
Abstract
Background
The global population of older individuals is growing, and ageing is a key risk factor for atherosclerotic cardiovascular diseases. Abnormal accumulation of senescent cells can cause potentially deleterious effects on the organism with age. As a vital marker of cellular senescence, the senescence-associated secretory phenotype (SASP) is a novel mechanism to link cellular senescence with atherosclerosis.
Main body
In this review, we concretely describe the characteristics of the SASP and its regulation mechanisms. Importantly, we provide novel perspectives on how the SASP can promote atherosclerosis. The SASP from different types of senescent cells have vital roles in atherosclerosis progression. As a significant mediator of the harmful effects of senescent cells, it can play a pro-atherogenic role by producing inflammation and immune dysfunction. Furthermore, the SASP can deliver senescence signals to the surrounding vascular cells, gradually contributing to the development of atherosclerosis. Finally, we focus on a variety of novel therapeutic strategies aimed to reduce the burden of atherosclerosis in elderly individuals by targeting senescent cells and inhibiting the regulatory mechanisms of the SASP.
Conclusion
This review systematically summarizes the multiple roles of the SASP in atherosclerosis and can contribute to the exploration of new therapeutic opportunities.
Similar content being viewed by others


Introduction
Ageing has long been considered a major risk factor for many chronic and fatal diseases [1], including cardiovascular diseases (CVDs) [2], neurodegenerative diseases and cancer [3]. Atherosclerosis as a chronic inflammatory disease of the blood vessel walls is the main potential cause of CVD [4]. It is well known that there are many different risk factors for atherosclerosis such as high blood pressure, low-density lipoprotein (LDL) cholesterol, diabetes and smoking [5]. At the same time, increasing evidence suggests that ageing is one of the greatest risk factors for atherosclerosis [6]. Because ageing is a modifiable factor, it can be viable to slow down the development of age-related diseases by regulating basic ageing mechanisms. One of the mechanisms is cell senescence, which can lead to chronic inflammation through the SASP [7].
The SASP contributes to the secretion of inflammatory cell cytokines and chemokines that induce local and systemic inflammatory responses, immune system activation, tissue damage and fibrosis, and cell apoptosis and dysfunction. Moreover, the SASP can also induce the enlargement of local and systemic senescence to neighbouring cells via paracrine or endocrine mechanisms [8]. Furthermore, a variety of molecules involved in the SASP can serve as promoters and biomarkers of cardiovascular diseases including atherosclerosis [9]. Recent clinical trials have clearly demonstrated a causal relationship between inflammation and human atherosclerosis [10]. Atherosclerosis is considered a chronic inflammatory disease, and atherosclerotic plaques present with cell senescence [11]. Cell senescen ce and atherosclerosis have multiple common aetiological stimuli, but senescent cells are not just simple bystanders. Senescent cells from atherosclerotic plaques are lack proliferation, overexpress P16INK4A, P53, P21 [12,13,14], and increase the activity of senescence-associated beta-galactosidase (SAβG) [15]. They can also establish the SASP, which can cause increased secretion of various inflammatory cell cytokines, chemokines and matrix-degrading proteases [16]. Notably, there is evidence that the SASP, as a source of chronic inflammation and some plaque instability factors, is involved in the pathogenesis and development of atherosclerosis [17].
The SASP from senescent cells exerts many pro-atherogenic effects, which may involve vascular remodelling, plaque formation and rupture. There is evidence that plaque-rich arteries contain various typical SASP components, including matrix metalloproteinases and multiple inflammatory factors. However, these phenomena are not present in normal adjacent blood vessels [17]. Senescent cells in blood vessels with the SASP release various inflammatory cytokines (interleukin-6 and interleukin-8) and growth factors (such as VEGF, PDGF, chemokines and MMPs) [89], which are responsible for transferring senescence to adjacent cells, thus leading to persistent low-level chronic inflammation, defined as inflammatory ageing. CANTOS has shown that targeting inflammation has a significant therapeutic effect on atherosclerotic diseases [90]. Furthermore, inflammatory ageing promotes the development of catastrophic atherosclerotic thrombotic complications by increasing platelet reactivity and predisposing patients to plaque rupture and erosion [91]. There is evidence that human atherosclerotic plaques contain cells with multiple senescence markers related to the severity of the diseases [89]. Cellular senescence is considered a vital source of the inflammatory response in atherosclerosis and CVDs [9].
Senescent cells present in atherosclerotic lesions from LdLR − / − mice fed a low-fat diet (LFD) have high transcription levels of p16Ink4a, and p19Arf. At the same time, various typical SASP components of the plaque-rich aortic arch, including matrix metalloproteinases MMP-3 and MMP-13, and inflammatory factors IL-1α and TNF-α, are increasing [17]. However, these phenomena are absent in normal adjacent vessels or aortas [17]. Senescent vascular cells are characterized by the SASP and release various inflammatory cytokine growth factors, chemokines and MMPs, some of which are known as cardiovascular risk factors [18]. Furthermore, senescent vascular cells exhibit the SASP that enables them to communicate with the microenvironment of other cells and promote senescent regeneration and embryonic development of adjacent cells and tissues [20].
Advanced atherosclerosis has a large number of cells with senescent characteristics that support the pro-inflammatory state, leading to the formation of necrotic cores, which ultimately lead to the fragility and rupture of the plaque, formation of thrombosis and acute vascular occlusion. This is because the expression of cells in advanced atherosclerotic plaques further aggravates the SASP of inflammation and simultaneously produces metalloproteases that degrade the extracellular matrix and further destroy the stability of atherosclerotic plaques [92]. Specifically, the senescent cells in the lesion showed expression of vital SASP cytokines and effector factors of inflammation, monocyte chemotaxis and proteolysis, including IL-1α, MCP-1, MMP-12 and MMP-13. This indicates that senescent cells can directly affect the development of core atherosclerosis through specific secretion factors [17]. For example, the SASP directly drives inflammation through IL-1α translocation to the cell surface and activates neighbouring VSMCs, ECs and macrophages, causing the spread of inflammation and promoting atherosclerosis by secondary pro-inflammatory cytokines [16]. Thus, removing senescent cells from advanced plaques prevents plaque enlargement and adaptive poor plaque remodelling processes connected with plaque instability, including thinning the fibrous cap and elastic fibre degeneration [17].
Effect of the SASP on atherosclerosis via different senescent cells
According to experiments with human samples and animal models, the accumulation of senescent cardiovascular cells plays an irreplaceable role in the promotion and development of cardiovascular ageing (Fig. 3) [20], aggravating the development of atherosclerosis and other cardiovascular and cerebrovascular diseases [20, 27]. The short-term accumulation of senescent cells in tissues is important to the stability of the internal environment and organ function, while the long-term accumulation of senescent cells in tissues leads to an imbalance of the internal environment and organ dysfunction. This reverse effect of cell senescence is due to the SASP [61], [93].

Various senescent cells contributions to the mechanisms of atherosclerosis
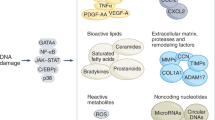
Furthermore, the paracrine signalling of the SASP has many negative influences including tissue remodelling, tumour generation and inflammatory ageing [94]. One of the most important mechanisms is inflammatory ageing. Inflammatory ageing contributes to the activation of leukocytes, ECs, and VSMCs, thereby accelerating vascular senescence and atherosclerosis due to chronic activation of inflammasomes and reduction of endogenous anti-inflammatory mechanisms [91]. We summarize a series of studies that suggest that different types of senescent cells play vital roles in the progression of atherosclerosis through the SASP factor (Table 1).
The “necrotic” core of late atherosclerotic plaques consists of lipids, foam cells, and debris. The fiber cap around the necrotic core is protective, which consists mainly of VSMCs. The stability of atherosclerotic plaques depends on the thickness of the fiber cap and the extent of inflammation of the fiber cap. At later stages, vascular cells such as VSMCs, ECs, monocytes and macrophages undergo senescence and release multiple cytokines as part of the SASP, which augment the preexisting inflammation.
Senescence of endothelial cells
Endothelial cell senescence and endothelial cell dysfunction [111] are closely associated with the subsequent development and progression of CVD [52, 112, 113. And an increase in senescent ECs causes various diseases associated with arterial thrombosis [110. Endothelial cell senescence directly destroys the endothelial barrier by destroying cell proliferation, permeability, and motility, which may lead to endothelial cell erosion and intraplaque haemorrhage [9]. Additionally, there is evidence that ECs display strong SA β-gal activity, an important feature of senescent cells, in human coronary atherosclerotic lesions [13].
Endothelial cell senescence is associated with a significant increase in the production or release of a series of inflammatory factors and chemokines, known as the SASP [114]. Senescent ECs can produce more inflammatory factors such as interleukin-1, interleukin-6, interleukin-8, interleukin-15, MCP-1, TNF, and other mediators that promote atherosclerosis and thrombosis, and express cell adhesion molecules such as ICAM-1, and PAI-1 [52]. In addition, senescent ECs with the SASP express adhesion molecules including VCAM-1 and ICAM-1 and release many cell cytokines, causing the recruitment of monocytes. Then, circulating monocytes invade the subendothelial space, absorb ox-LDL and convert into foam cells [96]. Both foam cells and ECs with the SASP release excess chemotactic proteins, including interleukin-1, tumour necrosis factor, and MCP-1, which lead to the recruitment of immune cells and the formation of atherosclerotic plaques [95]. ECs with the SASP can lead to thrombosis via the activation of PAI-1, a known marker of senescence [95, 98]. Senescent HUVECs secrete many factors, such as interleukin-6, interleukin-8, and MCP-1, and growth factors such as VEGF, TGF and MMPs, compared with young cells [115].
Kotla S and his colleagues confirmed that D-flow-induced senescence and activation of ECs play an important role in promoting atherosclerosis. Their study found that TERF2IP-dependent gene expression and its role in D-flow-induced SASP Telomere Repeat-Binding Factor 2 (TERF2) Interacting Protein (TERF2IP) is a complex on the telomere that regulates the SASP [116]. Age-related vasculopathy, including atherosclerosis, is a major risk factor for vascular diseases (VDs). A chronic aseptic low-grade inflammatory state, also known as inflammation, is a feature of the elderly and is involved in the development of VD. It is becoming a vital mediator of inflammation and VD. This is because microRNA34a expression increases with vascular ageing, and it causes VSMCs and ECs to acquire the SASP [109]. It has been reported that senescent cells have this specific secretory phenotype that alters the homeostasis of surrounding tissues and the environment. For example, senescent endothelial cells and VSMCs have been shown to promote the expression of a variety of inflammatory factors, including IL-6 and IL-8. At the same time, senescent smooth muscle cells also release MMP-9 and generate less collagen, thus promoting vascular remodelling and atherosclerotic plaque instability [117].
