
Abstract
Objective
To use genome-wide association study (GWAS) by subtraction, a method for deriving novel GWASs from existing summary statistics, to derive genome-wide summary statistics for paternal smoking.
Result
A GWAS by subtraction was implemented using a weighted linear model that defined the child-genotype paternal-phenotype association as the child-genotype child-phenotype association minus the child-genotype maternal-phenotype association. We first use the laws of inherence to derive the weighted linear model. We then implemented the linear model to create a GWAS of paternal smoking by subtracting the summary statistics from a GWAS of maternal smoking from the summary statistics of a GWAS of the index individual’s smoking. We used a Monte-Carlo simulation to validate the model and showed that this approach performed similarly in terms of bias to performing a traditional GWAS of paternal smoking. Finally, we validated the summary statistics in a Mendelian randomisation analysis by demonstrating an association of genetically predicted paternal smoking with paternal lung cancer and emphysema.
Similar content being viewed by others


Introduction
Genome-wide association studies (GWAS) are a common way of estimating genotype–phenotype associations [1]. In a GWAS, the association of each variant with the phenotype is estimated in a hypothesis-free manner [2]. GWASs typically only include ‘common’ genetic variants which occur in at least 1% of the study population. One application of GWAS summary statistics is as a source of genotype–phenotype associations for two-sample Mendelian randomisation (MR) analyses [3,4,5]. MR is an epidemiological design which leverages the random inheritance of genetic variants to justify the assumptions of the instrumental variable framework.
The implications of parental smoking on child outcomes are of public health importance. Because half of an individual’s genotype is a random sample of half of their mother’s genotype, one should be able to find robust associations between the child’s genotype and the maternal phenotype due to the associations between the maternal genotype and maternal phenotype and the maternal genotype and offspring’s genotype. Published GWASs summary statistics of maternal smoking allow for the investigation of the effect of an individual’s parental smoking on their (the child’s) outcomes in what has been dubbed ‘proxy gene-environment MR’ [6].
Paternal smoking is also an exposure of interest. For example, studies looking at the effect of maternal smoking on offspring birth outcomes have used paternal smoking as a negative control [7]. However, there are no published GWASs of paternal smoking. GWAS by subtraction (GWAS-BS) is a recent method for deriving novel GWASs from existing summary statistics [8]. In a traditional GWAS-BS, two or more GWASs are combined using structural equation modelling. An alternative is to use a weighted linear model (WLM) [9]. A WLM is created by combining the GWAS summary statistics using an a priori linear model (e.g., y = x1 + 2*x2—3 where x1 and x2 are the single-nucleotide polymorphism (SNP) effects being combined). Because the amount of genetic overlap between parents and children is known a priori, WLM has already been used to adjust GWAS summary statistics for dynastic effects (e.g., the association between the maternal genotype and child phenotype due to the direct inherence of the genetic variants by the child rather than in utero effects) [8].
Here we used a WLM and GWASs of lifetime smoking and maternal smoking to derive a GWAS of paternal smoking (Fig. 1). In brief, because the offspring’s genetic liability to smoke is half due to the inertance of the maternal genetic liability, and half due to the inherence of the paternal genetic liability, by subtracting the maternal genetic liability estimated in a GWAS from the child’s genetic liability estimated in a GWAS using a WLM we created GWAS summary statistics for paternal smoking.

Study overview figure
Main text
Derivation of the linear model used in the GWAS by subtraction
An individual’s genetic risk is the average of their parent’s genetic risk. Hence:
where Bcg-cp is the association between the child’s genotype and child’s phenotype (i.e. his/her genetic liability towards the phenotype), Bfg-fp is the association between the father’s genotype and father’s phenotype, and Bmg-mp is the association between the maternal genotype and maternal phenotype.
The association between the child’s genotype and the parental phenotype is the product of the association between the child’s genotype and the parental genotype, and the parental genotype and the parental phenotype. Therefore:
where Bcg-fp is the association between the child’s genotype and the father’s phenotype, and Bcg-fg is the association between the child’s genotype and the father’s phototype.
Because the child inherits half of each parent’s genetic liability, Bcg-fg will be equal to 0.5. Therefore:
Therefore, by combining [1, 4, 5] we get:
Using the rules of propagation of error, the standard error of this effect would therefore be
where SEcg-cp is the standard error in the association between the child’s genotype and child’s phenotype, and SEcg-mp is the standard error in the association between the child’s genotype and maternal phenotype. A key of variable names in the equations can be found in Table 1.
Validation of the linear model
To validate this linear model, we ran a simulation. We report our simulations using the ADEMP (aims, data-generating mechanisms, estimands, methods, and performance measures) approach [10]:
Aims
To validate the proposed WLM as a method for producing unbiased estimates of the association between the child’s genotype and the father’s phenotype.
