
Abstract
Background
Lignocellulose is an important raw material for biomass-to-energy conversion, and it exhibits a complex but inefficient degradation mechanism. Microbial degradation is promising due to its environmental adaptability and biochemical versatility, but the pathways used by microbes for lignin degradation have not been fully studied. Degradation intermediates and complex metabolic pathways require more study.
Results
A novel actinomycete DF3-3, with the potential for lignin degradation, was screened and isolated. After morphological and molecular identification, DF3-3 was determined to be Streptomyces thermocarboxydus. The degradation of alkali lignin reached 31% within 15 days. Manganese peroxidase and laccase demonstrated their greatest activity levels, 1821.66 UL−1 and 1265.58 UL−1, respectively, on the sixth day. The highest lignin peroxidase activity was 480.33 UL−1 on the fourth day. A total of 19 lignin degradation intermediates were identified by gas chromatography–mass spectrometry (GC–MS), including 9 aromatic compounds. Genome sequencing and annotation identified 107 lignin-degrading enzyme-coding genes containing three core enzymatic systems for lignin depolymerization: laccases, peroxidases and manganese peroxidase. In total, 7 lignin metabolic pathways were predicted.
Conclusions
Streptomyces thermocarboxydus strain DF3-3 has good lignin degradation ability. Degradation products and genomics analyses of DF3-3 show that it has a relatively complete lignin degradation pathway, including the β-ketoadipate pathway and peripheral reactions, gentisate pathway, anthranilate pathway, homogentisic pathway, and catabolic pathway for resorcinol. Two other pathways, the phenylacetate–CoA pathway and the 2,3-dihydroxyphenylpropionic acid pathway, are predicted based on genome data alone. This study provides the basis for future characterization of potential biotransformation enzyme systems for biomass energy conversion.
Similar content being viewed by others
Background
Lignocellulosic biomass is an easily available, low cost and renewable feedstock for biofuel which is an important direction for the development of renewable energy. Currently, the process for pretreatment of lignocellulosic biofuel requires removal or delocalization of lignin, which might generate aromatic compounds that inhibit enzymatic hydrolysis and fermentation [1]. Due to its rich aromatic content, lignin is a valuable waste from the biomass industry [2]. Researchers worldwide are focusing on lignin and its components for conversion into value-added products. Lignin is a complex aromatic heteropolymer derived from the condensation of hydroxyphenylpropane monomers and comprises various ether bonds and carbon–carbon bonds [3]. It is mainly composed of three basic monomers: guaiacyl (G) units, syringyl (S) units and p-hydroxyphenyl (H) units [4, 5]. Microorganisms that degrade lignocellulose are widely distributed in nature, and known examples include bacteria and fungi. Among them, fungi and some bacteria have mainly been studied, and white-rot fungi, and brown rot fungi in particular have obvious degradation effects on lignin [6, 7], but the development of industrial applications has been difficult [8]. Some bacteria also participate in lignin degradation. Several typical lignin-degrading bacteria, such as Rhodococcus, Pseudomonas, Sphingobium and Sphingomonas, have been identified [9,10,11,12]. Streptomyces are among the identified bacteria capable of degrading lignin [13]. Pasti et al. [14] isolated 11 strains of actinomycetes from the intestines of termites and analysed their ability to degrade lignocellulose, lignin and carbohydrates. Other researchers have also screened streptomyces from the soil that degrade lignin [15,16,17].
There are many kinds of enzymes involved in the degradation of lignin, including laccase (Lac), lignin peroxidase (LiP), manganese peroxidase (MnP), multifunctional peroxidase (VP), and dye decolouring peroxidase (DyPs) [18]. The specific mechanism for biological degradation of lignin needs to be studied further. Lignin is rich in high-value degradation intermediates, such as vanillin, guaiacol, catechin and protocatechin [19]. At the same time, the study of the metabolic mechanism of lignin and its transformation and utilization is an important part of the research needed for production of “second-generation biofuels” [20].
Hundreds of lignin derivatives have been identified in studies of bacterial degradation of lignin [21]. Because of the complex degradation mechanism, the interpretation of metabolic pathways and intermediate products is an important part of understanding the degradation of lignin. With the development of sequencing technology and bioinformatics, genomics research has become an important method for studying degradation mechanisms. Researchers are paying increasing attention to analysing metabolism of intermediate products through annotations of related degradation genes. Pseudomonas putida is a lignin-degrading bacterium that has been studied earlier. Lin et al. [22] identified several lignin-degrading enzymes, including haem peroxidase, from its genome and constructed five lignin metabolic pathways. Masai et al. [23] conducted a series of studies on lignin degradation and related genes in Sphingomonas paucimobilis SYK-6 and established a relatively complete lignin degradation metabolic pathway. Niewerth et al. [24] determined and analysed the whole genome sequence of Arthrobacter sp. Rue61a in soil, and the results showed that it has an aromatic degradation pathway that utilizes the characteristic products of lignin degradation, reflecting the saprophytic capabilities and nutritional diversity of organisms.
