
Abstract
The tumor microenvironment (TME) is characterized by discrete changes in metabolic features of cancer and immune cells, with various implications. Cancer cells take up most of the available glucose to support their growth, thereby leaving immune cells with insufficient nutrients to expand. In the relative absence of glucose, T cells switch the metabolic program to lipid-based sources, which is pivotal to T-cell differentiation and activation in nutrient-stressed TME. Although consumption of lipids should provide an alternative energy source to starving T cells, a literature survey has revealed that it may not necessarily lead to antitumor responses. Different subtypes of T cells behave differently in various lipid overload states, which widely depends upon the kind of free fatty acids (FFA) engulfed. Key lipid metabolic genes provide cytotoxic T cells with necessary nutrients for proliferation in the absence of glucose, thereby favoring antitumor immunity, but the same genes cause immune evasion in Tmem and Treg. This review aims to detail the complexity of differential lipid metabolism in distinct subtypes of T cells that drive the antitumor or pro-tumor immunity in specific TME states. We have identified key drug targets related to lipid metabolic rewiring in TME.
Similar content being viewed by others

Introduction
Our bodies utilize lipids for various fundamental processes, ranging from the major energy source to the synthesis of vital macromolecules such as cholesterol, membrane phospholipids, and hormones. Like most cells, T cells are heavily dependent on lipid consumption for energy needs, but their naiveté, activation, and effector functions are influenced by the subtype and quantity of lipid intake [1]. A number of surface and intracellular proteins, such as differentiation 36 (CD36), fatty acid-binding protein (FABP), fatty acid transporter protein (FATP), and sterol regulatory-element binding proteins (SREBPs), are responsible for processing lipids in T cells. Under normal circumstances, quiescent T cells process lipids into more energy-efficient oxidative phosphorylation (OXPHOS), which is replaced by aerobic glycolysis once T cells are activated. A complex cascade of co-stimulatory triggers channel T cells into T-regulatory cells (Tregs) and helper T cells (Th cells), each with differential metabolic shifts. As the immune response approaches an end, activated T cells undergo apoptosis or are converted to nondividing T memory cells that revert to OXPHOS [2, 3].
The tumor microenvironment (TME) is a complex space characterized by multiple cell types and their interwoven interactions, predominantly favoring cancerous growth. The interdependence of nutrients, vasculature, and metabolic demands actively shapes the cellular fate in TME. The specialized metabolic switches, nutrient preferences, cellular growth demands, and secretion of various intrinsic and extrinsic factors prime the particular kind of cell in TME to behave in a specific manner [4, 5]. For example, growth signals and upregulation of metabolic features in tumor cells make them more suitable for proliferation, while immune cells undergo tumor evasion after metabolic switch at the same time [6, 18,19,20], while naïve T cells and memory T cells (Tmem) maintain fatty acid oxidation (FAO) as the default metabolic program. FAS has to go through various steps mediated by key enzymes before lipid synthesis can occur. Some of these enzymes affect the rewiring of T cells. For instance, it has been found that CD8+ T cells cannot expand without SREBP signaling during viral infection; however, it was expendable for homeostatic growth. SREBPs in Teffs induce the expression of enzymes fatty acid synthetase (FASN), acetyl-CoA carboxylase (ACC) and hydroxy-methyl-glutaryl-CoA reductase (HMGCR) [17]. Seon Ah Lim et al. have reinforced these observations in a mouse model of TME, showing that inhibition of SREBP-dependent lipid synthesis and metabolic reprogramming in Tregs initiates a robust antitumor response without causing autoimmune disorders. It has been further proven that depletion of an obligatory SREBP factor SCAP curtails tumor growth and upgrades anti-PD-1 immunotherapy [21]. Similarly, Yang-An Wen et al. confirmed the tumor growth suppression after the knockdown of either SREBP1 or SREBP2 target genes required for lipid biosynthesis [22]. At least theoretically, depletion of SREBPs in cancer cells and Tregs yields anticancer benefits; however, deletion of SREBPs in Teffs can affect their blast, which may lead to immune evasion. These SREBPs might be context-dependent drug targets. Further research is required to ascertain them in all subsets of T cells and cancer cells in complex TME.
In addition to SREBPs, ACC is another widely studied lipogenic gene in this direction, which has led to similar results. For instance, Luciana Berod and colleagues found that TH17 cells, but not Tregs, rely on the de novo lipid synthesis mediated by ACC1. Tregs do not follow this scheme as they tend to utilize exogenous FFAs. T cell–specific deletion of ACC1 in mice is able to ameliorate autoimmune disease through anti-inflammatory actions [23]. Similarly, CD8 + T-cell expansion and proliferation are severely curtailed in the absence of ACC1 and subsequently restored by exogenous fatty acid supply [24]. Inhibition and activation of ACC1 favor peripheral Tregs and Th17 cell differentiation, respectively, under TME [25].
FAS required for proliferation is heavily based on acetyl-CoA produced from glycolysis. Inhibition of FAS-related enzymes such as SREBPs, ACC1, or downstream targets leads to defective effector T-cell responses [26]. Although modulation of SREBPs or downstream pathways is rarely attempted in cancer, they may be good therapeutic targets for the future.