Data-generating mechanisms
We simulated both the inherited maternal and paternal genotypes as two independent but identically distributed one-level binomial distributions
For the paternal and maternal inherited genetic variants respectively.
The child’s genotype was then defined as
The genotype–phenotype association was defined as
The child’s phenotype was then the product of both the parental variants and a random normal error:
The maternal genotype was defined as
The maternal and paternal phenotypes were then respectively defined as:
Estimand
The association between the child’s genotype and the paternal phenotype.
Methods
We compare two methods for estimating the association between the child’s genotype with the child’s phenotype and the maternal phenotype by (1) regressing the paternal phenotype on the child’s genotype. This method would produce results analogous to those of a traditional GWAS of paternal smoking. (2) Regressing the maternal phenotype on the child’s genotype and the child’s phenotype on the child’s genotype and combining these using the proposed WLM.
Performance measure
We then calculated the mean bias and Monte-Carlo standard error in the two estimates.
Additional simulations
To further validate the model for a wider range of settings we additionally ran the simulation using the beta and minor allele frequency values for the 126 genome wide significant SNPs from the Wootton et al. GWAS of smoking as the beta (i.e. B above) minor allele frequency values (i.e. probabilities for Fi, Mi, Fp, and Mg above) for the simulation. We additionally ran the simulation using the minimum, 1st quartile, mean, median, 3rd quartile, and maximum values of the above two parameters from the GWAS (Additional file 1: Table S1).
Results of the simulation
The mean bias by directly regressing the paternal phenotype on the child’s genotype was 0.001 (95% CI 0.003 to − 0.001), while the mean bias in the WLM was 0.000 (95% CI 0.002 to − 0.002). This implies that our WLM should perform similarly, in terms of bias, to a GWAS of paternal smoking.This conclusion was reinforced by the additional simulations (Additional file 1: Table S1).
Creation and validation of GWAS summary statistics
We implemented the above WLM using the GWAS of lifetime smoking by Wootton and colleagues and the GWAS of maternal smoking during pregnancy by Elsworth and colleagues [11, 12]. Both GWAS were created using the Medical Research Council Integrated Epidemiology Unit (MRC IEU) GWAS pipeline, which is described in detail elsewhere [13]. To have comparable units in both GWASs we converted both GWAS to have units on the standardised mean difference scale. We standardised the lifetime smoking summary statistics by dividing the beta and standard error of the summary statistics by the standard deviation of lifetime smoking (0.6940093). We standardised the maternal smoking GWAS summary statistics by first converting them into log odds ratios by the effect estimate and standard error by (121634/397732)*(1-(121634/397732)) (i.e., p[1-p], where p is the prevalence of maternal smoking during pregnancy), and then convert the log odds into a standardised mean difference by dividing them by \(\left(\pi *{3}^{-0.5}\right)\) [13, 14] (Additional file 2: Fig S1).
The resulting GWAS had a genomic control inflation factor (λGC) of 1.091 (SE = 0.027). Figure 2 presents the Manhattan and QQ plot for the GWAS. The λGC and QQ plot imply that the test statistics are larger than what would be expected by chance, and therefore the potential presence of residual bias in our summary statistics. The reduction in the number of hits in the Manhattan plot when compared to the maternal smoking and lifetime smoking GWAS implies that the WLM has reduced power compared to these GWASs. Both the GWASs used the UK Biobank (UKB), and had adjusted for the UKB genoty** chip. Following general advice, we additionally created a secondary GWAS of paternal smoking from the same GWASs, but without adjusting for genoty** chip. However, not adjusting for genoty** chip appears to result in more biased estimates (see the Additional file for more details).

Manhattan and QQ plots for the GWAS of paternal smoking
Validation of the GWAS summary statistics
We validated the GWAS summary data by testing using MR that variants predicting paternal smoking associated with paternal lung cancer and emphysema/bronchitis. In brief, we selected SNPs with a 5 × 10–6 association in our paternal smoking GWAS. The p-value threshold was chosen to boost the number of SNPs included in the analysis, and therefore power while ensuring reasonably strong instrument strength. As a sensitivity analysis, we also selected SNPs at 5 × 10–7 and 5 × 10–8 p-value thresholds. We clumped with an r2 of 0.001 and kb of 10,000. We additionally implemented the False Discovery Rate Inverse Quantile Transformation Winner’s curse correction on the exposure GWASs to correct for the effect of Winner’s curse [15]. We then used the Elsworth and colleagues UKB GWAS in the MRC IEU OpenGWAS platform as a source of paternal outcome data [11]. Details on genoty**, quality control, and phenoty** can be found in the original publications and on the UKB website (https://biobank.ndph.ox.ac.uk/ukb/search.cgi).