Several Streptomyces strains, such as Streptomyces viridosporus T7A [25] and Streptomyces setonii 75Vi2 [26], have been reported to degrade lignin. However, the actual catabolic pathways of lignin derivatives and the responsible enzymes and genes have not been investigated using molecular methods with Streptomyces. In our present study, a novel isolate, DF3-3, identified as Streptomyces thermocarboxydus, was found to degrade lignin. Alkali lignin is a model compound with a structure similar to that of lignocellulose and is often used as a raw material for lignin degradation studies [27]. This study used alkali lignin to investigate the characteristics of alkali lignin degradation by Streptomyces thermocarboxydus strain DF3-3. GC–MS combined with genomics was used to identify the genes responsible for lignin degradation and explore the metabolic pathway for lignin degradation by Streptomyces thermocarboxydus strain DF3-3.
Results
Morphological and physiological characteristics of DF3-3
A strain of actinomycete named DF3-3 was isolated from the greening litter of Bei**g University of Agriculture. The scribing form of the plate shows a rough white surface, the upper and lower surfaces are inconsistent in colour, and the hyphae in the base are obvious on Gause's medium (Fig. 1a). When DF3-3 was inoculated on Gause’s guaiacol medium, a clear colour reaction appeared. When inoculated on Gause’s–Azure B medium, a transparent fading circle appeared (Fig. 1b, c). This shows that DF3-3 has the ability to degrade lignin [28]. Microscopy was used to observe the morphology of DF3-3 (Fig. 1d). DF3-3 grew luxuriantly on the plate, hyphae were developed, and aerial hyphae were slender. The spore filaments were spiral-shaped with obvious characteristics of Streptomyces. Figure 1d shows a scanning electron micrograph of DF3-3 grown on Gause's medium. Mature spore chains were moderately long, with 40 to 80 spores per chain. The single spores were oval or cylindrical with diameters of 0.5 to 0.7 μm and lengths of 1.1 to 1.3 μm and have rough surfaces.

DF3-3 colour reaction and morphological characteristics. a Colony morphology streaked on a plate of DF3-3. b Transparent fading circle on Gause’s–Azure B medium. c Colour reaction on Gause’s–guaiacol medium. d Scanning electron micrograph of DF3-3
While kee** the conditions of the basal medium otherwise unchanged, different nitrogen sources and carbon sources were added to observe the growth of DF3-3 with different nitrogen sources. The results are shown in Table 1. DF3-3 could grow on six common nitrogen sources including ammonium chloride, potassium nitrate, ammonium sulfate, ammonium tartrate, acrylamide and peptone, and could grow on sixteen common carbon sources including glucose, mannose, melibiose, starch, maltose, α-D-methyl glucoside, trehalose, cellobiose, xylose, salicin, glycerol, sodium malate, sodium succinate, sodium malonate, sodium tyrosinate,Amylase. However, it could not grow on other eleven common carbon sources (Table 1). DF3-3 was inoculated onto the Gause’s liquid medium, and incubated, respectively, at different temperatures and pH conditions, the optimum temperature range of 30–35 °C and optimum pH 7.5–8.5 were found by examining the strain weights after 7 days (Additional file 1: Fig. S2).
Molecular identification
Genome de novo sequencing was performed on DF3-3 cells using second- and third-generation sequencing methods, namely, Illumina HiSeq + PacBio, and the gene location and sequence information of the samples were obtained through de novo assembly and gene prediction. According to the whole genome sequencing results (Table 2), strain DF3-3 has a chromosome with a total genome length of 7,311,713 bp. GeneMarkS predicted and annotated a total of 6929 coding sequences (CDSs), with a G + C content of 72.24%.
At present, 95% of the average nucleotide identity (ANI) is often used as the standard for species classification and species clustering [29]. The whole genomes of eight strains with high homology to strain DF3-3 were selected and compared and analysed with DF3-3 using ANI. The results are shown in Fig. 2. The similarity between DF3-3 and Streptomyces thermocarboxydus reached 98.96%, and it can now be identified as Streptomyces thermocarboxydus.

ANI analysis results for DF3-3 and other bacteria
Biodegradation of alkali lignin by DF3-3
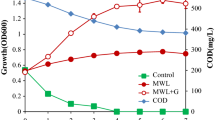
To investigate lignin degradation by strain DF3-3, cells were incubated at 30 °C in medium with alkali lignin as the carbon source. The growth curve and degradation rate of alkali lignin are shown in Fig. 3. The growth rate of DF3-3 increased significantly in 1–7 days, then, maintained a small fluctuation in 8–14 days. The degradation rate of alkaline lignin maintained a relatively uniform increase in days 1–14, eventually, the degradation rate reached a maximum value of 31% on day 15. The degradation of lignin by microorganisms requires a relatively slow process to reach a significant level. Fungi are more efficient in the breakdown of lignin than bacteria in which delignification is slower and more limited [30], the white-rot fungus Phanerochaete chrysosporium was used for degradation of lignin, and the efficiency reached approximately 20% on day 15 [31], so DF3-3 exhibited a better performance. This result is also similar to the degradation results seen with some Streptomyces strains, such as S. viridosporus T7A (lignin loss 30.9%) and S. setonii 75Vi2 (lignin loss 34.1%) [26, 32].

