
Abstract
Smoking is the major cause of cardiovascular diseases and cancer. It induces oxidative stress, leading to DNA damage and cellular senescence. Senescent cells increase the expression and release of pro-inflammatory molecules and matrix metalloproteinase, which are known to play a vital role in the initiation and progression of cardiovascular diseases and metastasis in cancer. The current study investigated the smoking induced cellular senescence and employed colchicine that blocked senescence in endothelial cells exposed to tobacco smoke condensate. Colchicine prevented oxidative stress and DNA damage in tobacco smoke-condensate-treated endothelial cells. Colchicin reduced β-gal activity, improved Lamin B1, and attenuated cell growth arrest markers P21 and P53. Colchicine also ameliorated the expression of SASP factors and inhibited the activation of NF-kB and MAPKs P38 and ERK. In summary, colchicine inhibited tobacco smoke condensate-induced senescence in endothelial cells by blocking the activation of NF-kB and MAPKs P38 and ERK.
Graphical Abstract

Similar content being viewed by others

Introduction
Tobacco smoking is one of the major preventable causes of premature deaths globally and is a potentially acquired risk factor for cardiovascular diseases (CVDs), chronic obstructive pulmonary disease, and cancer [1,2,3]. The scientific literature suggests tobacco smoking to be a causative agent for CVDs and cancers [2]. According to the World Health Organization, tobacco kills more than 8 million people each year worldwide, including around 1.3 million people who do not smoke but are exposed to second-hand smoke.
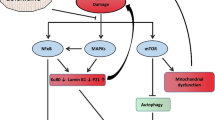
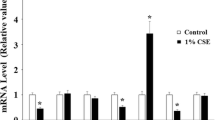
There are more than 7000 chemicals in cigarette smoke, of which 250 are harmful, and 69 are well-established carcinogens [3, 4]. The constituents of cigarette smoke generate oxidative stress, which can subsequently cause DNA damage, leading to stress-induced premature cellular senescence [4,5,6]. Cellular senescence is characterized by cell cycle arrest, macromolecular damage, metabolic dysfunction, and a shift in the expression of secondary markers [7, 8]. Transient cellular senescence can exert a beneficial impact, such as wound healing and tumor suppression [6, 9]. However, the chronic accumulation of senescent cells can impair wound healing, accelerate aging, promote inflammation, and give rise to chronic diseases such as CVDs, cancer, and neurodegenerative diseases [9,10,18]. Therefore, it can be postulated that through mitigating the expression of SASP factors (Fig. 3; Table 2) [23,24,25,26,27], colchicine can suppress the initiation and progression of CVDs [22, 23, 25] and metastasis, angiogenesis, and growth in cancers [42].
Our pathway analysis showed that tobacco smoke condensate activated NF-κB (Fig. 4) and MAPKs P38 and ERK (Fig. 5). It has already been reported that DNA damage, oxidative stress, and ethanol can activate these pathways [21, 26, 27, 38, 39]. Activation of these pathways has been linked to cellular senescence, and inhibiting these pathways could block cellular senescence [21]. After activation, these pathways enhance the transcription, protein expression, and stability of senescence-driving proteins such as P53 and P21 [21, 37, 43, 44]. Moreover, MAPKs P38 and ERK increase the levels of SASP factors through NF-κB transcriptional activity [21]. The activation of P38 is needed for NF-κB transcriptional activity in senescence [45]. Interestingly, colchicine, without significantly affecting p-mTOR expression, increased the relative protein expression of mTOR downstream signalling molecules p-S6 and p-4EBP-1 (Fig. 6). These two downstream signalling molecules have the opposite effect on life span [46,47,48]. S6K deficiency in mice and flies extended mean life span [46, 48] and 4EBP-1 overexpression in flies increased life span [47]. Therefore, the net effect of colchicine through mTOR pathway activation would be challenging to determine. Taken together, it is very likely that by blocking NF-κB, P38- and ERK (Figs. 4 and 5) [15, 21, 26, 27, 44], colchicine suppressed senescence and the expression of SASP factors in endothelial cells (Figs. 2 and 3) treated with tobacco smoke condensate.