We implemented the MR analysis using the TwoSampleMR R package [16, 17]. We harmonised the data, and allowed TwoSampleMR to removed palindromic SNPs whose effect allele could not be inferred using based on minor allele frequency. SNP effects were combined using the inverse-variance weighted (IVW) meta-analysis with multiplicative random effects.
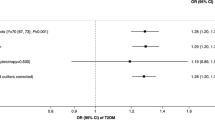
Our paternal GWAS had relatively strong instruments (mean F = 24 from 26 SNPs). As expected, in our primary analysis we found positive associations between each standard deviation of genetically proxied paternal smoking and the log odds of paternal lung cancer (risk difference per standard deviation (SD) increase in smoking = 0.754 (se = 0.362, p = 0.034) and emphysema/bronchitis (risk difference per SD increase in smoking = 1.014 (se = 0.285, p = 0.0004). Our secondary analyses using more stringent p-value thresholds to select SNPs found results in the same direction, but larger beta values. Using a 5 × 10–7 p-value threshold (N SNPs = 12, F = 27) the log odds of paternal lung cancer (risk difference per SD increase in smoking = 1.050 (se = 0.726, p = 0.148) and emphysema/bronchitis (risk difference per SD increase in smoking = 1.411 (se = 0.510, p = 0.006). Using a 5 × 10–7 p-value threshold (N SNPs = 3, F = 35) the log odds of paternal lung cancer (risk difference per SD increase in smoking = 1.452 (se = 2.774, p = 0.601) and emphysema/bronchitis (risk difference per SD increase in smoking = 3.132 (se = 0.943, p = 0.0009). The lack of a significant effect for lung cancer with the more stringent p-values probably reflects the reduction of power from using fewer SNPs, and that there are around 20% fewer lung cancer cases than emphysema/bronchitis cases (37,443 vs 46,263).
Limitations
To the best of our knowledge, this is the first application of WLMs to derive GWAS summary statistics for one parent when observations have only been made on the offspring and another parent.
Both our WLM and a direct GWAS of paternal smoking could be affected by assortative mating. Specifically, we would expect a direct GWAS of parental smoking to be inflated by assortative mating because the other parent’s genotype would confound the association with the offspring’s genotype. The WLM will then also be biased to the extent that the inflation of the SNP effects in the GWAS of parental smoking is not proportional to the inflation of the GWAS of lifetime smoking. In addition, our WLM may also be biased by residual population structure in either the lifetime smoking or maternal smoking GWASs, or indirect genetic effects in the lifetime smoking GWAS [18].
We created this GWAS primarily for use within a Mendelian randomisation design. Multivariable MR (MVMR) is an extension of MR to include multiple exposures. Estimation of the (conditional) instrument strength for an MVMR analysis requires knowing the correlation between the exposures, or assuming that the correlation is zero. The latter is less conservative, however, because we lack the phenotype exposure data the prior cannot be estimated. An alternative may be to use the correlations of maternal smoking with the relevant phenotypes as a proxy. Doing so should produce more conservative estimates of the instruments than assuming no correlation. The correlation between maternal and paternal smoking can, however, be estimated from the existing literature on assortative mating [19].
Finally, a limitation of linear models is that they can result in under-fitting of data, e.g. due to non-differential measurement error. We are, however, not aware of other external GWAS or biobank data form which we could further validate our GWAS by comparing our results too.
In this research note we have described and validated the creation of a GWAS of paternal smoking via a GWAS-by-subtraction. We hope that they will further facilitate the study of intergenerational effects.
Availability of data and materials
The code and GWAS summary statistics used in this study is available from https://doi.org/10.17605/OSF.IO/SPKMT.
References
Benn M, Nordestgaard BG. From genome-wide association studies to mendelian randomization: novel opportunities for understanding cardiovascular disease causality, pathogenesis, prevention, and treatment. Cardiovasc Res. 2018. https://doi.org/10.1093/cvr/cvy045/487705.
Mitchell R, Hemani G, Dudding T, Corbin L, Harrison S, Paternoster L. UK Biobank Genetic Data: MRC-IEU Quality Control, version 2. https://research-information.bris.ac.uk/en/datasets/uk-biobank-genetic-data-mrc-ieu-quality-control-version-2
Woolf B, Di Cara N, Moreno-Stokoe C, Skrivankova V, Drax K, Higgins JPT, et al. Investigating the transparency of reporting in two-sample summary data Mendelian randomization studies using the MR-base platform. Int J Epidemiol. 2022;46(6):815.
Bowden J, Del Greco MF, Minelli C, Davey Smith G, Sheehan NA, Thompson JR. Assessing the suitability of summary data for two-sample Mendelian randomization analyses using MR-Egger regression: the role of the I2 statistic. Int J Epidemiol. 2016;45(6):1961–74.
Burgess S, Foley CN, Zuber V. Inferring causal relationships between risk factors and outcomes from genome-wide association study data. Annu Rev Genomics Hum Genet. 2018;19(1):303–27.