Senescence of vascular smooth muscle cells
VSMC senescence is closely related to a variety of cardiovascular diseases, including atherosclerosis, and has an important promoting effect [101, 118, 119]. Moreover, senescent VSMCs usually drive the instability of atherosclerotic plaques, leading to myocardial ischaemia and cerebral ischaemia [110. Senescence of VSMCs is associated with an enlarged necrotic core and plaque calcification. It has been reported that senescent VSMCs in atherosclerosis have the SASP, so they can not only promote inflammation by releasing inflammatory cytokines and chemokines in an interleukin-1-dependent way, but also recruit immune cell aggregation to accelerate the progression of atherosclerosis [102].
VSMCs from human atherosclerosis are characterized by high expression levels of genes associated with senescent cells, which explains why the cells have the SASP [12]. Senescent VSMCs acquire the phenotype secreted by osteoblasts and activate several osteogenic pathways such as osteoprotegerin (OPG), a key factor of the SASP and a CVD risk factor, which can promote plaque calcification [9]. Senescent VSMCs, together with SASP, lead to chronic vascular inflammation, loss of arterial function, and age-related conditions [102]. Senescent human VSMCs secrete a variety of SASP factors including interleukin-6, interleukin-8, and MCP-1, in an IL-1α-dependent manner, but the expression of anti-inflammatory factors, such as RANTES and IL-1R2, is decreased. Notably, the SASP molecules released by these senescent VSMCs can promote the recruitment of monocytes and macrophages.
Moreover, we found that senescent human vascular smooth muscle cells can actively promote the occurrence of atherosclerosis and plaque rupture by establishing the SASP. The SASP is characterized by sustained secretion of a variety of inflammatory factors and chemokines, which are regulated by the autocrine stimulation of senescent vascular smooth muscle cells by IL-1α. These powerful chemotactic signals lead to the aggregation of monocytes and lymphocytes, while secreted IL-1α leads to the activation of adjacent normal VSMCs and ECs, leading to pro-inflammatory cytokines secretion and amplification of adhesion receptor expression. Senescent VSMCs also release less collagen and more MMP-9, which degrades collagen in the matrix and weakens the plaque [101]. Consequently, VSMCs senescence not only promotes atherosclerotic progression, but also suppresses plaque repair.
Senescent VSMCs can promote chronic inflammation associated with atherosclerosis by an interleukin-1α-driven senescence-related secretory phenotype and initiating neighbouring cells into a pre-atherosclerotic state. These data indicate the need to block interleukin-1α to inhibit this potentially important chronic inflammatory source in atherosclerosis [101].
Senescence of T cell
In addition to vascular system cells, senescent immune cells found in the walls of blood vessels also contribute to the development of atherosclerosis [79]. Senescent T cells secrete abundant pro-inflammatory factors such as tumour necrosis factor (TNF) and osteopontin, reminiscent of the SASP [106]. Lymphocytes (T cells) with shortened telomere length are more evident than myeloid cells in patients with coronary heart disease compared with age-matched controls, suggesting that T cells may play a vital role in ageing and telomere-mediated atherosclerosis [120].
The expression of CD27 and CD28 proteins is strongly downregulated in senescent T cells, while the secretion of pro-inflammatory factors such as IL-6 and TNF-α is increased, suggesting the presence of the SASP state [68]. Elevated levels of Th1 and Th17, cells and their cytokines interleukin-17, interleukin-21, and interleukin-23 are found in carotid atherosclerotic plaques, and these factors are associated with the promotion of atherosclerosis and plaque vulnerability [121]. Senescent T cells with CD4, CD28null, and CD45 re-expression are found to be a highly active subset that expressed increased tumour necrosis factor, INF-c, and CXCR-3. Moreover, many studies have shown that ageing affects memory T cells in unstable atherosclerotic plaques [79]. Meanwhile, CD4+ and CD8+ T-EMRA cells are considered to be predictors of cardiovascular-related mortality in elderly people [122].
In addition to the pro-inflammatory phenotype, T-EMRA cells show many characteristics of cellular senescence, such as decreased proliferation, mitochondrial dysfunction, increased secretion of tumour necrosis factor and interferon-γ, and increased p38 MAPK signaling [123]. Moreover, T-EMRA cells show atypical cytotoxic activity against plaque endothelial cells, potentially leading to plaque erosion [9].There is increasing evidence that senescent T cells, mainly cytotoxic CD8+ T cells, are also involved in the pathogenesis of CVD. In atherosclerosis and ACS, CD8+CD28− T cell expansion is found in cohorts of CMV infected individuals and constitutes a risk factor for vascular dysfunction. Furthermore, during the development of atherosclerosis, CD8+ CD28null CD27− senescent T cells in inflammatory vascular walls continuously produce IFN-γ, a factor of the SASP, which activates macrophages to release MMPs to degrade the extracellular matrix [107]. Senescent T cells are present in peripheral blood mononuclear cells of humans [121]with atherosclerosis [124] and myocardial ischaemia [125].
Senescence of macrophage
Some studies have shown that senescent immune cells show characteristics of the SASP, such as T cells [123, 126, 127], B cells [128] and macrophages [129]. The pro-inflammatory SASP properties of immune cells, particularly macrophages, appear to play an important role in maintaining low-grade chronic defect states in tissues [126]. Macrophages are the main senescent cells present with high levels of SA-β-Gal staining and the production of the SASP [17]. At the late stage of macrophage senescence, the net effect is the expression of the SASP, which aggravates the senescence of macrophages and releases outside the cells to impair the function of the surrounding cells. This process is known as “paracrine senescence” [53], which can cause more extensive inflammation [108]. Moreover, lncRNAs induce the SASP expression, that is, the upregulation of matrix metalloproteinase, interleukin-1β, interleukin-6 and TNF-α, and the down expression of IL-4 and IL-10, leading to inflammation, which is one of the signs of senescent macrophages [11, 108].
The involvement of macrophages in senescence and chronic inflammatory diseases is known as “macrophage senescence”. This concept highlights the critical role of the innate immune system, in particular pro-inflammatory macrophages as promotors and enhancers of premature senescence and in chronic diseases including CVD, and a recent study on the presence of pro-inflammatory M1 macrophages in plaque atherosclerosis leading to ACS [121]. Proatherogenic inflammatory factors including IL-6, IL-12, and reactive oxygen species (ROS), can promote the oxidative stress response of plaques [121]. Alternatively, studies have shown that senescent CD14+ CD16+ monocytes are capable of increasing inflammatory responses and interactions with endothelial cells. Thus, the accumulation of monocytes in senescence can promote the progression of chronic inflammation-related diseases including atherosclerosis [130]. In advanced atherosclerotic plaques, senescent macrophages work by increasing the formation of MMP-3 and MMP-13 [17], causing unstable features of atherosclerotic plaques including elastic fibre defects and thinning of the fibrous cap in the artery and the aorta [17, 110].
It has been reported that senescent macrophages formed in a BRD4-dependent epigenetic way are one of the crucial factors that drive the development of atherosclerosis. The chromosomal redistribution of BRD4 promotes the expression of senescence-associated proteins and the SASP genes regulated by NF-κB activation. Inhibition of BRD4 by inhibitors including siBRD4, JQ-1 or I-BET762 prevents the senescence of macrophages and lipid accumulation by reducing the expression of the SASP proteins in autocrine and paracrine senescence. This study is important for understanding the pathophysiology of age-related diseases and can guide future research on targeted drug treatment in the clinic [129].
Effect of the SASP on atherosclerosis through immune dysfunction
Immunosenescence and inflammation
Whether cell senescence is good or bad depends largely on the time, short-term types seem to be beneficial because the immune system can efficiently clear away senescent cells (Fig. 4) [25]. A healthy immune system is absolutely necessary to clear senescent cells [131]. Senescent cells can efficiently mobilize immune cells to cause their own clearance, but clearance is delayed or impaired with age [11, 132,133,134]. It has been reported that the SASP has some benefits for tissue remodelling and repair [32], and can stimulate the immune system to clear away senescent cells. However, this protective process may eventually become less efficient with age and lead to increased inflammatory responses [79] and age-related diseases including atherosclerosis.

The effect of the SASP factors on the immune response. a Effects of the SASP factors secreted by short-term types of cell senescence on immune cells. b Effects of the SASP factors secreted by long-term types of cell senescence on immune cells. c The SASP plays an immunosuppressive role by promoting the proliferation of MDSCs
Immunosenescence is a phenomenon in which the immune system gradually declines with age [68]. When the chemotaxis and targeting of immune cells such as NK cells, macrophages and T cells to the increased number of senescent cells are reduced [24], age-related phagocytosis of neutrophils and macrophages is impaired, leading to the accumulation of senescent cells [135]. This finally leads to increased generation of the SASP, which promotes inflammation and the progression of age-related diseases [7]. Notably, high levels of pro-inflammatory factors promote the progression of inflammation, increase endothelial damage, and vascular remodelling damage, and ultimately lead to atherosclerosis [92, 121]. Immunosenescence is associated with chronic low-grade inflammation [136]. Immune monitoring decreases with age, for example, “older” T cells release more pro-inflammatory factors, including interleukin and tumor necrosis factor than “younger” T cells [137]. The accumulation of increasingly inefficient ageing-associated T cells may explain the susceptibility to fatal infections as well as the progression of noncommunicable diseases, including cancer or arteriosclerosis [106]. The major pathological processes of age-associated cardiovascular diseases, including endothelial dysfunction and arteriosclerosis, are primarily associated with immunosenescence and inflammation and can lead to atherosclerosis [138].