Conclusion
Tobacco smoking is one of the potential acquired causative risk factors for chronic and age-related diseases. Colchicine prevented tobacco smoke condensate-induced DNA damage and senescence in endothelial cells exposed to smoke condensate. It mitigated the expression of SASP factors in endothelial cells treated with tobacco smoke condensate. Colchicine blocked tobacco smoke-condensate-induced activation of NF-κB, P38, and ERK. These findings suggest that by suppressing the activation of NF-κB and MAPKs, colchicine inhibited senescence in endothelial cells treated with tobacco smoke condensate. The current findings will have implications in cardiovascular diseases.
Materials and methods
Cell Culture
Three different HUVEC models were procured from Promocell (Heidelberg, Germany). Endothelial cell medium (C-22,010, Promocell, Heidelberg, Germany) containing endothelial growth factors (C-39,215, Promocell, Heidelberg, Germany) was used to maintain endothelial cells at 37 °C in a humidified environment at 37 °C and 5% CO2. Upon arrival, the cells were thawed and seeded in T75 culture flasks. The cells were passaged when they reached 80–90% confluence. For passaging, the cells were washed with PBS and incubated with trypsin for 4 min at 37 °C in a humidified environment at 37 °C and 5% CO2. The cells were seeded at a density of 5000 cells/cm2 in new cell culture plates. All experiments were performed at passage 7. The cells were treated with either 50 µg tobacco smoke condensate, 50 nm colchicine or tobacco smoke condensate combined with colchicine. Untreated cells were used as control. All experiments were performed with three biological replicates except Lamin B1 and DCFH-DA staining, where one HUVEC cell model was used.
Tobacco smoke condensate preparation
Commercially available cigarettes were smoked through ethanol. After that, ethanol was evaporated at room temperature. Tobacco smoke condensate was weighed, and 100 mg/mL of tobacco smoke condensate was dissolved in DMSO.
DCFH-DA staining
To investigate the accumulation of cellular ROS, 5000 cells/cm2 endothelial cells were seeded in a 96-well plate. The next day, the medium was changed with a new medium either containing 50 µg/mL tobacco smoke condensate, 50 nM colchicine, or 50 µg/mL smoke condensate combined with 50 nM colchicine. Endothelial cell medium alone was used as a control. After 2 h of treatment, 10 µM of the fluorescence probe 2,7-dichlorofluorescein diacetate (DCFH-DA, D6883, Sigma-Aldrich, MO, USA) was added to the cells. The cells were incubated with DCFG-DA at 37 °C for 30 min in the dark. The cells were washed three times with a serum-free medium. The images were taken with a fluorescence microscope and analyzed with image J.
Immunofluorescence staining
The cells were treated with different conditions (as described in the previous section) for 2 h for 8-OHDG staining and 24 h for Lamin B1 staining. After washing cells thrice with PBS, the cells were fixed with 4% paraformaldehyde for 10 min at RT. After washing cells thrice with PBS, for permeabilization, the cells were treated with 0.2% Triton™ X-100 at RT for 10 min. For blocking, the cells were incubated with 5% bovine serum albumin (BSA) at RT for 1 h. The cells were incubated overnight with primary antibodies 8-OHDG (Supplementary table S1) and Lamin B1 (Supplementary table S1) at 4° C. The next day, after washing cells thrice with PBS, the cells were incubated with secondary antibodies (Supplementary table S1) for 1 h at RT. HOECHST (Sigma-Aldrich) was used for nuclear staining. The images were captured using a Leica DMi8 Inverted Microscope and the compatible LAS-X Life Science Microscope Software (Leica Application Suite X) Platform. The images were analyzed using ImageJ (version 1.53c) (National Institutes of Health, Bethesda, MD, USA).