Growth curve and alkaline lignin degradation curve for DF3-3. Average values of three replicates are shown with the standard error of the mean as error bars
Analysis of lignin-degrading enzymes
Lignin molecules are not easily taken passively into the cell; therefore, Streptomyces thermocarboxydus DF3-3 might produce extracellular enzymes for synergistic degradation. Three major types of enzymes responsible for the degradation of lignin are lignin peroxidase (LiP), manganese peroxidase (MnP) and laccase (Lac) [33]. MnPs oxidize Mn(II) to Mn(III), and Mn(III) oxidizes phenolic compounds and generates phenoxy radicals that in turn undergo a variety of reactions, resulting in depolymerization. In the presence of Mn(II), MnP oxidizes nonphenolic lignin model compounds via peroxidation of unsaturated lipids. LiP is the most effective peroxidase and can oxidize phenolic and nonphenolic compounds, amines, aromatic ethers, and polycyclic aromatics [34]. Lac is a copper oxidoreductase [35] that can degrade refractory polyphenols and nonphenolics in lignin, and the expression of its coding genes in bacteria has also been reported [36]. The activities of these three enzymes from DF3-3 are shown in Fig. 4. MnP and Lac activity increased constantly during the initial 6 days, with maxima of 1821.66 U/L and 1265.58 U/L seen at day 6, followed by slight decreases from day 7. Lip activity was maintained at a low level, with a maximum of 480.33 U/L on day 4. These results indicated that MnP and Lac play crucial roles during the entire process of alkaline lignin degradation by DF3-3.

Enzyme activities of LiP, Lac, and MnP of DF3-3 during 11 days of incubation. Average values of three replicates are shown with the standard errors of the mean shown as error bars
Aromatic intermediates identified by GC–MS analysis
Using ethyl acetate as the solvent for GC–MS determination [71]. Organic acids, phenoxy free radicals and various transition metal coordination complexes can act as diffusion media of electrons and react directly with lignin, causing bond breakage [72]. A large number of similar low molecular weight compounds were detected in the lignin degradation products of DF3-3, which may be involved in both degradation agents and products during the degradation process. Low molecular weight compounds, containing phenolic hydroxyl, methoxy, amine, ketone, aldehyde, carboxyl and other functional groups, can generate phenoxy radicals to oxidize the non-phenolic residues of lignin in the presence of redox enzymes [73]. The roles of these compounds in the degradation of lignin by DF3-3 need further investigation.
Conclusions
Based on the above data and analyses, we isolated a bacterial strain identified as Streptomyces thermocarboxydus strain DF3-3 from greening litter and concluded that it degraded alkaline lignin, and the degradation efficiency reached 31% within 15 days. In total, 19 alkaline lignin degradation intermediates were identified by GC–MS, and 107 possible lignin-degrading enzyme encoding genes in the DF3-3 genome were annotated, 7 pathways for metabolism of lignin and its intermediates were predicted, including the β-ketoadipate pathway and peripheral reactions, gentisate pathway, anthranilate pathway, catabolic pathway for resorcinol, homogentisic pathway, phenylacetate–CoA pathway, and the 2,3-dihydroxyphenylpropionic acid pathway. Intermediates in the first five metabolic pathway were detected by GC–MS. The degradation products and genomics analyses show that DF3-3 has a relatively complete lignin degradation pathway.
Methods
Sampling, isolation and screening of bacterial strain
Samples were collected from greening litter of Bei**g University of Agriculture (40.0947°N, 116.3151°E). One gram of sample was placed in a 50-mL sterile centrifuge tube containing 10 mL of sterile distilled water and shaken at 120 rpm overnight. Next, 10−1 and 10−2 serial dilutions of each sample suspension were spread as 0.1-mL aliquots on Gause’s synthetic medium with the formula (g/L): 0.5 NaCl; 1 KNO3; 0.5 K2HPO4·3H2O; 0.5 MgSO4·7H2O; 0.01 FeSO4·7H2O; 20 soluble starch [8]. The plates were incubated at 30 °C for 1 week, and distinct colonies were picked and subcultured for further analysis. Gause’s guaiacol medium and Gause’s–Azure B medium used for lignin degradation screening contained 0.1% guaiacol and 0.1% aniline blue, respectively, added to Gause’s medium. Different external nitrogen sources (20 g/L), such as acrylamide and potassium nitrate, and additional carbon sources (1 g/L), such as glucose and mannose, were used to replace soluble starch culture strains in studies of their utilization of nitrogen sources and carbon sources. The culture medium for detecting lignin degradation and enzyme activity was kraft lignin–MSM medium (3 g of kraft lignin L, 2 g of [NH4]2SO4, 1 g of K2HPO4, 1 g of KH2PO4, 0.2 g of MgSO4, 0.1 g of CaCl2, 0.05 g of FeSO4, and 0.02 g of MnSO4 in 1 L distilled water, pH 7.0).
Scanning electron microscope observations
The shapes of the bacteria were observed by scanning electron microscopy. A cover glass was inserted into the solid medium to cultivate the strain, and the insert was removed after the bacterial body climbed onto the glass slide. The precipitate was washed by adding a phosphate buffer solution (pH 7.2), added to 2.5% glutaraldehyde, fixed at room temperature for 2–4 h, and then placed in a refrigerator at 4 °C overnight. After elution with a 30–95% ethanol gradient, the material was rinsed with tert-butanol, then 20 μL of tert-butanol was added and the mixture was put into a refrigerator at − 20 ℃ until it froze and solidified. Using critical point drying (HITACHI HCP-2 Critical Point Dryer) and gold sputter coating (Eiko IB-3 ion plating machine), the sample was observed by scanning electron microscopy (SEM, JSM-6360LV, JEOL, Japan) [74].