Notably, immunosenescence and the SASP are found in the elderly and in many age-associated diseases, including cardiovascular diseases as well as atherosclerosis, metabolic diseases, tumour promotion, and neurodegenerative diseases [121]. Chronic low-grade inflammation and immune senescence promote the development of multiple diseases mentioned above, so research on immune senescence could bring feasible solutions to the prevention and treatment of age-related diseases. Mitigating inflammation to inhibit the occurrence of age-associated diseases and the decrease in the immune response should be the focus of future research [106]. And attempts are being made to unite different strategies, such as anti-inflammatory drugs and immune checkpoint inhibitors, to optimally block inflammation [139]. For example, studies have shown that long-term and low-dose resveratrol reversed immune ageing and inflammation in elderly mice because it improves age-related elevations of 8-hydroxy-2′-deoxyguanosine, a label of oxidative DNA impairment [140].
Immunosuppressive and immunosuppressive cells
Senescence cells through the SASP induce immune cell senescence and dysfunction, leading to excessive accumulation of senescent cells [110]. This is the immunosuppressive function of the SASP [141, 142]. An increase in senescent cells and the SASP cytokines can chronically damage the resident tissues of senescent cells and inhibit the function of immune cells [24]. SASP-related secretions include some chemokines that induce bone marrow-derived inhibitory cells (MDSCs) by promoting the proliferation of bone marrow [143]. MDSCs are the main immunosuppressive cells that suppress the immunological response [144, 145].
Studies have shown that Tregs and MDSCs release immunosuppressive factors, including interleukin-10 and TGF-b, which may inhibit the activation of T cells through membrane-bound PD-1 or PD-L1 connections [143, 146]. There is also ample evidence that immunosuppressive cells, such as MDSCs and Tregs, suppress the immunomodulatory ability and cytotoxic activity of NK and CD8 + T cells [147,148,149]. Thereby allowing senescent cells, including immune cells and nonimmune cells, to accumulate in the tissue during senescence. CD8+ CTLs and CD4+ Th1-like cells produce cytotoxic pro-inflammatory cytokines, including IFN-γ, and Th2-like cells produce IL-4 and TGF-β may be responsible for the elimination of many senescent somatic cells [107]. Overall, according to the above studies as indicated that, MDSCs may have an important role in the production of an immunosuppressive environment in chronic inflammatory illness.
The SASP of senescent cells induces an inflammatory state, activating immunosuppression. Immunosuppressive cells inhibit the monitoring and removal of senescent cells, thus forming a feed-forward loop between cell senescence and immune suppression, which not only contributes to the senescence progression itself but also promotes the development of age-associated illness. There is a large amount of evidence that some age-associated illnesses are related to the immunosuppression of MDSCs, such as atherosclerosis, liver steatosis, osteoporosis and type 2 diabetic nephropathy [150]. MDSC-induced immunosuppression weakens the clearance of senescent cells and interferes with energy metabolism and the production of tissue proteins [143]. The cooperation between senescent cells and immunosuppressed MDSCs regulates chronic inflammatory illness and promotes inflammation during senescence [143]. The discovery and study of therapies to remove senescent cells seem to be vital to improve the function of the immune system in elderly individuals. Many approaches are currently found to promote the immune removal of the abundant senescent cell burden in elderly individuals and age-associated diseases.
Effect of the SASP on information transmission between senescent cells
In the vascular system, senescent cells may influence the effect and phenotype of surrounding cells in a variety of ways [151]. Senescent cells can communicate by direct contact between cells [152, 153], and cell fusion [154], by forming cytoplasmic bridges [155], through extracellular vesicle (EV) signal transduction [156] and through the SASP [57]. The SASP state occurs in senescent cells, and may maintain and amplify senescence via autocrine and paracrine regulatory loops. It can not only exacerbate its own SASP, but also induce the SASP in surrounding cells [91].
The SASP enables senescent vascular cells to send messages to surrounding cells and the microenvironment, influencing cell senescence, tissue regeneration, and embryonic development of neighbouring cells [20, 157]. Endothelial cells are connected through gaps, so the signals for inducing senescence can be sent between cells. The release of the SASP factors may promote paracrine senescence. For example, TGF-β family members, VEGF, and chemokines including CCL2 and CL20 may send senescence to normal adjacent cells [53], thus affecting their functions. Through the above mechanisms, senescent cells can lead to endothelial damage, flawed barrier function, elevated pro-inflammatory states, and pathological remodelling of the vessels during senescence [151], and then promote the occurrence of atherosclerosis. In addition, interactions between monocytes and endothelial cells are well-known to play an important role in inflammatory atherosclerosis. There is evidence that TNF-induced endothelial cells can generate more EVs composed of chemotaxis mediators, including ICAM-1, CXCL-10 and CCL-5, which have been taken up by monocytes and can benefit the expression of pro-inflammatory markers such as interleukin-6, and interleukin-8 and their adhesion and movement [158].
The SASP is a method of cellular communication during senescence. The classical SASP consists of soluble cytokines, growth factors and ECM remoulding enzymes. However, the emerging SASP factors and other ways of what we call nonclassical intercellular communication have also been described during senescence [159]. Recently, comprehensive quantitative protein omics characterization of the SASP has revealed additional and various SASP factors that are secreted as soluble factors and exosomes, some of which have been indicated to be abundant in the plasma of humans during senescence and age-related illness. Moreover, exosomes were recently thought to be vital mediators of the paracrine senescence effect of the SASP [32]. There are clear studies that ageing expands the number of exosomes in the blood circulation and tissues, and in particular, the release of immunosuppressed exosomes is increased from senescent cells [160]. When they spread to neighbouring cells, they can enhance immunosuppression within recipient immunocytes, for example, tissue residence and recruitment of immunocytes. This indicates that the increase in immunosuppression with age weakens the removal of senescent cells, and then enhances senescence progression in tissues [160]. Among the markers of ageing, the SASP, especially the EV signal related to the SASP, plays a vital role in the transmission of ageing signals in paracrine or endocrine ways (Fig. 5). EVs, various small vesicles with lipid bilayers, can be secreted from various cells and have been detected in almost all humoral fluids [161]. Their communication between cells and organs suggests their involvement in the transmission of senescence signals in age-related diseases. They have a damaged influence on downstream targets at the immune, inflammatory, gene expression and metabolic levels [61]. In age-related diseases, senescent cells play a vital role in the transmission of the SASP factors to adjacent cells and tissues, with different EVs being representative. CVDs are the most common age-related diseases that manifest as tissue, cell, molecular and functional changes of the heart with age. A large number of circulating premature ageing and anti-ageing mocules from various systems simultaneously coordinate the ageing progression of the cardiovascular system. Moreover, they may lead to vascular ageing, impaired angiogenesis and promote chronic inflammatory response, pathological remodelling, atherosclerosis, and myocardial damage [61].

The SASP is an important mediator of the pathophysiological functions of senescent cells
Intrinsic and extrinsic factors contribute to cellular senescence. Then, senescent cells secrete pro-inflammatory cytokines as part of the SASP, this process leads to the increased inflammation in atherosclerotic plaques. Meanwhile, the SASP enables senescent cells to communicate with other cells and promotes senescence and inflammation via paracrine action, eventually lead to vascular-related diseases
For example, in nonalcoholic fatty liver disease and cardiovascular disease models, EVs can shuttle between the liver and the vasculature, indicating their role in long-distance interorgan communication. MiR-1-containing lipid hepatocyte-derived EVs inhibit KLF4 expression, while activating the NF-kappa B signalling pathway, thus participating in the development of atherosclerosis by promoting endothelial cell inflammation and senescence [162]. Age-related EVs significantly induce vascular calcification and are a hazardous factor for atherosclerosis and myocardial damage. EVs obtained from senescent ECs and plasma of many old patients contain high levels of Annexin, bone morphogenetic protein-2 and Ca2 + , which can promote the progression and proliferation of calcified aortic SMCs in humans. Monocyte-endothelial cell interactions also play an important role in inflammatory atherosclerosis [61]. EV-derived miRNAs from atherogenic macrophages, especially miR-146a, can promote the progression of atherosclerosis by decreasing cell removal and supporting macrophage entrapment in the arterial wall [163].
Treatment of targeted senescent cells
Antiaging interventions should attract more attention because of the limited efficacy in treating atherosclerosis, cancer, and neurodegenerative diseases. The use of senolytics offers an alternative antiaging approach that offers major health benefits. In animal models of atherosclerosis, many antiaging drugs are promising candidates for improving human health because reducing senescent cells can delay ageing and delay the development of chronic diseases [164]. Multiple drugs and molecules play an anti-aging role in atherosclerosis (Table 2). In principle, sensory therapy methods may be extensively divided into two parts: the senolytic strategy (an antiaging strategy that selectively removes senescent cells) and the senomorphic strategy (an antiaging strategy that inhibits the SASP without affecting cell death).
Anti-atherosclerosis by targeting senescent cells
Large numbers of SAβG-positive vascular smooth muscle cells, endothelial cells and macrophages are found in aged arteries and plaques in atherosclerosis compared with surrounding young cells and normal cells, indicating that atherosclerosis connects with premature cell senescence. Cellular senescence promotes many stages of the progression of atherosclerosis. It is a key driver of atherosclerosis formation and maturation, with evidence suggesting that selective removal of these cells by drugs is promising for the treatment of atherosclerosis [17]. Strong preclinical studies indicate that removing p16ink4a expressing senescent cells can significantly extend lifespan in mouse models [151]. Senescent cells can badly influence adjacent cells by altered secretion phenotypes: the SASP. The SASP promotes senescence in normal cells and causes the development and progression of age-associated disorders, including atherosclerosis. Thus, eliminating senescent cells may delay age-related degeneration and prolong life [176].
Senescent cells are a vital source of local, low-grade chronic inflammation and plaque instability. Therefore, it is also a promising upstream treatment target. Targeting senescent cells or the progression of senescence has been explored to improve atherosclerosis, reduce inflammation, and enhance plaque stability [9]. Selective removal of P16INK4A-active senescent cells in transgenic animal models of accelerated ageing (INKATTAC and P16-3MR mice) extended the healthy period [19, 177, 178]. Moreover, the removal of senescent cells in versions of this genetic model or therapy mouse with special senolytics can extend the health span [179, 180], restore vascular reactivity [170], and stabilize atherosclerotic plaques [17]. Senescent vascular cells not only acquire the SASP, but also exhibit the production of ROS. Pharmacological activators of Nrf2, such as resveratrol, can attenuate ROS-induced DNA damage and hope to inhibit ROS-induced senescence in cardiovascular cells [151]. Emerging studies ablating senescent cells using genetic systems or drugs suggest that eliminating senescent cells reduces inflammation within tissues [19, 181]. Many initial studies indicate that senolytic medicine can suppress atherogenesis such as dasatinib, the polyphenols quercetin and fisetin, and the Bcl-2 or Bcl-XL inhibitor navitoclax (ABT263). They can efficiently and selectively remove senescent cells from the vascular system [17, 151].