Western blot
For protein analysis, HUVECs were treated with 50 µg/mL smoke condensate, 50 nM colchicine, or 50 µg/mL smoke condensate combined with 50 nM colchicine for 24 h. Untreated endothelial cells were used as controls. RIPA buffer was used for total protein extraction. DC Protein Assay Kit (500–0116, Bio-Rad, Hercules, CA, USA) was used to quantify protein concentration. Subsequently, 30 µg of total protein under reducing conditions was loaded onto a 12% sodium dodecyl sulfate-polyacrylamide gel. For the first 20 min, electrophoresis was conducted at 60 Volts, followed by 110 Volts for 30–60 min. The separated proteins were then transferred onto a 0.45 μm pore –size nitrocellulose membrane at 250 mA for 120 min. The membranes were blocked for one hour with a 5% bovine serum albumin (BSA) solution in 0.05% TBST to minimize nonspecific binding. After that, the membranes were incubated with primary antibodies (see Supplementary Table S1) in 5% BSA overnight at 4 °C on a shaking platform. Afterward, the membranes underwent 3 × 10 min washes with TBST and were subsequently exposed to secondary antibodies diluted in 0.05% TBST (refer to Supplementary Table S1) for one hour at room temperature. Densitometry analysis was performed using NIHImageJ with β-actin correction.
Quantitative polymerase chain reaction (qPCR)
For qPCR analysis, the endothelial cells were treated as described in the previous section. Total RNA was extracted using the Nucleo Spin RNA kit (740955.50, MACHEREY-NAGEL, Düren, Germany) according to the manufacturer’s instructions. A total of 1.2 µg of RNA was utilized for reverse transcription, accomplished using the MMLV Reverse Transcriptase kit (M1701, Promega, Walldorf, Germany), Random Hexamer Primers (48,190,011, Thermo Fisher), and RiboLock RNase Inhibitor (EO0384, Thermo Fisher). The qPCR was run using total cDNA combined with AceQ SYBR qPCR Master Mix (Q111-03, Vayzme, Nan**g, China) and primers (Supplementary Table 2) on a Bio-Rad thermal cycler. The thermal cycling program consisted of an initial denaturation step at 95 °C for 8 min, followed by 40 cycles of 95 °C for 15 s, 58.9 °C for 30 s, and 72 °C for 30 s, concluding with a melting curve analysis. To calculate relative mRNA expressions, data were normalized to β-actin expression, and the relative expression levels were quantified using the comparative ΔCT method.
Senescence associated β-Galactosidase staining
For senescence associate beta-galactosidase (SA-β-Gal) staining, endothelial cells were treated for 24 h as described in the previous sections. SA β-Gal staining was performed using the Senescence Cells Histochemical Staining Kit (GALS, Sigma, MO, USA) following the manufacturer’s instructions. The cells were incubated with SA–-β-galactosidase staining solution at 37 °C for seven hours. The staining solution was aspirated and the cells were overlaid with 70% glycerol in PBS. After staining, the cells were stored at 4 °C. The images were captured using a Leica DMi8 Inverted Microscope and the compatible LAS-X Life Science Microscope Software (Leica Application Suite X) Platform. The images were analyzed using ImageJ (version 1.53c) (National Institutes of Health, Bethesda, MD, USA).
Statistical analysis
We analyzed the data on PRISM using one-way ANOVA followed by Tukey’s post hoc test.
Data availability
The datasets supporting the conclusions of this article are included within the article and its additional file.
References
Kotlyarov S. The role of smoking in the mechanisms of Development of Chronic Obstructive Pulmonary Disease and Atherosclerosis. Int J Mol Sci 2023, 24.
Kopp W. Pathogenesis of (smoking-related) non-communicable diseases-evidence for a common underlying pathophysiological pattern. Front Physiol. 2022;13:1037750.