Strain growth curve determination
To assess the growth of bacteria, an equal quantity of bacteria was inserted into Gause’s liquid medium and cultured on a shaker. The culture solution was removed and centrifuged every 24 h. The supernatant was discarded, and the filter paper was placed into an oven. The mixture was dried to a constant weight, and the filter paper and the bacteria were weighed. The weight of the filter paper was compared with the weight of the bacteria. All assays were performed with three replicates.
Biodegradation of alkali lignin
To determine the lignin loss from alkaline lignin cause by various strains, samples (1.5 mL) were centrifuged at 12,000 ×g for 10 min. One millilitre of supernatant was diluted by adding 2 mL of phosphate buffer (pH 7.6). The lignin concentration was determined by measuring the absorbance at 280 nm with a UV–Vis spectrophotometer (Shimadzu UN-1900i) [75]. The calculated standard curve for lignin was y = 0.0786x–0.0245, R2 = 0.9988.
Enzyme assay
Samples were centrifuged at 12,000 rpm for 5 min, and the supernatant was used for lignin peroxidase (Lip), laccase and manganese peroxidase (MnP) enzyme assays. Laccase activity was determined by monitoring the oxidation of ABTS at 420 nm (ε420 = 36,000 M−1 cm−1). A lignin peroxidase assay was carried out using peroxidase oxidation of Azure B. LiP activity was determined by measuring the absorbance at 651 nm (ε651 = 48.8 M−1 cm−1). Manganese peroxidase activity was determined from the change in absorbance occurring when Mn2+ is oxidized to Mn3+ and forms a complex with malonate, which produced absorbance at 270 nm (ε270 = 11,590 M−1 cm−1) [76].
Genome sequencing and functional annotation
The genome of DF3-3 was sequenced at Major Biomedical Technology Co., Ltd. (Shanghai, China). Genomic DNA was extracted using a Wizard® Genomic DNA Purification Kit (Promega). Purified genomic DNA was quantified by a TBS-380 fluorometer (Turner BioSystems Inc., Sunnyvale, CA). The genome was sequenced by adopting the second-generation + third-generation sequencing method of Illumina HiSeq + PacBio, with a shotgun library of 400 bp insertion size. Assembly software canu, SPAdes, etc. was used for three-generation sequence assembly [77], and GeneMarkS software was used to predict the coding sequence (CDS) in the genome [78]. The prediction and annotation of genes were carried out using Prodigal Son (prokaryotic dynamic programming gene discovery algorithm). GeneMarkS was used to predict the plasmid genome. tRNAscan-SE v2.0 software was used to predict the tRNA contained in the genome, and Barrnap software was used to predict the rRNA contained in the genome. Functional annotation of the predicted coding gene was carried out by comparison with 6 major databases (NR, Swiss-Prot, Pfam, EggNOG, GO and KEGG) [79,80,81,82].
Alkali lignin degradation products determined by GC–MS
DF3-3 was inoculated in 100 ml of medium with AL as the carbon source and cultured on a shaker for 7 days. Samples were collected every 24 h, and a number of control groups was set up. The sample was centrifuged (10,000 rpm, 15 min) to remove the bacteria, the supernatant was acidified with HCl to pH 2–3, and it was thoroughly extracted with a threefold volume of ethyl acetate. The extract was rotary evaporated to 10 ml at 37 °C and dried with anhydrous Na2SO4. After evaporating the solvent in a nitrogen stream, 100 µl of the organic layer was derivatized. Then, 100 μl of dioxane and 10 μl of pyridine were added to the sample and vortexed, and 50 μl of bis(trimethylsilyl)trifluoroacetamide (BSTFA) was added. The mixed solution was placed in a water bath at 80 °C for 45 min and shaken regularly. The silanized sample was tested by GC–MS [83].
The analytical column was a DB-5 capillary column (30 m length, 0.25 mm inner diameter, 0.25 mm film thickness). The carrier gas was helium. The column temperature was initially 50 °C (5 min), then it was raised to 280 °C (10 °C/min, holding time of 5 min). The transmission line and ion source temperatures were 200 and 250 °C, respectively. The solvent delay time was 4.0 min. The injection volume was 1 μl. Electron ionization mass spectra were recorded in the range 30–550 (m/z) in full scan mode.
Availability of data and materials
The additional data generated during this study are available in the Additional file.
Abbreviations
- AL:
-
Alkali lignin
- GC–MS:
-
Gas chromatography–mass spectrometry
- MnP:
-
Manganese peroxidase
- Lac:
-
Laccase
- LiP:
-
Lignin peroxidase
References
Zeng Y, Zhao S, Yang S, Ding SY. Lignin plays a negative role in the biochemical process for producing lignocellulosic biofuels. Curr Opin Biotechnol. 2014;27:38–45.
Kumar M, Singhal A, Thakur IS. Comparison of submerged and solid state pretreatment of sugarcane bagasse by Pandoraea sp. ISTKB: enzymatic and structural analysis. Bioresour Technol. 2016;203:18–25.
Kamimura N, Sakamoto S, Mitsuda N, Masai E, Kajita S. Advances in microbial lignin degradation and its applications. Curr Opin Biotechnol. 2019;56:179–86.