A study indicates that phosphate binder prevents phosphate-induced cell senescence of VSMCs and vascular calcification in a modified [182], and Sirtuin 1 inhibits hyperphosphatemia-induced calcification of VSMCs [183]. It has been reported that macrophage telomerase reverse transcriptase (TERT) damage leads to a senescent phenotype [184], and that TERT is overexpressed in atherosclerotic plaques. Thus, the induction of TERT expression can inhibit the senescence of macrophages and may play a vital anti-atherosclerosis role. In addition, some of the currently available drugs and compounds, including antioxidants, statins, ACE inhibitors and ARBs, may delay premature ageing and delay atherosclerosis by changing ROS and oxidative DNA damage [5]. Although anti-aging treatments are expected to treat age-associated illnesses including atherosclerosis, many difficulties remain to be solved [119]. Mouse experiments have shown that the removal of senescent cells can delay the onset and development of atherosclerosis and may also delay the development of established plaques [17]. However, whether they are sufficient to reverse the atherosclerotic plaque developed in humans remains to be studied [119].
Anti-atherosclerosis by inhibiting the regulatory pathway of the SASP
Senomorphic treatment inhibits the pro-inflammatory properties of senescent cells and normalizes the process of the SASP in the ageing environment by regulating many biochemical pathways such as m-TOR, p38-MAPK, NF-kappa B, JAK/STAT, ROCK and glucocorticoid receptors [61]. The component of the SASP varies depending on the cell types of the senescent cells, the special promoters that led to senescence and the hormonal milieu. Therefore, hormones and medicines, including rapamycin, metformin, JAK1/JAK2 inhibitors and glucocorticoids may inhibit the SASP [8].
For example, rapamycin may inhibit the SASP from senescent fibroblasts, possibly by inhibiting m-Tor-dependent IL-1α mRNA translation and the JAK/STAT3 pathway [185]. In addition to having anti-inflammatory properties, rapamycin can inhibit the m-TOR pathway [186] to play an antiaging role [187]. Meanwhile, many inhibitors of NF-κB, including BMS-06 and vinpocetine, have been shown to inhibit the progression of atherosclerosis in mice [188]. Emerging experimental evidence indicates that p38 MAPK inhibitors inhibit the SASP, protect cells from senescence and inhibit senescent paracrine signalling in vitro [189]. Statins are effective medicines to delay age-related inflammation in the arterial tube wall and slow the progression of atherosclerosis. It has been shown that statins play a role in delaying endothelial senescence and T cell senescence by p38-mediated SASP inhibition and cell cycle regulation [190]. Specific agents can also directly target the mechanisms of DNA damage and DDR to reduce atherosclerosis. Chloroquine plays a role in slowing atherosclerosis through p53-dependent ATM signalling in animal models [5].
We found that stress-induced DNA damage mainly caused the SASP through the formation of CCFs. Of interest, we also suggest that metformin and rapamycin-induced autophagy activation usefully reduced the number and level of CCFs and inhibited the activation of the cGAS-sting-NF-κB-SASP cascade and cell senescence. These effects of autophagy activation factors explained that autophagic lysosomal function contributed to the removal of CCF and the inhibition of the SASP, further supporting the role of the lysosomal inhibitor buffamycin A1 in blocking autophagy-mediated CCF clearance and ageing inhibition [191]. Moreover, metformin has been reported to improve the inflammaging profile of TH17 cells by enhancing T cell autophagy in vitro and in humans and improving the biological function of mitochondria [192].
Anti-atherosclerosis via extracellular vesicles (EVs)
There is increasing evidence that EVs released by senescent cells have special characteristics and help regulate the phenotype of receptor cells, similar to SASP-like factors. Therefore, EVs released by senescent cells, that is, senescence-associated EVs, appear to be a special SASP factor [193]. In the senescence microenvironment, senescent cells secrete EVs that can transmit senescence signals in an amplified manner. Moreover, they can be cell-free therapeutic tools. Not only did they have no risk of inducing tumours, they also showed a lower immune rejection. EVs are emerging candidates for anti-ageing treatments [61].
Therapeutic EVs come from natural, modified or artificial sources. EVs derived from stem cells contain a variety of substances that can perform antioxidant stress and anti-inflammatory activities and ultimately achieve anti-ageing effects. Emerging evidence suggests that EVs donated by centenarians can have many powerful antioxidants to maintain cell, tissue, and system balance. In fibroblasts of centenarians, EVs include high levels of the enzyme RNAseH2C, which can prevent the DNA damage-induced inflammatory response and decrease the age-related chronic inflammatory response [194]. Senescent endothelial cells can damage the integrity of the vascular endothelium, leading to vascular ageing and a series of disorders, including atherosclerosis. In vitro experiments showed that MSC-sEV could reduce the ageing biomarkers of oxidative stress-induced senescent ECs, reduce the SASP, and rescue angiogenesis, migration and other dysfunction in oxidative stress-induced senescent endothelial cells [195].
EVs take an active part in every stage of the progression of atherosclerosis, and contribute to the formation of thrombi which can lead to a major adverse cardiovascular event. However, certain EVs can have protective biological functions in response to environmental and some stimuli [196]. For example, after treatment with calcium and phosphate, macrophages promote vascular calcification by releasing calcified MVs rich in S100A9 and conjunctin V. In contrast, exosomes from MSCs carrying SIRT6 can target VSMCs and prevent the calcification of blood vessels [196, 197]. EC-derived EVs inhibit the activation of monocytes by delivering miR-10a and targeting the NF-kappa B pathway. It is evidence that injecting this EV into mice can inhibit the activation of monocytes [198].
Conclusion
Various senescent cells can develop the SASP, which can change the cell microenvironment and produce a chronic inflammatory response, causing tissue damage during ageing. Furthermore, the SASP is also a vital factor for the occurrence and development of multiple ageing-related diseases, particularly atherosclerosis. Therefore, the prevention of accelerated cellular senescence and the SASP represents an important therapeutic opportunity, and understanding the mechanisms responsible for this change is essential for the promotion of prevention and therapy of atherosclerosis and other age-associated diseases. In this review, we have highlighted some potential strategies that can mitigate the detrimental effects of senescent cells and the SASP. Although there are many unresolved difficulties and challenges, we suggest that more experiments are needed to study the real nature of the role played by the SASP in cellular senescence and atherosclerosis. In the future, we believe that more studies will be conducted to develop effective treatments and identify novel potential therapeutic targets for ageing-related diseases, especially atherosclerosis, in an ageing population.
Availability of data and materials
Not applicable.
Abbreviations
- SASP:
-
Senescence-associated secretory phenotype
- CVD:
-
Cardiovascular disease
- LDL:
-
Low-density lipoprotein
- SaβG:
-
Senescence-associated beta galactosidase
- VEGF:
-
Vascular endothelial growth factor
- PDGF:
-
Platelet derived growth factor
- MMPs:
-
Metalloproteases
- EV:
-
Extracellular vesicle
- TNF:
-
Tumor necrosis factor
- ECM:
-
Extracellular matrix
- DAMPs:
-
Damage-associated molecular patterns
- M-TOR:
-
Mammalian target of rapamycin
- DDR:
-
DNA damage reaction
- CCFs:
-
Cytoplasmic chromatin fragments
- VSMCs:
-
Vascular smooth muscle cells
- ECs:
-
Endothelial cells
- ICAM-1:
-
Intercellular adhesion molecule-1
- VCAM-1:
-
Vascular cell adhesion molecule-1
- ET-1:
-
Endothelin-1
- ROS:
-
Reactive oxygen species
- HUVEC:
-
Human umbilical vein endothelial cells
- OPG:
-
Osteoprotegerin
- TERF 2:
-
Telomere repeat-binding factor 2
- MDSCs:
-
Marrow-derived inhibitory cells
- TERT:
-
Telomerase reverse transcriptase
- ACS:
-
Acute coronary syndrome
- VDs:
-
Vascular diseases
References
Niccoli T, Partridge L. Ageing as a risk factor for disease. Curr Biol. 2012;22(17):R741–52.
Shakeri H, Lemmens K, Gevaert AB, De Meyer GRY, Segers VFM. Cellular senescence links aging and diabetes in cardiovascular disease. Am J Physiol Heart Circ Physiol. 2018;315(3):H448–62.
Baker DJ, Petersen RC. Cellular senescence in brain aging and neurodegenerative diseases: evidence and perspectives. J Clin Invest. 2018;128(4):1208–16.
Saigusa R, Winkels H, Ley K. T cell subsets and functions in atherosclerosis. Nat Rev Cardiol. 2020;17(7):387–401.
Wang JC, Bennett M. Aging and atherosclerosis: mechanisms, functional consequences, and potential therapeutics for cellular senescence. Circ Res. 2012;111(2):245–59.
Tyrrell DJ, Blin MG, Song J, Wood SC, Zhang M, Beard DA, et al. Age-associated mitochondrial dysfunction accelerates atherogenesis. Circ Res. 2020;126(3):298–314.
Tchkonia T, Zhu Y, van Deursen J, Campisi J, Kirkland JL. Cellular senescence and the senescent secretory phenotype: therapeutic opportunities. J Clin Invest. 2013;123(3):966–72.
Khosla S, Farr JN, Tchkonia T, Kirkland JL. The role of cellular senescence in ageing and endocrine disease. Nat Rev Endocrinol. 2020;16(5):263–75.
Stojanovic SD, Fiedler J, Bauersachs J, Thum T, Sedding DG. Senescence-induced inflammation: an important player and key therapeutic target in atherosclerosis. Eur Heart J. 2020;41(31):2983–96.
Libby P. Targeting inflammatory pathways in cardiovascular disease: the inflammasome, interleukin-1, interleukin-6 and beyond. Cells. 2021;10(4):951.
Rea IM, Gibson DS, McGilligan V, McNerlan SE, Alexander HD, Ross OA. Age and age-related diseases: role of inflammation triggers and cytokines. Front Immunol. 2018;9:586.