Dahdah A, Jaggers RM, Sreejit G, Johnson J, Kanuri B, Murphy AJ, Nagareddy PR. Immunological insights into cigarette Smoking-Induced Cardiovascular Disease Risk. Cells 2022, 11.
Seo YS, Park JM, Kim JH, Lee MY. Cigarette smoke-Induced reactive oxygen species formation: a concise review. Antioxid (Basel) 2023, 12.
Paudel KR, Panth N, Manandhar B, Singh SK, Gupta G, Wich PR, Nammi S, MacLoughlin R, Adams J, Warkiani ME et al. Attenuation of cigarette-smoke-Induced oxidative stress, senescence, and inflammation by Berberine-Loaded Liquid Crystalline nanoparticles: in Vitro Study in 16HBE and RAW264.7 cells. Antioxid (Basel) 2022, 11.
Kumari R, Jat P. Mechanisms of Cellular Senescence: cell cycle arrest and Senescence Associated Secretory phenotype. Front Cell Dev Biol. 2021;9:645593.
Gonzalez-Gualda E, Baker AG, Fruk L, Munoz-Espin D. A guide to assessing cellular senescence in vitro and in vivo. FEBS J. 2021;288:56–80.
Gorgoulis V, Adams PD, Alimonti A, Bennett DC, Bischof O, Bishop C, Campisi J, Collado M, Evangelou K, Ferbeyre G, et al. Cellular Senescence: defining a path Forward. Cell. 2019;179:813–27.
Huang W, Hickson LJ, Eirin A, Kirkland JL, Lerman LO. Cellular senescence: the good, the bad and the unknown. Nat Rev Nephrol. 2022;18:611–27.
Song P, An J, Zou MH. Immune Clearance of Senescent Cells to Combat Ageing and Chronic diseases. Cells 2020, 9.
**ao S, Qin D, Hou X, Tian L, Yu Y, Zhang R, Lyu H, Guo D, Chen XZ, Zhou C, Tang J. Cellular senescence: a double-edged sword in cancer therapy. Front Oncol. 2023;13:1189015.
Sharma R, Diwan B, Sharma A, Witkowski JM. Emerging cellular senescence-centric understanding of immunological aging and its potential modulation through dietary bioactive components. Biogerontology. 2022;23:699–729.
Wang Y, Luo G, Chen J, Jiang R, Zhu J, Hu N, Huang W, Cheng G, Jia M, Su B, et al. Cigarette smoke attenuates phagocytic ability of macrophages through down-regulating milk fat globule-EGF factor 8 (MFG-E8) expressions. Sci Rep. 2017;7:42642.
Song P, Zhao Q, Zou MH. Targeting senescent cells to attenuate cardiovascular disease progression. Ageing Res Rev. 2020;60:101072.
Sun Y, Wang X, Liu T, Zhu X, Pan X. The multifaceted role of the SASP in atherosclerosis: from mechanisms to therapeutic opportunities. Cell Biosci. 2022;12:74.
Sweeney M, Cook SA, Gil J. Therapeutic opportunities for senolysis in cardiovascular disease. FEBS J. 2023;290:1235–55.
Zhao H, Wu L, Yan G, Chen Y, Zhou M, Wu Y, Li Y. Inflammation and tumor progression: signaling pathways and targeted intervention. Signal Transduct Target Ther. 2021;6:263.
Young D, Das N, Anowai A, Dufour A. Matrix Metalloproteases as Influencers of the Cells’ Social Media. Int J Mol Sci 2019, 20.
Khan D, Cornelius JF, Muhammad S. The role of NF-kappaB in intracranial aneurysm pathogenesis: a systematic review. Int J Mol Sci 2023, 24.
Khan D, Li X, Hashimoto T, Tanikawa R, Niemela M, Lawton M, Muhammad S. Current mouse models of intracranial aneurysms: analysis of pharmacological agents used to Induce aneurysms and their impact on Translational Research. J Am Heart Assoc 2024:e031811.