Ralph J, Lundquist K, Brunow G, Lu F, Kim H, Schatz PF, et al. Lignins: natural polymers from oxidative coupling of 4-hydroxyphenyl- propanoids. Phytochem Rev. 2004;3:29–60.
Abdelaziz OY, Brink DP, Prothmann J, Ravi K, Sun M, García-Hidalgo J, et al. Biological valorization of low molecular weight lignin. Biotechnol Adv. 2016;34:1318–46.
Tuor U, Winterhalter K, Fiechter A. Enzymes of white-rot fungi involved in lignin degradation and ecological determinants for wood decay. J Biotechnol. 1995;41:1–17.
Sánchez C. Lignocellulosic residues: biodegradation and bioconversion by fungi. Biotechnol Adv. 2009;27:185–94.
Yang YS, Zhou JT, Lu H, Yuan YL, Zhao LH. Isolation and characterization of Streptomyces spp. strains F-6 and F-7 capable of decomposing alkali lignin. Environ Technol. 2012;33:2603–9.
Mcleod MP, Warren RL, Hsiao W, Araki N, Myhre M, Fernandes C, et al. The complete genome of Rhodococcus sp. RHA1 provides insights into a catabolic powerhouse. Proc Natl Acad Sci USA. 2006;103:15582–7.
Masai E, Kamimura N, Kasai D, Oguchi A, Ankai A, Fukui S, et al. Complete genome sequence of Sphingobium sp. strain SYK-6, a degrader of lignin-derived biaryls and monoaryls. J Bacteriol. 2012;194:534–5.
Lin L, Cheng Y, Pu Y, Sun S, Li X, ** M, et al. Systems biology-guided biodesign of consolidated lignin conversion. Green Chem. 2016;18:5536–47.
Janshekar H, Fiechter A. On the bacterial degradation of lignin. Eur J Appl Microbiol Biotechnol. 1982;14:47–50.
Bugg TD, Ahmad M, Hardiman EM, Singh R. The emerging role for bacteria in lignin degradation and bio-product formation. Curr Opin Biotechnol. 2011;22:394–400.
Pasti MB, Pometto AL, Nuti MP, Crawford DL. Lignin-solubilizing ability of actinomycetes isolated from termite (Termitidae) gut. Appl Environ Microbiol. 1990;56:2213–8.
Vishwanatha BT, Babu G, Malagi SB, Dandin CJ, Nayaka S. Isolation, identification and characterization of streptomyces sp. SN-2. Biosci Biotechnol Res Asia. 2017;14:1401–7.
Feng H, Sun Y, Zhi Y, Mao L, Luo Y, Wei X, et al. Lignocellulose degradation by the isolate of Streptomyces griseorubens JSD-1. Funct Integr Genom. 2015;15:163–73.
Feng H, Zhi Y, Shi W, Mao L, Zhou P. Isolation, identification and characterization of a straw degrading Streptomyces griseorubens JSD-1. Afr J Microbiol Res. 2013;7:2730–5.
Lambertz C, Ece S, Fischer R, Commandeur U. Progress and obstacles in the production and application of recombinant lignin-degrading peroxidases. Bioengineered. 2016;7:145–54.
Cragg SM, Beckham GT, Bruce NC, Bugg TDH, Distel DL, Dupree P, et al. Lignocellulose degradation mechanisms across the tree of life. Curr Opin Chem Biol. 2015;29:108–19.
Bugg TDH, Ahmad M, Hardiman EM, Rahmanpour R. Pathways for degradation of lignin in bacteria and fungi. Nat Prod Rep. 2011;28:1883–96.
Zakzeski J, Bruijnincx PC, Jongerius AL, Weckhuysen BM. The catalytic valorization of lignin for the production of renewable chemicals. Chem Rev. 2010;110:3552–99.
Lin L, Wang X, Cao L, Xu M. Lignin catabolic pathways reveal unique characteristics of dye-decolorizing peroxidases in Pseudomonas putida. Environ Microbiol. 2019;21:1847–63.
Masai E, Katayama Y, Fukuda M. Genetic and biochemical investigations on bacterial catabolic pathways for lignin-derived aromatic compounds. J Agric Chem Soc Jpn. 2007;71:1–15.
Niewerth H, Schuldes JR, Parschat K, Kiefer P, Vorholt JA, Daniel R, et al. Complete genome sequence and metabolic potential of the quinaldine-degrading bacterium Arthrobacter sp. Rue61a. BMC Genom. 2012;13:534.
Ramachandra M, Crawford DL, Hertel G. Characterization of an extracellular lignin peroxidase of the lignocellulolytic actinomycete Streptomyces viridosporus. Appl Environ Microbiol. 1988;54:3057–63.
Antai SP, Crawford DL. Degradation of softwood, hardwood, and grass lignocelluloses by two streptomyces strains. Appl Environ Microbiol. 1981;42:378–80.
Chakar FS, Ragauskas AJ. Review of current and future softwood kraft lignin process chemistry. Ind Crops Prod. 2004;20:131–41.
Sumiati T, Suryadi H, Harmita H, Sutriyo S. Isolation of white rot fungi from rotten wood from bogor botanical garden in Indonesia and its ligninolytic enzymes activity. Pharm J. 2022;14:68.