Gorenne I, Kavurma M, Scott S, Bennett M. Vascular smooth muscle cell senescence in atherosclerosis. Cardiovasc Res. 2006;72(1):9.
Minamino T, Miyauchi H, Yoshida T, Ishida Y, Yoshida H, Komuro I. Endothelial cell senescence in human atherosclerosis: role of telomere in endothelial dysfunction. Circulation. 2002;105(13):1541–4.
Morgan RG, Ives SJ, Lesniewski LA, Cawthon RM, Andtbacka RHI, Noyes RD, et al. Age-related telomere uncap** is associated with cellular senescence and inflammation independent of telomere shortening in human arteries. Am J Physiol Heart Circ Physiol. 2013;305(2):H251–8.
Matthews C, Gorenne I, Scott S, Figg N, Kirkpatrick P, Ritchie A, et al. Vascular smooth muscle cells undergo telomere-based senescence in human atherosclerosis: effects of telomerase and oxidative stress. Circ Res. 2006;99(2):156–64.
Bennett MR, Clarke MCH. Basic research: killing the old: cell senescence in atherosclerosis. Nat Rev Cardiol. 2016;14(1):8–9.
Childs BG, Baker DJ, Wijshake T, Conover CA, Campisi J, van Deursen JM. Senescent intimal foam cells are deleterious at all stages of atherosclerosis. Science. 2016;354(6311):472–7.
Stojanović SD, Fuchs M, Kunz M, **ao K, Just A, Pich A, et al. Inflammatory drivers of cardiovascular disease: molecular characterization of senescent coronary vascular smooth muscle cells. Front Physiol. 2020;11:520.
Baker DJ, Childs BG, Durik M, Wijers ME, Sieben CJ, Zhong J, et al. Naturally occurring p16(Ink4a)-positive cells shorten healthy lifespan. Nature. 2016;530(7589):184–9.
Song P, Zhao Q, Zou M-H. Targeting senescent cells to attenuate cardiovascular disease progression. Ageing Res Rev. 2020;60: 101072.
Coppé J-P, Patil CK, Rodier F, Sun Y, Muñoz DP, Goldstein J, et al. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol. 2008;6(12):2853–68.
He S, Sharpless NE. Senescence in health and disease. Cell. 2017;169(6):1000–11.
Coppé J-P, Desprez P-Y, Krtolica A, Campisi J. The senescence-associated secretory phenotype: the dark side of tumor suppression. Annu Rev Pathol. 2010;5:99.
Prata LGPL, Ovsyannikova IG, Tchkonia T, Kirkland JL. Senescent cell clearance by the immune system: emerging therapeutic opportunities. Semin Immunol. 2018;40: 101275.
Zhu X, Chen Z, Shen W, Huang G, Sedivy JM, Wang H, et al. Inflammation, epigenetics, and metabolism converge to cell senescence and ageing: the regulation and intervention. Signal Transduct Target Ther. 2021;6(1):245.
Demaria M, Ohtani N, Youssef SA, Rodier F, Toussaint W, Mitchell JR, et al. An essential role for senescent cells in optimal wound healing through secretion of PDGF-AA. Dev Cell. 2014;31(6):722–33.
Childs BG, Li H, van Deursen JM. Senescent cells: a therapeutic target for cardiovascular disease. J Clin Invest. 2018;128(4):1217–28.
Davalos AR, Kawahara M, Malhotra GK, Schaum N, Huang J, Ved U, et al. p53-dependent release of Alarmin HMGB1 is a central mediator of senescent phenotypes. J Cell Biol. 2013;201(4):613–29.
Wiley CD, Liu S, Limbad C, Zawadzka AM, Beck J, Demaria M, et al. SILAC analysis reveals increased secretion of hemostasis-related factors by senescent cells. Cell Rep. 2019;28(13):3329.
Basisty N, Kale A, Jeon OH, Kuehnemann C, Payne T, Rao C, et al. A proteomic atlas of senescence-associated secretomes for aging biomarker development. PLoS Biol. 2020;18(1): e3000599.
Lei Q, Liu T, Gao F, **e H, Sun L, Zhao A, et al. Microvesicles as potential biomarkers for the identification of senescence in human mesenchymal stem cells. Theranostics. 2017;7(10):2673–89.
Di Micco R, Krizhanovsky V, Baker D, d’Adda di Fagagna F. Cellular senescence in ageing: from mechanisms to therapeutic opportunities. Nat Rev Mol Cell Biol. 2021;22(2):75–95.
Salminen A, Kauppinen A, Kaarniranta K. Emerging role of NF-κB signaling in the induction of senescence-associated secretory phenotype (SASP). Cell Signal. 2012;24(4):835–45.
Kuilman T, Michaloglou C, Vredeveld LCW, Douma S, van Doorn R, Desmet CJ, et al. Oncogene-induced senescence relayed by an interleukin-dependent inflammatory network. Cell. 2008;133(6):1019–31.
Orjalo AV, Bhaumik D, Gengler BK, Scott GK, Campisi J. Cell surface-bound IL-1alpha is an upstream regulator of the senescence-associated IL-6/IL-8 cytokine network. Proc Natl Acad Sci USA. 2009;106(40):17031–6.
Acosta JC, O’Loghlen A, Banito A, Guijarro MV, Augert A, Raguz S, et al. Chemokine signaling via the CXCR2 receptor reinforces senescence. Cell. 2008;133(6):1006–18.
Sun Y, Coppé J-P, Lam EWF. Cellular senescence: the sought or the unwanted? Trends Mol Med. 2018;24(10):871–85.
Chien Y, Scuoppo C, Wang X, Fang X, Balgley B, Bolden JE, et al. Control of the senescence-associated secretory phenotype by NF-κB promotes senescence and enhances chemosensitivity. Genes Dev. 2011;25(20):2125–36.
Freund A, Patil CK, Campisi J. p38MAPK is a novel DNA damage response-independent regulator of the senescence-associated secretory phenotype. EMBO J. 2011;30(8):1536–48.
Kang C, Xu Q, Martin TD, Li MZ, Demaria M, Aron L, et al. The DNA damage response induces inflammation and senescence by inhibiting autophagy of GATA4. Science. 2015;349(6255):aaa5612.
Herranz N, Gallage S, Mellone M, Wuestefeld T, Klotz S, Hanley CJ, et al. mTOR regulates MAPKAPK2 translation to control the senescence-associated secretory phenotype. Nat Cell Biol. 2015;17(9):1205–17.
Laberge R-M, Sun Y, Orjalo AV, Patil CK, Freund A, Zhou L, et al. MTOR regulates the pro-tumorigenic senescence-associated secretory phenotype by promoting IL1A translation. Nat Cell Biol. 2015;17(8):1049–61.
Rodier F, Campisi J. Four faces of cellular senescence. J Cell Biol. 2011;192(4):547–56.
Kuilman T, Michaloglou C, Mooi WJ, Peeper DS. The essence of senescence. Genes Dev. 2010;24(22):2463–79.
Tyrrell DJ, Goldstein DR. Ageing and atherosclerosis: vascular intrinsic and extrinsic factors and potential role of IL-6. Nat Rev Cardiol. 2021;18(1):58–68.
Rovillain E, Mansfield L, Caetano C, Alvarez-Fernandez M, Caballero OL, Medema RH, et al. Activation of nuclear factor-kappa B signalling promotes cellular senescence. Oncogene. 2011;30(20):2356–66.
Huggins CJ, Malik R, Lee S, Salotti J, Thomas S, Martin N, et al. C/EBPγ suppresses senescence and inflammatory gene expression by heterodimerizing with C/EBPβ. Mol Cell Biol. 2013;33(16):3242–58.
Herranz N, Gil J. Mechanisms and functions of cellular senescence. J Clin Invest. 2018;128(4):1238–46.
Lopes-Paciencia S, Saint-Germain E, Rowell M-C, Ruiz AF, Kalegari P, Ferbeyre G. The senescence-associated secretory phenotype and its regulation. Cytokine. 2019;117:15–22.
Tilstra JS, Robinson AR, Wang J, Gregg SQ, Clauson CL, Reay DP, et al. NF-κB inhibition delays DNA damage-induced senescence and aging in mice. J Clin Invest. 2012;122(7):2601–12.
Song S, Lam EWF, Tchkonia T, Kirkland JL, Sun Y. Senescent cells: emerging targets for human aging and age-related diseases. Trends Biochem Sci. 2020;45(7):578–92.
Pantsulaia I, Ciszewski WM, Niewiarowska J. Senescent endothelial cells: potential modulators of immunosenescence and ageing. Ageing Res Rev. 2016;29:13–25.
Acosta JC, Banito A, Wuestefeld T, Georgilis A, Janich P, Morton JP, et al. A complex secretory program orchestrated by the inflammasome controls paracrine senescence. Nat Cell Biol. 2013;15(8):978–90.
Gareus R, Kotsaki E, Xanthoulea S, van der Made I, Gijbels MJJ, Kardakaris R, et al. Endothelial cell-specific NF-kappaB inhibition protects mice from atherosclerosis. Cell Metab. 2008;8(5):372–83.
Song D, Fang G, Mao S-Z, Ye X, Liu G, Miller EJ, et al. Selective inhibition of endothelial NF-κB signaling attenuates chronic intermittent hypoxia-induced atherosclerosis in mice. Atherosclerosis. 2018;270:68–75.
Niu N, Xu S, Xu Y, Little PJ, ** Z-G. Targeting mechanosensitive transcription factors in atherosclerosis. Trends Pharmacol Sci. 2019;40(4):253–66.
Birch J, Gil J. Senescence and the SASP: many therapeutic avenues. Genes Dev. 2020;34(23–24):1565–76.
Rodier F, Coppé J-P, Patil CK, Hoeijmakers WAM, Muñoz DP, Raza SR, et al. Persistent DNA damage signalling triggers senescence-associated inflammatory cytokine secretion. Nat Cell Biol. 2009;11(8):973–9.
Mahmoudi M, Gorenne I, Mercer J, Figg N, Littlewood T, Bennett M. Statins use a novel Nijmegen breakage syndrome-1-dependent pathway to accelerate DNA repair in vascular smooth muscle cells. Circ Res. 2008;103(7):717–25.