Anerillas C, Abdelmohsen K, Gorospe M. Regulation of senescence traits by MAPKs. Geroscience. 2020;42:397–408.
Roubille F, Merlet N, Busseuil D, Ferron M, Shi Y, Mihalache-Avram T, Mecteau M, Brand G, Rivas D, Cossette M, et al. Colchicine reduces atherosclerotic plaque vulnerability in rabbits. Atheroscler Plus. 2021;45:1–9.
Sun X, Duan J, Gong C, Feng Y, Hu J, Gu R, Xu B. Colchicine ameliorates dilated Cardiomyopathy Via SIRT2-Mediated suppression of NLRP3 inflammasome activation. J Am Heart Assoc. 2022;11:e025266.
Yang M, Lv H, Liu Q, Zhang L, Zhang R, Huang X, Wang X, Han B, Hou S, Liu D et al. Colchicine Alleviates Cholesterol Crystal-Induced Endothelial Cell Pyroptosis through Activating AMPK/SIRT1 Pathway. Oxid Med Cell Longev 2020, 2020:9173530.
Zhang B, Huang R, Yang D, Chen G, Chen Y, Han J, Zhang S, Ma L, Yang X. Combination of Colchicine and Ticagrelor Inhibits Carrageenan-Induced Thrombi in Mice. Oxid Med Cell Longev 2022, 2022:3087198.
Zhou H, Khan D, Gerdes N, Hagenbeck C, Rana M, Cornelius JF, Muhammad S. Colchicine protects against ethanol-Induced Senescence and Senescence-Associated Secretory phenotype in endothelial cells. Antioxidants. 2023;12:960.
Zhou H, Khan D, Hussain SM, Gerdes N, Hagenbeck C, Rana M, Cornelius JF, Muhammad S. Colchicine prevents oxidative stress-induced endothelial cell senescence via blocking NF-kappaB and MAPKs: implications in vascular diseases. J Inflamm (Lond). 2023;20:41.
Tardif JC, Kouz S, Waters DD, Bertrand OF, Diaz R, Maggioni AP, Pinto FJ, Ibrahim R, Gamra H, Kiwan GS, et al. Efficacy and safety of low-dose colchicine after myocardial infarction. N Engl J Med. 2019;381:2497–505.
Nidorf SM, Fiolet ATL, Mosterd A, Eikelboom JW, Schut A, Opstal TSJ, The SHK, Xu XF, Ireland MA, Lenderink T, et al. Colchicine in patients with chronic coronary disease. N Engl J Med. 2020;383:1838–47.
Pennings GJ, Reddel CJ, Traini M, Campbell H, Chen V, Kritharides L. Colchicine inhibits ROS generation in response to glycoprotein VI stimulation. Sci Rep. 2021;11:11965.
** S, Weaver DT. Double-strand break repair by Ku70 requires heterodimerization with Ku80 and DNA binding functions. EMBO J. 1997;16:6874–85.
Evangelisti C, Rusciano I, Mongiorgi S, Ramazzotti G, Lattanzi G, Manzoli L, Cocco L, Ratti S. The wide and growing range of lamin B-related diseases: from laminopathies to cancer. Cell Mol Life Sci. 2022;79:126.
Shimi T, Butin-Israeli V, Adam SA, Hamanaka RB, Goldman AE, Lucas CA, Shumaker DK, Kosak ST, Chandel NS, Goldman RD. The role of nuclear lamin B1 in cell proliferation and senescence. Genes Dev. 2011;25:2579–93.
Freund A, Laberge RM, Demaria M, Campisi J. Lamin B1 loss is a senescence-associated biomarker. Mol Biol Cell. 2012;23:2066–75.
Quaas M, Muller GA, Engeland K. p53 can repress transcription of cell cycle genes through a p21(WAF1/CIP1)-dependent switch from MMB to DREAM protein complex binding at CHR promoter elements. Cell Cycle. 2012;11:4661–72.
Engeland K. Cell cycle regulation: p53-p21-RB signaling. Cell Death Differ. 2022;29:946–60.