Yang Q, Millard LAC, Davey SG. Proxy gene-by-environment Mendelian randomization study confirms a causal effect of maternal smoking on offspring birthweight, but little evidence of long-term influences on offspring health. Int J Epidemiol. 2020;49(4):1207–18.
Taylor AE, Carslake D, de Mola CL, Rydell M, Nilsen TIL, Bjørngaard JH, et al. Maternal smoking in pregnancy and offspring depression: a cross cohort and negative control study. Sci Rep. 2017;7(1):12579.
Investigating the genetic architecture of noncognitive skills using GWAS-by-subtraction | Nature Genetics. https://www.nature.com/articles/s41588-020-00754-2
Maternal and fetal genetic effects on birth weight and their relevance to cardio-metabolic risk factors | Nature Genetics. https://www.nature.com/articles/s41588-019-0403-1
Morris TP, White IR, Crowther MJ. Using simulation studies to evaluate statistical methods. Stat Med. 2019;38(11):2074–102.
Elsworth B, Lyon M, Alexander T, Liu Y, Matthews P, Hallett J, et al. The MRC IEU OpenGWAS data infrastructure. bioRxiv. 2020. https://doi.org/10.1101/2020.08.10.244293v1.
Wootton RE, Richmond RC, Stuijfzand BG, Lawn RB, Sallis HM, Taylor GMJ, et al. Evidence for causal effects of lifetime smoking on risk for depression and schizophrenia: a Mendelian randomisation study. Psychol Med. 2020;50(14):2435–43.
Ruth Mitchell E. MRC IEU UK Biobank GWAS pipeline version 2. data.bris. 2019. https://data.bris.ac.uk/data/dataset/pnoat8cxo0u52p6ynfaekeigi
Higgins J, Thomas J, Chandler J, Cumpston M, Li T, Page M, et al. Cochrane Handbook for Systematic Reviews of Interventions version 6.3. Cochrane; 2022. www.training.cochrane.org/handbook.
Bigdeli TB, Lee D, Riley BP, Vladimirov V, Fanous AH, Kendler KS, et al. FIQT: a simple, powerful method to accurately estimate effect sizes in genome scans. bioRxiv. 2015. https://doi.org/10.1101/019299v1.
R Core Team. R: A language and environment for statistical computing. R Foundation for Statistical Computin. 2021. https://www.R-project.org/
Hemani G, Zheng J, Elsworth B, Wade KH, Haberland V, Baird D, et al. The MR-base platform supports systematic causal inference across the human phenome. Elife. 2018;7:e34408.
Kong A, Thorleifsson G, Frigge ML, Vilhjalmsson BJ, Young AI, Thorgeirsson TE, et al. The nature of nurture: effects of parental genotypes. Science. 2018;359(6374):424–8.
Langley K, Heron J, Davey Smith G, Thapar A. Maternal and paternal smoking during pregnancy and risk of ADHD symptoms in offspring: testing for intrauterine effects. Am J Epidemiol. 2012;176(3):261–8.
Acknowledgements
This work was carried out using the computational facilities of the Advanced Computing Research Centre, University of Bristol—http://www.bris.ac.uk/acrc/.
Funding
Benjamin Woolf is funded by an Economic and Social Research Council (ESRC) South West Doctoral Training Partnership (SWDTP) 1 + 3 PhD Studentship Award (ES/P000630/1). BW, HS and MM work in the MRC Integrative Epidemiology Unit that is supported by the University of Bristol and UK Medical Research Council (MC_UU_00011/1, MC_UU_00011/3, MC_UU_00011/7). DG is funded by the British Heart Foundation Centre of Research Excellence (RE/18/4/34215) at Imperial College London. This research was funded by United Kingdom Research and Innovation Medical Research Council (MC_UU_00002/7). For the purpose of open access, the author has applied a Creative Commons Attribution (CC BY) licence to any Author Accepted Manuscript version arising from this submission.
Author information
Authors and Affiliations
Contributions
BW and DG conceived and designed the study. All authors contributed to the writing of the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
UKB received ethical approval from the North West Multi-Centre Research Ethics Committee (REC reference 11/NW/0382). All participants provided written informed consent to participate in the study. Data from the UKB are fully anonymised.
Consent for publication
Not applicable.
Competing interests
DG is employed part-time by Novo Nordisk. The other authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1.
Table S1. Results of the additional simulations.
Additional file 2.
Details of the paternal GWAS not adjusted for UK Biobank genoty** chip. Figure S1. Manhattan and QQ plots for the two paternal smoking GWAS.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Woolf, B., Sallis, H.M., Munafò, M.R. et al. Deriving GWAS summary estimates for paternal smoking in UK biobank: a GWAS by subtraction. BMC Res Notes 16, 159 (2023). https://doi.org/10.1186/s13104-023-06438-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13104-023-06438-4