaped by the environment and correlates with metabolic outcomes in response to diet. Commun Biol. 2021;4(1):281. https://doi.org/10.1038/s42003-021-01820-z.
Sun Y, Zuo T, Cheung CP, Gu W, Wan Y, Zhang F, et al. Population-level configurations of gut mycobiome across 6 ethnicities in urban and rural China. Gastroenterology. 2021;160(1):272–86.e11. https://doi.org/10.1053/j.gastro.2020.09.014.
Santus W, Rana AP, Devlin JR, Kiernan KA, Jacob CC, Tjokrosurjo J, et al. Mycobiota and diet-derived fungal xenosiderophores promote Salmonella gastrointestinal colonization. Nat Microbiol. 2022;7(12):2025–38. https://doi.org/10.1038/s41564-022-01267-w.
Funding
This work was supported by the European Union’s Horizon 2020 research and innovation program under the Marie Sklodowska-Curie Action, Innovative Training Network [grant number 812969].
Author information
Authors and Affiliations
Contributions
Z.X. and C.M. conceived and designed the study. Z.X. led the sample collection study. Z.X. performed the enrichment and DNA extraction experiments. Z.X. and A.C. performed the bioinformatics analysis. C.M. and C.D. supervised the study. Z.X., A.C., and C.M. wrote the original draft. Z.X., A.C., C.M., and C.D. reviewed and edited the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The study was approved by the local Ethics Committee of the Vall d’Hebron University Hospital, Barcelona (Project identification code: PR(AG)84/2020). All participants signed a consent form.
Consent for publication
All participants consented independently when donating samples. All data obtained and generated during the study were kept confidential.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Data S1.
A total of 97 sequences in FASTA format were obtained from NCBI.
Additional file 2: Data S2.
For creating the HMM of the SSU, 100 sequences were recovered and used through the same process as with the LSU.
Additional file 3: Data S3.
The strains and their randomly attributed abundances.
Additional file 4: Figure S1.
Workflow of generating simulated sequencing reads for mock communities. The colored dots represent the species in the in silico mock community. The genomes of the species were used directly as the input of InSilicoSeq for mimicking the shotgun sequencing. The ITS sequences of the species were replicated with their corresponding CN-MD before input to the InSilicoSeq for mimicking the ITS sequencing.
Additional file 5: Figure S2.
Workflow diagram of the enrichment protocol, Created with BioRender.com.
Additional file 6: Data S4.
This observation is not in agreement with a previous study that reported a high number of ITS copies, ranging from 14 to 1442.
Additional file 7: Data S5.
The comparison between the two values did not show significant differences.
Additional file 8: Data S6.
The resulting CN-MD ranged from 7 to 170, with an average of 60.
Additional file 9: Data S7.
The resulting CN-MD ranged from 7 to 170, with an average of 60.
Additional file 10: Figure S3.
PcoA plot of the expected mock community profiling and the recovered mock community profiling from shotgun sequencing and ITS sequencing.
Additional file 11: Figure S4.
Total number of reads in each sample (a), and fungal reads in each sample with and without enrichment (b).
Additional file 12: Figure S5.
Enrichment efficiency in bacteria.
Additional file 13: Figure S6.
Genus level fungal microbiome community compositions grouped by different timepoints (a), and Genus level bacterial microbiome community compositions grouped by different timepoints (b).
Additional file 14: Figure S7.
Bacterial taxonomic compositions are associated with habitual diet. Heatmap of all the detected significant correlations between bacterial taxonomic compositions and diet categories. The asterisk indicates that the correlation index for the corresponding species metadata pair is significant.
Additional file 15: Figure S8.
Fungal functional compositions are associated with habitual diet. Heatmap of all the detected significant correlations between fungal functional compositions and diet categories. The asterisk indicates that the correlation index for the corresponding species metadata pair is significant.
Additional file 16: Figure S9.
Bacterial functional compositions are associated with habitual diet. Heatmap of all the detected significant correlations between bacterial functional compositions and diet categories. The asterisk indicates that the correlation index for the corresponding species metadata pair is significant.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
**e, Z., Canalda-Baltrons, A., d’Enfert, C. et al. Shotgun metagenomics reveals interkingdom association between intestinal bacteria and fungi involving competition for nutrients. Microbiome 11, 275 (2023). https://doi.org/10.1186/s40168-023-01693-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40168-023-01693-w