Strzeszewska A, Alster O, Mosieniak G, Ciolko A, Sikora E. Insight into the role of PIKK family members and NF-кB in DNAdamage-induced senescence and senescence-associated secretory phenotype of colon cancer cells. Cell Death Dis. 2018;9(2):44.
Yin Y, Chen H, Wang Y, Zhang L, Wang X. Roles of extracellular vesicles in the aging microenvironment and age-related diseases. J Extracell Vesicles. 2021;10(12): e12154.
Tedgui A, Mallat Z. Cytokines in atherosclerosis: pathogenic and regulatory pathways. Physiol Rev. 2006;86(2):515–81.
Benigni A, Cassis P, Remuzzi G. Angiotensin II revisited: new roles in inflammation, immunology and aging. EMBO Mol Med. 2010;2(7):247–57.
Cheng ZJ, Vapaatalo H, Mervaala E. Angiotensin II and vascular inflammation. Med Sci Monit. 2005;11(6):RA194–205.
Salazar G. NADPH oxidases and mitochondria in vascular senescence. Int J Mol Sci. 2018;19(5):1327.
Martínez de Toda I, Vida C, Sanz San Miguel L, De la Fuente M. Function, oxidative, and inflammatory stress parameters in immune cells as predictive markers of lifespan throughout aging. Oxid Med Cell Longev. 2019;2019:4574276.
Lanna A, Henson SM, Escors D, Akbar AN. The kinase p38 activated by the metabolic regulator AMPK and scaffold TAB1 drives the senescence of human T cells. Nat Immunol. 2014;15(10):965–72.
Salminen A. Immunosuppressive network promotes immunosenescence associated with aging and chronic inflammatory conditions. J Mol Med (Berl). 2021;99(11):1553–69.
Raught B, Gingras AC, Sonenberg N. The target of rapamycin (TOR) proteins. Proc Natl Acad Sci USA. 2001;98(13):7037–44.
Wullschleger S, Loewith R, Hall MN. TOR signaling in growth and metabolism. Cell. 2006;124(3):471–84.
Schmelzle T, Hall MN. TOR, a central controller of cell growth. Cell. 2000;103(2):253–62.
Noda T, Ohsumi Y. Tor, a phosphatidylinositol kinase homologue, controls autophagy in yeast. J Biol Chem. 1998;273(7):3963–6.
Carroll B, Nelson G, Rabanal-Ruiz Y, Kucheryavenko O, Dunhill-Turner NA, Chesterman CC, et al. Persistent mTORC1 signaling in cell senescence results from defects in amino acid and growth factor sensing. J Cell Biol. 2017;216(7):1949–57.
Dominick G, Bowman J, Li X, Miller RA, Garcia GG. mTOR regulates the expression of DNA damage response enzymes in long-lived Snell dwarf, GHRKO, and PAPPA-KO mice. Aging Cell. 2017;16(1):52–60.
Correia-Melo C, Marques FDM, Anderson R, Hewitt G, Hewitt R, Cole J, et al. Mitochondria are required for pro-ageing features of the senescent phenotype. EMBO J. 2016;35(7):724–42.
Evangelisti C, Cenni V, Lattanzi G. Potential therapeutic effects of the MTOR inhibitors for preventing ageing and progeria-related disorders. Br J Clin Pharmacol. 2016;82(5):1229–44.
Park JH, Lee NK, Lim HJ, Ji ST, Kim Y-J, Jang WB, et al. Pharmacological inhibition of mTOR attenuates replicative cell senescence and improves cellular function via regulating the STAT3-PIM1 axis in human cardiac progenitor cells. Exp Mol Med. 2020;52(4):615–28.
Walters HE, Deneka-Hannemann S, Cox LS. Reversal of phenotypes of cellular senescence by pan-mTOR inhibition. Aging (Albany NY). 2016;8(2):231–44.
Machado-Oliveira G, Ramos C, Marques ARA, Vieira OV. Cell senescence, multiple organelle dysfunction and atherosclerosis. Cells. 2020;9(10):2146.
Vizioli MG, Liu T, Miller KN, Robertson NA, Gilroy K, Lagnado AB, et al. Mitochondria-to-nucleus retrograde signaling drives formation of cytoplasmic chromatin and inflammation in senescence. Genes Dev. 2020;34(5–6):428–45.
Loo TM, Miyata K, Tanaka Y, Takahashi A. Cellular senescence and senescence-associated secretory phenotype via the cGAS-STING signaling pathway in cancer. Cancer Sci. 2020;111(2):304–11.
Glück S, Guey B, Gulen MF, Wolter K, Kang T-W, Schmacke NA, et al. Innate immune sensing of cytosolic chromatin fragments through cGAS promotes senescence. Nat Cell Biol. 2017;19(9):1061–70.
Yang H, Wang H, Ren J, Chen Q, Chen ZJ. cGAS is essential for cellular senescence. Proc Natl Acad Sci USA. 2017;114(23):E4612–20.
Dou Z, Ghosh K, Vizioli MG, Zhu J, Sen P, Wangensteen KJ, et al. Cytoplasmic chromatin triggers inflammation in senescence and cancer. Nature. 2017;550(7676):402–6.
McHugh D, Gil J. Senescence and aging: causes, consequences, and therapeutic avenues. J Cell Biol. 2018;217(1):65–77.
López-Otín C, Blasco MA, Partridge L, Serrano M, Kroemer G. The hallmarks of aging. Cell. 2013;153(6):1194–217.
Franceschi C, Campisi J. Chronic inflammation (inflammaging) and its potential contribution to age-associated diseases. J Gerontol A Biol Sci Med Sci. 2014;69(Suppl 1):S4–9.
Libby P. Inflammation in atherosclerosis. Nature. 2002;420(6917):868–74.
Ghamar Talepoor A, Khosropanah S, Doroudchi M. Partial recovery of senescence in circulating follicular helper T cells after Dasatinib treatment. Int Immunopharmacol. 2021;94: 107465.
Ridker PM, Everett BM, Thuren T, MacFadyen JG, Chang WH, Ballantyne C, et al. Antiinflammatory therapy with canakinumab for atherosclerotic disease. N Engl J Med. 2017;377(12):1119–31.
Liberale L, Montecucco F, Tardif JC, Libby P, Camici GG. Inflamm-ageing: the role of inflammation in age-dependent cardiovascular disease. Eur Heart J. 2020;41(31):2974–82.
Ferrucci L, Fabbri E. Inflammageing: chronic inflammation in ageing, cardiovascular disease, and frailty. Nat Rev Cardiol. 2018;15(9):505–22.
Ovadya Y, Landsberger T, Leins H, Vadai E, Gal H, Biran A, et al. Impaired immune surveillance accelerates accumulation of senescent cells and aging. Nat Commun. 2018;9(1):5435.
Uyar B, Palmer D, Kowald A, Murua Escobar H, Barrantes I, Möller S, et al. Single-cell analyses of aging, inflammation and senescence. Ageing Res Rev. 2020;64: 101156.
Banerjee P, Kotla S, Reddy Velatooru L, Abe RJ, Davis EA, Cooke JP, et al. Senescence-associated secretory phenotype as a hinge between cardiovascular diseases and cancer. Front Cardiovasc Med. 2021;8: 763930.
Weber C, Noels H. Atherosclerosis: current pathogenesis and therapeutic options. Nat Med. 2011;17(11):1410–22.
Donato AJ, Gano LB, Eskurza I, Silver AE, Gates PE, Jablonski K, et al. Vascular endothelial dysfunction with aging: endothelin-1 and endothelial nitric oxide synthase. Am J Physiol Heart Circ Physiol. 2009;297(1):H425–32.
McDonald AP, Meier TR, Hawley AE, Thibert JN, Farris DM, Wrobleski SK, et al. Aging is associated with impaired thrombus resolution in a mouse model of stasis induced thrombosis. Thromb Res. 2010;125(1):72–8.
Camaré C, Pucelle M, Nègre-Salvayre A, Salvayre R. Angiogenesis in the atherosclerotic plaque. Redox Biol. 2017;12:18–34.
Ruscetti M, Morris JP, Mezzadra R, Russell J, Leibold J, Romesser PB, et al. Senescence-induced vascular remodeling creates therapeutic vulnerabilities in pancreas cancer. Cell. 2020;181(2):424.
Gardner SE, Humphry M, Bennett MR, Clarke MCH. Senescent vascular smooth muscle cells drive inflammation through an interleukin-1α-dependent senescence-associated secretory phenotype. Arterioscler Thromb Vasc Biol. 2015;35(9):1963–74.
Chi C, Li D-J, Jiang Y-J, Tong J, Fu H, Wu Y-H, et al. Vascular smooth muscle cell senescence and age-related diseases: state of the art. Biochim Biophys Acta Mol Basis Dis. 2019;1865(7):1810–21.
Stojanović SD, Fiedler J, Bauersachs J, Thum T, Sedding DG. Senescence-induced inflammation: an important player and key therapeutic target in atherosclerosis. Eur Heart J. 2020;41(31):2983–96.
Wang M, Kim SH, Monticone RE, Lakatta EG. Matrix metalloproteinases promote arterial remodeling in aging, hypertension, and atherosclerosis. Hypertension. 2015;65(4):698–703.
Van Campenhout A, Golledge J. Osteoprotegerin, vascular calcification and atherosclerosis. Atherosclerosis. 2009;204(2):321–9.
Mittelbrunn M, Kroemer G. Hallmarks of T cell aging. Nat Immunol. 2021;22(6):687–98.
Thomas R, Wang W, Su D-M. Contributions of age-related thymic involution to immunosenescence and inflammaging. Immun Ageing. 2020;17:2.
Nie L, Zhang P, Wang Q, Zhou X, Wang Q. lncRNA-triggered macrophage inflammaging deteriorates age-related diseases. Mediators Inflamm. 2019;2019:4260309.
Raucci A, Macrì F, Castiglione S, Badi I, Vinci MC, Zuccolo E. MicroRNA-34a: the bad guy in age-related vascular diseases. Cell Mol Life Sci. 2021. https://doi.org/10.1007/s00018-021-03979-4.
Song P, An J, Zou M-H. Immune clearance of senescent cells to combat ageing and chronic diseases. Cells. 2020;9(3):671.
Kim Y-M, Youn S-W, Sudhahar V, Das A, Chandhri R, Cuervo Grajal H, et al. Redox regulation of mitochondrial fission protein Drp1 by protein disulfide isomerase limits endothelial senescence. Cell Rep. 2018;23(12):3565–78.