Saha K, Adhikary G, Kanade SR, Rorke EA, Eckert RL. p38delta regulates p53 to control p21Cip1 expression in human epidermal keratinocytes. J Biol Chem. 2014;289:11443–53.
Li X, Khan D, Rana M, Hänggi D, Muhammad S. Doxycycline attenuated ethanol-Induced Inflammaging in endothelial cells: implications in alcohol-mediated vascular diseases. Antioxidants. 2022;11:2413.
Zhou H, Li X, Rana M, Cornelius JF, Khan D, Muhammad S. mTOR inhibitor Rapalink-1 prevents ethanol-Induced Senescence in endothelial cells. Cells. 2023;12:2609.
Feng Y, Ye D, Wang Z, Pan H, Lu X, Wang M, Xu Y, Yu J, Zhang J, Zhao M, et al. The role of Interleukin-6 Family members in Cardiovascular diseases. Front Cardiovasc Med. 2022;9:818890.
Su JH, Luo MY, Liang N, Gong SX, Chen W, Huang WQ, Tian Y, Wang AP. Interleukin-6: a Novel Target for Cardio-Cerebrovascular diseases. Front Pharmacol. 2021;12:745061.
Wang L, Lankhorst L, Bernards R. Exploiting senescence for the treatment of cancer. Nat Rev Cancer. 2022;22:340–55.
Bulavin DV, Saito S, Hollander MC, Sakaguchi K, Anderson CW, Appella E, Fornace AJ Jr. Phosphorylation of human p53 by p38 kinase coordinates N-terminal phosphorylation and apoptosis in response to UV radiation. EMBO J. 1999;18:6845–54.
Nicolae CM, O’Connor MJ, Constantin D, Moldovan GL. NFkappaB regulates p21 expression and controls DNA damage-induced leukemic differentiation. Oncogene. 2018;37:3647–56.
Freund A, Patil CK, Campisi J. p38MAPK is a novel DNA damage response-independent regulator of the senescence-associated secretory phenotype. EMBO J. 2011;30:1536–48.
Selman C, Tullet JM, Wieser D, Irvine E, Lingard SJ, Choudhury AI, Claret M, Al-Qassab H, Carmignac D, Ramadani F, et al. Ribosomal protein S6 kinase 1 signaling regulates mammalian life span. Science. 2009;326:140–4.
Zid BM, Rogers AN, Katewa SD, Vargas MA, Kolipinski MC, Lu TA, Benzer S, Kapahi P. 4E-BP extends lifespan upon dietary restriction by enhancing mitochondrial activity in Drosophila. Cell. 2009;139:149–60.
Kapahi P, Zid BM, Harper T, Koslover D, Sapin V, Benzer S. Regulation of lifespan in Drosophila by modulation of genes in the TOR signaling pathway. Curr Biol. 2004;14:885–90.
Acknowledgements
We are thankful to Michael Hewera for drawing graphical abstract.
Funding
Open Access funding enabled and organized by Projekt DEAL. We are thankful to Stiftung Neurochirurgische Forschung (DGNC), EANS Research Funds, Forschungskommission HHU Düsseldorf, James und Elisabeth Cloppenburg, Peek & Cloppenburg Düsseldorf Fund and BMBF to S. Muhammad.
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Contributions
Conceptualization: SM; Methodology: DK, HZ, JV, VAK; Formal analysis and investigation: DK, HZ; Writing - original draft preparation: DK; Writing - review and editing: RKK, SM; Funding acquisition: SM; Resources: SM; Supervision: SM.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Khan, D., Zhou, H., You, J. et al. Tobacco smoke condensate-induced senescence in endothelial cells was ameliorated by colchicine treatment via suppression of NF-κB and MAPKs P38 and ERK pathways activation. Cell Commun Signal 22, 214 (2024). https://doi.org/10.1186/s12964-024-01594-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12964-024-01594-x