Jain C, Rodriguez RL, Phillippy AM, Konstantinidis KT, Aluru S. High throughput ANI analysis of 90K prokaryotic genomes reveals clear species boundaries. Nat Commun. 2018;9:5114.
Sigoillot J-C, Berrin J-G, Bey M, Lesage-Meessen L, Levasseur A, Lomascolo A, et al. Fungal strategies for lignin degradation. Adv Bot Res. 2012;61:263–308.
Zhao M, Zhang C, Zeng G, Huang D, Xu P, Cheng M. Growth, metabolism of Phanerochaete chrysosporium and route of lignin degradation in response to cadmium stress in solid-state fermentation. Chemosphere. 2015;138:560–7.
Crawford DL, Pometto AL, Crawford RL. Lignin degradation by Streptomyces viridosporus: isolation and characterization of a new polymeric lignin degradation intermediate. Appl Environ Microbiol. 1983;45:898–904.
Kumar M, Mishra A, Singh SS, Srivastava S, Thakur IS. Expression and characterization of novel laccase gene from Pandoraea sp. ISTKB and its application. Int J Biol Macromol. 2018;115:308–16.
Pérez J, Munoz-Dorado J, De la Rubia T, Martinez J. Biodegradation and biological treatments of cellulose, hemicellulose and lignin: an overview. Int Microbiol. 2002;5:53–63.
de Gonzalo G, Colpa DI, Habib MHM, Fraaije MW. Bacterial enzymes involved in lignin degradation. J Biotechnol. 2016;236:110–9.
Wu J, Kim KS, Lee JH, Lee YC. Cloning, expression in Escherichia coli, and enzymatic properties of laccase from Aeromonas hydrophila WL-11. J Environ Sci. 2010;22:635–40.
Zhu D, Zhang P, **e C, Zhang W, Sun J, Qian WJ, et al. Biodegradation of alkaline lignin by Bacillus ligniniphilus L1. Biotechnol Biofuels. 2017;10:44.
Chen YH, Chai LY, Zhu YH, Yang ZH, Zheng Y, Zhang H. Biodegradation of kraft lignin by a bacterial strain Comamonas sp. B-9 isolated from eroded bamboo slips. J Appl Microbiol. 2012;112:900–6.
Raj A, Reddy MMK, Chandra R, Purohit HJ, Kapley A. Biodegradation of kraft-lignin by Bacillus sp. isolated from sludge of pulp and paper mill. Biodegradation. 2007;18:783–92.
Ksibi M, Amor SB, Cherif S, Elaloui E, Houas A, Elaloui M. Photodegradation of lignin from black liquor using a UV/TiO2 system. J Photochem Photobiol A Chem. 2003;154:211–8.
Shin KS, Lee YJ. Depolymerisation of lignosulfonate by peroxidase of the white-rot basidiomycete, Pleurotus ostreatus. Biotechnol Lett. 1999;21:585–8.
**ong YI, Zhao Y, Ni K, Shi Y, Xu Q. Characterization of ligninolytic bacteria and analysis of alkali-lignin biodegradation products. Pol J Microbiol. 2020;69:339–47.
Pérez-Pantoja D, De la Iglesia R, Pieper DH, González B. Metabolic reconstruction of aromatic compounds degradation from the genome of the amazing pollutant-degrading bacterium Cupriavidus necator JMP134. FEMS Microbiol Rev. 2008;32:736–94.
Toyama T, Momotani N, Ogata Y, Miyamori Y, Inoue D, Sei K, et al. Isolation and characterization of 4-tert-butylphenol-utilizing Sphingobium fuliginis strains from Phragmites australis rhizosphere sediment. Appl Environ Microbiol. 2010;76:6733–40.
Gabriel FL, Giger W, Guenther K, Kohler HPE. Differential degradation of nonylphenol isomers by Sphingomonas xenophaga Bayram. Appl Environ Microbiol. 2005;71:1123–9.
Gabriel FL, Cyris M, Jonkers N, Giger W, Guenther K, Kohler HPE. Elucidation of the ipso-substitution mechanism for side-chain cleavage of α-quaternary 4-nonylphenols and 4-t-butoxyphenol in Sphingobium xenophagum Bayram. Appl Environ Microbiol. 2007;73:3320–6.
Janusz G, Pawlik A, Sulej J, Świderska-Burek U, Jarosz-Wilkołazka A, Paszczyński A. Lignin degradation: microorganisms, enzymes involved, genomes analysis and evolution. FEMS Microbiol Rev. 2017;41:941–62.
Xu G, Li F, Wang Q. Occurrence and degradation characteristics of dibutyl phthalate (DBP) and di-(2-ethylhexyl) phthalate (DEHP) in typical agricultural soils of China. Sci Total Environ. 2008;393:333–40.
Schühle K, Jahn M, Ghisla S, Fuchs G. Two similar gene clusters coding for enzymes of a new type of aerobic 2-aminobenzoate (anthranilate) metabolism in the bacterium Azoarcus evansii. J Bacteriol. 2001;183:5268–78.
Liu D, Yan X, Si M, Deng X, Min X, Shi Y, et al. Bioconversion of lignin into bioplastics by Pandoraea sp. B-6: molecular mechanism. Environ Sci Pollut Res Int. 2019;26:2761–70.
Mohamed Mel S, Ismail W, Heider J, Fuchs G. Aerobic metabolism of phenylacetic acids in Azoarcus evansii. Arch Microbiol. 2002;178:180–92.