Bochenek ML, Schütz E, Schäfer K. Endothelial cell senescence and thrombosis: ageing clots. Thromb Res. 2016;147:36–45.
Regina C, Panatta E, Candi E, Melino G, Amelio I, Balistreri CR, et al. Vascular ageing and endothelial cell senescence: molecular mechanisms of physiology and diseases. Mech Ageing Dev. 2016;159:14–21.
Ungvari Z, Tarantini S, Donato AJ, Galvan V, Csiszar A. Mechanisms of vascular aging. Circ Res. 2018;123(7):849–67.
Korybalska K, Kawka E, Kusch A, Aregger F, Dragun D, Jörres A, et al. Recovery of senescent endothelial cells from injury. J Gerontol A Biol Sci Med Sci. 2013;68(3):250–7.
Kotla S, Le NT, Vu HT, Ko KA, Gi YJ, Thomas TN, et al. Endothelial senescence-associated secretory phenotype (SASP) is regulated by Makorin-1 ubiquitin E3 ligase. Metabolism. 2019;100: 153962.
Mistriotis P, Andreadis ST. Vascular aging: molecular mechanisms and potential treatments for vascular rejuvenation. Ageing Res Rev. 2017;37:94–116.
Bennett MR, Sinha S, Owens GK. Vascular smooth muscle cells in atherosclerosis. Circ Res. 2016;118(4):692–702.
Grootaert MOJ, Moulis M, Roth L, Martinet W, Vindis C, Bennett MR, et al. Vascular smooth muscle cell death, autophagy and senescence in atherosclerosis. Cardiovasc Res. 2018;114(4):622–34.
Spyridopoulos I, Hoffmann J, Aicher A, Brümmendorf TH, Doerr HW, Zeiher AM, et al. Accelerated telomere shortening in leukocyte subpopulations of patients with coronary heart disease: role of cytomegalovirus seropositivity. Circulation. 2009;120(14):1364–72.
Barbe-Tuana F, Funchal G, Schmitz CRR, Maurmann RM, Bauer ME. The interplay between immunosenescence and age-related diseases. Semin Immunopathol. 2020;42(5):545–57.
Spyridopoulos I, Martin-Ruiz C, Hilkens C, Yadegarfar ME, Isaacs J, Jagger C, et al. CMV seropositivity and T-cell senescence predict increased cardiovascular mortality in octogenarians: results from the Newcastle 85+ study. Aging Cell. 2016;15(2):389–92.
Callender LA, Carroll EC, Beal RWJ, Chambers ES, Nourshargh S, Akbar AN, et al. Human CD8 EMRA T cells display a senescence-associated secretory phenotype regulated by p38 MAPK. Aging Cell. 2018;17(1): e12675.
Grahame-Clarke C. Human cytomegalovirus, endothelial function and atherosclerosis. Herpes. 2005;12(2):42–5.
Tae YuH, Youn J-C, Lee J, Park S, Chi H-S, Lee J, et al. Characterization of CD8(+)CD57(+) T cells in patients with acute myocardial infarction. Cell Mol Immunol. 2015;12(4):466–73.
Rodriguez IJ, Lalinde Ruiz N, Llano León M, Martínez Enríquez L, Montilla Velásquez MDP, Ortiz Aguirre JP, et al. Immunosenescence study of T cells: a systematic review. Front Immunol. 2020;11: 604591.
Garrido A, Cruces J, Ceprián N, Vara E, de la Fuente M. Oxidative-inflammatory stress in immune cells from adult mice with premature aging. Int J Mol Sci. 2019;20(3):769.
Frasca D, Diaz A, Romero M, Blomberg BB. Human peripheral late/exhausted memory B cells express a senescent-associated secretory phenotype and preferentially utilize metabolic signaling pathways. Exp Gerontol. 2017;87(Pt A):113–20.
Wang H, Fu H, Zhu R, Wu X, Ji X, Li X, et al. BRD4 contributes to LPS-induced macrophage senescence and promotes progression of atherosclerosis-associated lipid uptake. Aging (Albany NY). 2020;12(10):9240–59.
Merino A, Buendia P, Martin-Malo A, Aljama P, Ramirez R, Carracedo J. Senescent CD14+CD16+ monocytes exhibit proinflammatory and proatherosclerotic activity. J Immunol. 2011;186(3):1809–15.
Senovilla L, Galluzzi L, Zitvogel L, Kroemer G. Immunosurveillance as a regulator of tissue homeostasis. Trends Immunol. 2013;34(10):471–81.
Akbar AN. The convergence of senescence and nutrient sensing during lymphocyte ageing. Clin Exp Immunol. 2017;187(1):4–5.
Henson SM, Lanna A, Riddell NE, Franzese O, Macaulay R, Griffiths SJ, et al. p38 signaling inhibits mTORC1-independent autophagy in senescent human CD8+ T cells. J Clin Invest. 2014;124(9):4004–16.
McElhaney JE, Effros RB. Immunosenescence: what does it mean to health outcomes in older adults? Curr Opin Immunol. 2009;21(4):418–24.
Plowden J, Renshaw-Hoelscher M, Engleman C, Katz J, Sambhara S. Innate immunity in aging: impact on macrophage function. Aging Cell. 2004;3(4):161–7.
Varricchi G, Bencivenga L, Poto R, Pecoraro A, Shamji MH, Rengo G. The emerging role of T follicular helper (TFH) cells in aging: influence on the immune frailty. Ageing Res Rev. 2020;61: 101071.
Mondal AM, Horikawa I, Pine SR, Fujita K, Morgan KM, Vera E, et al. p53 isoforms regulate aging- and tumor-associated replicative senescence in T lymphocytes. J Clin Invest. 2013;123(12):5247–57.
Del Pinto R, Ferri C. Inflammation-accelerated senescence and the cardiovascular system: mechanisms and perspectives. Int J Mol Sci. 2018;19(12):3701.
Chambers ES, Akbar AN. Can blocking inflammation enhance immunity during aging? J Allergy Clin Immunol. 2020;145(5):1323–31.
Wong YT, Gruber J, Jenner AM, Tay FEH, Ruan R. Chronic resveratrol intake reverses pro-inflammatory cytokine profile and oxidative DNA damage in ageing hybrid mice. Age (Dordr). 2011;33(3):229–46.
Di Mitri D, Toso A, Chen JJ, Sarti M, Pinton S, Jost TR, et al. Tumour-infiltrating Gr-1+ myeloid cells antagonize senescence in cancer. Nature. 2014;515(7525):134–7.
Eggert T, Wolter K, Ji J, Ma C, Yevsa T, Klotz S, et al. Distinct functions of senescence-associated immune responses in liver tumor surveillance and tumor progression. Cancer Cell. 2016;30(4):533–47.
Salminen A, Kauppinen A, Kaarniranta K. Myeloid-derived suppressor cells (MDSC): an important partner in cellular/tissue senescence. Biogerontology. 2018;19(5):325–39.
Kwak Y, Kim H-E, Park SG. Insights into myeloid-derived suppressor cells in inflammatory diseases. arch immunol ther exp (warsz). 2015;63(4):269–85.
Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol. 2009;9(3):162–74.
Lu C, Redd PS, Lee JR, Savage N, Liu K. The expression profiles and regulation of PD-L1 in tumor-induced myeloid-derived suppressor cells. Oncoimmunology. 2016;5(12): e1247135.
Trzonkowski P, Szmit E, Myśliwska J, Myśliwski A. CD4+CD25+ T regulatory cells inhibit cytotoxic activity of CTL and NK cells in humans-impact of immunosenescence. Clin Immunol. 2006;119(3):307–16.
Hoechst B, Voigtlaender T, Ormandy L, Gamrekelashvili J, Zhao F, Wedemeyer H, et al. Myeloid derived suppressor cells inhibit natural killer cells in patients with hepatocellular carcinoma via the NKp30 receptor. Hepatology. 2009;50(3):799–807.
Ralainirina N, Poli A, Michel T, Poos L, Andrès E, Hentges F, et al. Control of NK cell functions by CD4 + CD25 + regulatory T cells. J Leukoc Biol. 2007;81(1):144–53.
Salminen A. Feed-forward regulation between cellular senescence and immunosuppression promotes the aging process and age-related diseases. Ageing Res Rev. 2021;67: 101280.
Ungvari Z, Tarantini S, Sorond F, Merkely B, Csiszar A. Mechanisms of vascular aging, a geroscience perspective: JACC focus seminar. J Am Coll Cardiol. 2020;75(8):931–41.
Nelson G, Wordsworth J, Wang C, Jurk D, Lawless C, Martin-Ruiz C, et al. A senescent cell bystander effect: senescence-induced senescence. Aging Cell. 2012;11(2):345–9.
Hoare M, Ito Y, Kang T-W, Weekes MP, Matheson NJ, Patten DA, et al. NOTCH1 mediates a switch between two distinct secretomes during senescence. Nat Cell Biol. 2016;18(9):979–92.
Chuprin A, Gal H, Biron-Shental T, Biran A, Amiel A, Rozenblatt S, et al. Cell fusion induced by ERVWE1 or measles virus causes cellular senescence. Genes Dev. 2013;27(21):2356–66.
Biran A, Zada L, Abou Karam P, Vadai E, Roitman L, Ovadya Y, et al. Quantitative identification of senescent cells in aging and disease. Aging Cell. 2017;16(4):661–71.
Takasugi M, Okada R, Takahashi A, Virya Chen D, Watanabe S, Hara E. Small extracellular vesicles secreted from senescent cells promote cancer cell proliferation through EphA2. Nat Commun. 2017;8:15729.
Muñoz-Espín D, Cañamero M, Maraver A, Gómez-López G, Contreras J, Murillo-Cuesta S, et al. Programmed cell senescence during mammalian embryonic development. Cell. 2013;155(5):1104–18.
Hosseinkhani B, Kuypers S, van den Akker NMS, Molin DGM, Michiels L. Extracellular vesicles work as a functional inflammatory mediator between vascular endothelial cells and immune cells. Front Immunol. 2018;9:1789.
Fafian-Labora JA, O’Loghlen A. Classical and nonclassical intercellular communication in senescence and ageing. Trends Cell Biol. 2020;30(8):628–39.