Ferrández A, Miñambres B, García B, Olivera ER, Luengo JM, García JL, et al. Catabolism of phenylacetic acid in Escherichia coli. Characterization of a new aerobic hybrid pathway. J Biol Chem. 1998;273:25974–86.
Brown ME, Chang MCY. Exploring bacterial lignin degradation. Curr Opin Chem Biol. 2014;19:1–7.
Masai E, Katayama Y, Kubota S, Kawai S, Yamasaki M, Morohoshi N. A bacterial enzyme degrading the model lignin compound beta-etherase is a member of the glutathione-S-transferase superfamily. FEBS Lett. 1993;323:135–40.
Masai E, Kubota S, Katayama Y, Kawai S, Yamasaki M, Morohoshi N. Characterization of the C alpha-dehydrogenase gene involved in the cleavage of beta-aryl ether by Pseudomonas paucimobilis. Biosci Biotechnol Biochem. 1993;57:1655–9.
Colpa DI, Fraaije MW, van Bloois E. DyP-type peroxidases: a promising and versatile class of enzymes. J Ind Microbiol Biotechnol. 2014;41:1–7.
Majumdar S, Lukk T, Solbiati JO, Bauer S, Nair SK, Cronan JE, et al. Roles of small laccases from Streptomyces in lignin degradation. Biochemistry. 2014;53:4047–58.
Ihssen J, Reiss R, Luchsinger R, Thöny-Meyer L, Richter M. Biochemical properties and yields of diverse bacterial laccase-like multicopper oxidases expressed in Escherichia coli. Sci Rep. 2015;5:10465.
Medema MH, Trefzer A, Kovalchuk A, van den Berg M, Müller U, Heijne W, et al. The sequence of a 1.8-mb bacterial linear plasmid reveals a rich evolutionary reservoir of secondary metabolic pathways. Genome Biol Evol. 2010;2:212–24.
Ahmad M, Taylor CR, Pink D, Burton K, Eastwood D, Bending GD, et al. Development of novel assays for lignin degradation: comparative analysis of bacterial and fungal lignin degraders. Mol BioSyst. 2010;6:815–21.
da Silva Coelho-Moreira J, Maciel GM, Castoldi R, da Silva Mariano S, Inácio FD, Bracht A, et al. Involvement of lignin-modifying enzymes in the degradation of herbicides. In: Andrew P, editor., et al., Herbicides: advances in research. London: Intech; 2013. p. 165–87.
Ferreira P, Hernández-Ortega A, Herguedas B, Rencoret J, Gutiérrez A, Martínez MJ, et al. Kinetic and chemical characterization of aldehyde oxidation by fungal aryl-alcohol oxidase. Biochem J. 2010;425:585–93.
Crawford DL, Pometto AL 3rd, Crawford RL. Production of useful modified lignin polymers by bioconversion of lignocellulose with Streptomyces. Biotechnol Adv. 1984;2:217–32.
Masai E, Katayama Y, Nishikawa S, Yamasaki M, Morohoshi N, Haraguchi T. Detection and localization of a new enzyme catalyzing the beta-aryl ether cleavage in the soil bacterium (Pseudomonas paucimobilis SYK-6). FEBS Lett. 1989;249:348–52.
Sonoki T, Obi T, Kubota S, Higashi M, Masai E, Katayama Y. Coexistence of two different O demethylation systems in lignin metabolism by Sphingomonas paucimobilis SYK-6: cloning and sequencing of the lignin biphenyl-specific O-demethylase (LigX) gene. Appl Environ Microbiol. 2000;66:2125–32.
Masai E, Harada K, Peng X, Kitayama H, Katayama Y, Fukuda M. Cloning and characterization of the ferulic acid catabolic genes of Sphingomonas paucimobilis SYK-6. Appl Environ Microbiol. 2002;68:4416–24.
Masai E, Sasaki M, Minakawa Y, Abe T, Sonoki T, Miyauchi K, et al. A novel tetrahydrofolate-dependent O-demethylase gene is essential for growth of Sphingomonas paucimobilis SYK-6 with syringate. J Bacteriol. 2004;186:2757–65.
Kersten PJ, Dagley S, Whittaker JW, Arciero DM, Lipscomb JD. 2-pyrone-4,6-dicarboxylic acid, a catabolite of gallic acids in Pseudomonas species. J Bacteriol. 1982;152:1154–62.
Kasai D, Masai E, Miyauchi K, Katayama Y, Fukuda M. Characterization of the 3-O-methylgallate dioxygenase gene and evidence of multiple 3-O-methylgallate catabolic pathways in Sphingomonas paucimobilis SYK-6. J Bacteriol. 2004;186:4951–9.
Wöhlbrand L, Kallerhoff B, Lange D, Hufnagel P, Thiermann J, Reinhardt R, et al. Functional proteomic view of metabolic regulation in “Aromatoleum aromaticum” strain EbN1. Proteomics. 2007;7:2222–39.
Acebes S, Ruiz-Dueñas FJ, Toubes M, Sáez-Jiménez V, Pérez-Boada M, Lucas MF, et al. Map** the long-range electron transfer route in ligninolytic peroxidases. J Phys Chem B. 2017;121:3946–54.
Arantes V, Milagres AM. Relevance of low molecular weight compounds produced by fungi and involved in wood biodegradation. Quim Nova. 2009;32:1586–95.
Acunzo F, Galli C, Gentili P, Sergi F. Mechanistic and steric issues in the oxidation of phenolic and non-phenolic compounds by laccase or laccase-mediator systems. The case of bifunctional substrates. New J Chem. 2006;30:583–91.
Castillo U, Myers S, Browne L, Strobel G, Hess WM, Hanks J, et al. Scanning electron microscopy of some endophytic streptomycetes in snakevine- Kennedia nigricans. Scanning. 2005;27:305–11.
Lara MA, Rodriguez-Malaver AJ, Rojas OJ, Holmquist O, González AM, Bullón J, et al. Black liquor lignin biodegradation by Trametes elegans. Int Biodeterior Biodegrad. 2003;52:167–73.
Kumar M, Singh J, Singh MK, Singhal A, Thakur IS. Investigating the degradation process of kraft lignin by β-proteobacterium, Pandoraea sp. ISTKB Environ Sci Pollut Res. 2015;22:15690–702.
Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, et al. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol. 2012;19:455–77.
Besemer J, Borodovsky M. GeneMark: web software for gene finding in prokaryotes, eukaryotes and viruses. Nucleic Acids Res. 2005;33:W451–4.
Bairoch A, Apweiler R. The SWISS-PROT protein sequence database and its supplement TrEMBL in 2000. Nucleic Acids Res. 2000;28:45–8.
Jensen LJ, Julien P, Kuhn M, von Mering C, Muller J, Doerks T, et al. eggNOG: automated construction and annotation of orthologous groups of genes. Nucleic Acids Res. 2008;36:D250–4.
Kanehisa M, Goto S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000;28:27–30.
Finn RD, Alex B, Jody C, Penelope C, Eberhardt RY, Eddy SR, et al. Pfam: the protein families database. Nucleic Acids Res. 2014;42:D222–30.
Raj A, Chandra R, Reddy MMK, Purohit HJ, Kapley A. Biodegradation of kraft lignin by a newly isolated bacterial strain, Aneurinibacillus aneurinilyticus from the sludge of a pulp paper mill. World J Microbiol Biotechnol. 2007;23:793–9.
Acknowledgements
The authors thank Bei**g Key Laboratory of New Technology in Agricultural Application for their support of the part of the work involving scanning electron microscopy. And genome analysis was performed using the free online platform of Majorbio Cloud Platform (www.majorbio.com).
Funding
This work was funded by the National Key Research and Development Program of China (code: 2017YFF0207800) and the Scientific Research Program of Bei**g Municipal Education Commission (code: KM201810020012,KN201910020017).
Author information
Authors and Affiliations
Contributions
YQL designed the experimental strategy. FYT, JC, YZ and XFJ carried out the experiments and performed the bioinformatics data analysis. FYT and JC wrote the manuscript. YQL supervised the overall research. This manuscript was proofread by all the authors. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Ethical approval and consent to participate are not required.
Consent for publication
All authors agree to the submission and publication of the manuscript in the journal Biotechnology for Biofuel.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Figure S1.
Schematic of the Streptomyces thermocarboxydus strain DF3-3 genome. The first and fourth circles of the circle diagram from the outside to the inside are the CDS on the positive chain and the negative chain, and different colors indicate different COG functional classifications. The second and third circles are CDS, tRNA and rRNA; on positive chain and negative chain, respectively. The fifth circle is GC content. The outward part indicates that the GC content in this region is higher than the average GC content of the whole genome. The higher the peak value, the greater the difference from the average GC content. The inward part indicates that the GC content in this region is lower than the average GC content of the whole genome. The sixth circle is GC-Skew value, and the specific algorithm is G-C/G+C, which can assist in judging leading strand and lag chain. In general, leading strand GC skew>0 and lag chain GC skew < 0 can also assist in judging replication origin (minimum cumulative offset) and end point (maximum cumulative offset), especially for circular genome. The innermost circle is the genome size marker. Figure S2. Growth of DF3-3 at different culture temperature and pH. a Growth at different temperatures. b Growth at different pH values. Table S1. The weight (g) of the growth curve and the degradation rate of the alkaline lignin of DF3-3. Table S2. Enzyme activities (U/L) of LiP, Lac, and MnP of DF3-3. Table S3. GCMS spectrum of products of alkaline lignin degradation by DF3-3. Table S4. Compounds at each peak and their relative contents. Table S5. GCMS spectra of products of alkaline lignin degradation by DF3-3. Table S6. Genes responsible for the β-ketoadipate pathway and peripheral reactions. Table S7. Genes responsible for gentisate pathways. Table S8. Genes responsible for pathways for anthranilate. Table S9. Genes responsible for pathways for resorcinol. Table S10. Genes responsible for catabolic pathways for homogentisic. Table S11. Genes responsible for pathways for phenylacetate–CoA. Table S12. Genes responsible for catabolic pathways for 2,3-dihydroxyphenylpropionate.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Tan, F., Cheng, J., Zhang, Y. et al. Genomics analysis and degradation characteristics of lignin by Streptomyces thermocarboxydus strain DF3-3. Biotechnol Biofuels 15, 78 (2022). https://doi.org/10.1186/s13068-022-02175-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13068-022-02175-1