Salminen A, Kaarniranta K, Kauppinen A. Exosomal vesicles enhance immunosuppression in chronic inflammation: impact in cellular senescence and the aging process. Cell Signal. 2020;75: 109771.
Saugstad JA, Lusardi TA, Van Keuren-Jensen KR, Phillips JI, Lind B, Harrington CA, et al. Analysis of extracellular RNA in cerebrospinal fluid. J Extracell vesicles. 2017;6(1):1317577.
Jiang F, Chen Q, Wang W, Ling Y, Yan Y, **a P. Hepatocyte-derived extracellular vesicles promote endothelial inflammation and atherogenesis via microRNA-1. J Hepatol. 2020;72(1):156–66.
Nguyen M-A, Karunakaran D, Geoffrion M, Cheng HS, Tandoc K, Perisic Matic L, et al. Extracellular vesicles secreted by atherogenic macrophages transfer microRNA to inhibit cell migration. Arterioscler Thromb Vasc Biol. 2018;38(1):49–63.
Martel J, Ojcius DM, Wu C-Y, Peng H-H, Voisin L, Perfettini J-L, et al. Emerging use of senolytics and senomorphics against aging and chronic diseases. Med Res Rev. 2020;40(6):2114–31.
Cao H, Jia Q, Yan L, Chen C, **ng S, Shen D. Quercetin suppresses the progression of atherosclerosis by regulating MST1-mediated autophagy in ox-LDL-induced RAW264.7 macrophage foam cells. Int J Mol Sci. 2019;20(23):6093.
Feng X, Chen W, Ni X, Little PJ, Xu S, Tang L, et al. Metformin, macrophage dysfunction and atherosclerosis. Front Immunol. 2021;12: 682853.
Schupp N, Schmid U, Heidland A, Stopper H. Rosuvastatin protects against oxidative stress and DNA damage in vitro via upregulation of glutathione synthesis. Atherosclerosis. 2008;199(2):278–87.
Oeseburg H, Iusuf D, van der Harst P, van Gilst WH, Henning RH, Roks AJM. Bradykinin protects against oxidative stress-induced endothelial cell senescence. Hypertension. 2009;53(2):417–22.
Yan L, Jia Q, Cao H, Chen C, **ng S, Huang Y, et al. Fisetin ameliorates atherosclerosis by regulating PCSK9 and LOX-1 in apoE mice. Exp Ther Med. 2021;21(1):25.
Roos CM, Zhang B, Palmer AK, Ogrodnik MB, Pirtskhalava T, Thalji NM, et al. Chronic senolytic treatment alleviates established vasomotor dysfunction in aged or atherosclerotic mice. Aging Cell. 2016;15(5):973–7.
Das A, Huang GX, Bonkowski MS, Longchamp A, Li C, Schultz MB, et al. Impairment of an endothelial NAD-HS signaling network is a reversible cause of vascular aging. Cell. 2018;173(1):74.
Xue L, Zhu W, Yang F, Dai S, Han Z, Xu F, et al. Appropriate dose of ethanol exerts anti-senescence and anti-atherosclerosis protective effects by activating ALDH2. Biochem Biophys Res Commun. 2019;512(2):319–25.
Grootaert MOJ, Finigan A, Figg NL, Uryga AK, Bennett MR. SIRT6 protects smooth muscle cells from senescence and reduces atherosclerosis. Circ Res. 2021;128(4):474–91.
Wang J, Uryga AK, Reinhold J, Figg N, Baker L, Finigan A, et al. Vascular smooth muscle cell senescence promotes atherosclerosis and features of plaque vulnerability. Circulation. 2015;132(20):1909–19.
Lv L, Ye M, Duan R, Yuan K, Chen J, Liang W, et al. Downregulation of Pin1 in human atherosclerosis and its association with vascular smooth muscle cell senescence. J Vasc Surg. 2018;68(3):873.
Matjusaitis M, Chin G, Sarnoski EA, Stolzing A. Biomarkers to identify and isolate senescent cells. Ageing Res Rev. 2016;29:12.
Baker DJ, Wijshake T, Tchkonia T, LeBrasseur NK, Childs BG, van de Sluis B, et al. Clearance of p16Ink4a-positive senescent cells delays ageing-associated disorders. Nature. 2011;479(7372):232–6.
Kirkland JL, Tchkonia T. Cellular senescence: a translational perspective. EBioMedicine. 2017;21:21–8.
Zhu Y, Tchkonia T, Pirtskhalava T, Gower AC, Ding H, Giorgadze N, et al. The Achilles’ heel of senescent cells: from transcriptome to senolytic drugs. Aging Cell. 2015;14(4):644–58.
Fuhrmann-Stroissnigg H, Ling YY, Zhao J, McGowan SJ, Zhu Y, Brooks RW, et al. Identification of HSP90 inhibitors as a novel class of senolytics. Nat Commun. 2017;8(1):422.
Jeon OH, Kim C, Laberge R-M, Demaria M, Rathod S, Vasserot AP, et al. Local clearance of senescent cells attenuates the development of post-traumatic osteoarthritis and creates a pro-regenerative environment. Nat Med. 2017;23(6):775–81.
Yamada S, Tatsumoto N, Tokumoto M, Noguchi H, Ooboshi H, Kitazono T, et al. Phosphate binders prevent phosphate-induced cellular senescence of vascular smooth muscle cells and vascular calcification in a modified, adenine-based uremic rat model. Calcif Tissue Int. 2015;96(4):347–58.
Takemura A, Iijima K, Ota H, Son B-K, Ito Y, Ogawa S, et al. Sirtuin 1 retards hyperphosphatemia-induced calcification of vascular smooth muscle cells. Arterioscler Thromb Vasc Biol. 2011;31(9):2054–62.
Liu S-C, Wang S-S, Wu M-Z, Wu D-C, Yu F-J, Chen W-J, et al. Activation of telomerase and expression of human telomerase reverse transcriptase in coronary atherosclerosis. Cardiovasc Pathol. 2005;14(5):232–40.
Wang R, Yu Z, Sunchu B, Shoaf J, Dang I, Zhao S, et al. Rapamycin inhibits the secretory phenotype of senescent cells by a Nrf2-independent mechanism. Aging Cell. 2017;16(3):564–74.
Tan P, Wang Y-J, Li S, Wang Y, He J-Y, Chen Y-Y, et al. The PI3K/Akt/mTOR pathway regulates the replicative senescence of human VSMCs. Mol Cell Biochem. 2016;422(1–2):1.
Sung JY, Lee KY, Kim J-R, Choi HC. Interaction between mTOR pathway inhibition and autophagy induction attenuates adriamycin-induced vascular smooth muscle cell senescence through decreased expressions of p53/p21/p16. Exp Gerontol. 2018;109:51–8.
Pamukcu B, Lip GYH, Shantsila E. The nuclear factor–kappa B pathway in atherosclerosis: a potential therapeutic target for atherothrombotic vascular disease. Thromb Res. 2011;128(2):117–23.
Hongo A, Okumura N, Nakahara M, Kay EP, Koizumi N. The effect of a p38 mitogen-activated protein kinase inhibitor on cellular senescence of cultivated human corneal endothelial cells. Invest Ophthalmol Vis Sci. 2017;58(9):3325–34.
Bennaceur K, Atwill M, Al Zhrany N, Hoffmann J, Keavney B, Breault D, et al. Atorvastatin induces T cell proliferation by a telomerase reverse transcriptase (TERT) mediated mechanism. Atherosclerosis. 2014;236(2):312–20.
Han X, Chen H, Gong H, Tang X, Huang N, Xu W, et al. Autolysosomal degradation of cytosolic chromatin fragments antagonizes oxidative stress-induced senescence. J Biol Chem. 2020;295(14):4451–63.
Bharath LP, Agrawal M, McCambridge G, Nicholas DA, Hasturk H, Liu J, et al. Metformin enhances autophagy and normalizes mitochondrial function to alleviate aging-associated inflammation. Cell Metab. 2020;32(1):44.
Kadota T, Fujita Y, Yoshioka Y, Araya J, Kuwano K, Ochiya T. Emerging role of extracellular vesicles as a senescence-associated secretory phenotype: insights into the pathophysiology of lung diseases. Mol Aspects Med. 2018. https://doi.org/10.1016/j.mam.2017.11.005.
Storci G, De Carolis S, Papi A, Bacalini MG, Gensous N, Marasco E, et al. Genomic stability, anti-inflammatory phenotype, and up-regulation of the RNAseH2 in cells from centenarians. Cell Death Differ. 2019;26(9):1845–58.
**ao X, Xu M, Yu H, Wang L, Li X, Rak J, et al. Mesenchymal stem cell-derived small extracellular vesicles mitigate oxidative stress-induced senescence in endothelial cells via regulation of miR-146a/Src. Signal Transduct Target Ther. 2021;6(1):354.
Giró O, Jiménez A, Pané A, Badimon L, Ortega E, Chiva-Blanch G. Extracellular vesicles in atherothrombosis and cardiovascular disease: friends and foes. Atherosclerosis. 2021;330:61–75.
Wei W, Guo X, Gu L, Jia J, Yang M, Yuan W, et al. Bone marrow mesenchymal stem cell exosomes suppress phosphate-induced aortic calcification via SIRT6-HMGB1 deacetylation. Stem Cell Res Ther. 2021;12(1):235.
Njock M-S, Cheng HS, Dang LT, Nazari-Jahantigh M, Lau AC, Boudreau E, et al. Endothelial cells suppress monocyte activation through secretion of extracellular vesicles containing antiinflammatory microRNAs. Blood. 2015;125(20):3202–12.
Acknowledgements
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as potential conflicts of interest.
Funding
This work was supported by grants from the National Natural Science Foundation of China (Grant Nos.82171299).
Author information
Authors and Affiliations
Contributions
All authors contributed to the study conception and design. Material preparation and analysis were performed by YS, XW, and TL. The first draft of the manuscript was written by YS and final manuscript was reviewed and edited by XP and XZ. All authors commented on previous versions of the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Its publication has been approved by all co-authors.
Competing interests
The author declares no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Sun, Y., Wang, X., Liu, T. et al. The multifaceted role of the SASP in atherosclerosis: from mechanisms to therapeutic opportunities. Cell Biosci 12, 74 (2022). https://doi.org/10.1186/s13578-022-00815-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13578-022-00815-5