FAO, a key bioenergetic pathway [27], displays a critical role in the development of tolerogenic dendritic cells (DCs) [28]. Interestingly, DCs of cancerous origin tend to accumulate oxidized lipids, thereby suppressing T-cell effector functions, which indirectly favors tumor progression [29]. It is speculated that accumulated fatty acids support FAO and therefore promote tolerogenicity in the cancer setting [30]. Furthermore, a carnitine palmitoyltransferase-1 A (CPT1A) inhibitor, etomoxir, has immunomodulatory actions on CD8+ Tmem cell differentiation [31]. However, Brenda Raud et al. have pointed out the flexible metabolic fuel choices of Tmem and found that CPT1A-mediated long chain fatty acid oxidation (LC-FAO) is expendable for the expansion of CD8+ T cell memory [32].
PD-L1/2 in T-cell metabolism in TME
Programmed death ligand-1/2 (PD-L1/2) belongs to CD28 family, and is predominantly expressed on tumors and tumor-infiltrating myeloid cells [33, 34]. PD-L1/2 has been suspected to play a negative role in TME by suppressing antitumor immunity by regulating inhibitory cascade on effector T cells [35, 36]. For example, a recent report has suggested that PD-1 participates in the metabolic reprogramming of activated T cells [37] by reducing Akt (protein kinase B) activation and subsequently inhibiting mammalian target rapamycin (mTOR) activity [37,38,39]. Indeed, reduced activation of mTOR in PD-1+ CD8 + T cells activates transcription factor forkhead box O1 (FoxO1), allowing the survival of exhausted CD8 T cells [39]. As glycolysis is necessary for T cells to obtain the required amount of energy for proliferation, the diversion from glycolysis to FAO likely sabotages antitumor immunity in TME. The preferential diversion from glycolysis to FAO leads to the longevity of Tmem cells, which possess substantial mitochondrial spare respiratory capacity (SRC) [40]. Consistently, inhibition of the PD-1 pathway in the early phases of a viral infection leads to raised mTOR signaling in virus-specific CD8 + T cells, resulting in quicker infection clearance [41]. Certainly, mTOR and Akt are key lipid regulators within the cell [42]. Similarly, inhibition of PD-1/PD-L1 leads to extensive cytotoxic T-cell infiltration into TME as reported in ex vivo, in vitro, and in vivo experimental models [43]. Blocking PD-1 in cancer patients leads to decreased tumor progression and improved survival [44, 45].
PD-1 has also been reported in T-cell exhaustion, which is counterproductive to immunity [46]. Exhaustion of PD-1–associated CD8 + T-cell existed in chronic viral infections in mice [47] and in clinical studies [48,49,50]. It is not surprising that, in 2014, FDA approved the first blocking antibody targeting PD-1 to treat metastatic melanoma, and up to August 2017, many drugs against the PD-1 pathway had been applied in various cancers [46]. However, a recent study has contradicted the holistic benefits of PD-1 blockade in TME as distinct responsiveness of T-cell subpopulations to PD-1 blockade was observed. The study observed that effector and central memory phenotypes were among the most affected T-cell subpopulations after PD-1 blockade but had different gene expression profiles with PD-L1 in comparison to PD-L2 [51].
T cells in TME of obese states
Although it is well established that obesity and lipid overload states are obvious causes of cancer and its progression [52,53,54,55], it has rarely been investigated how these conditions rewire the T-cell differentiation and metabolic switch in TME. Alison E Ringel et al. have recently addressed this important question and demonstrated how obesity shifts the metabolic program of TME to inhibit T-cell function and promote tumor growth. Researchers have systematically shown in a murine model that a high-fat diet (HFD) is differentially taken up by tumor cells in TME as compared to CD8 + T cells, which leads to modified fatty acid partitioning, diminished CD8+ T-cell infiltration, and promotion of tumorigenesis [56]. Of course, preferential fat consumption by tumor cells makes localized T cells less efficient as fat is required to raise the number and plasticity of T cells in TME.
There are several other genes and enzymes with limited evidence but with potential to be good drug targets. The consumption of fat is not only restricted to activate the T cells, but it is also evident that under normal circumstances de novo cardiolipin synthesis keeps the function of CD8 + T cells intact. Mauro Corrado and colleagues demonstrated poor T-cell antigenic responses in T cells deficient in the cardiolipin-synthesizing enzyme—protein tyrosine phosphatase, mitochondrial-1 (PTPMT1). PTPMT1-dependent cardiolipin synthesis is also important for mitochondrial fitness, especially during Tmem cell differentiation or nutrition scarcity [57]. Monoacylglycerol lipase (MGL) hydrolyzes monoglycerides into glycerol and fatty acids. It is abundantly present in tumor cells, and MGL knockout (KO) mice exhibit a reduced tumor size compared with control mice. Interestingly, the reduction in tumor progression is associated with a parallel upregulation in the number of CD8+ T cells. Furthermore, naïve CD8+ T cells exhibit enhanced tumoricidal activity in MGL KO mice [58]. P4HA2 is a metabolism-related gene that is upregulated in cervical cancer tissues, and negatively correlates with CD8 + T cells. Knockdown of P4HA2 suppresses lipid droplet storage in cancer cells [59]. Teresa Manzo and colleagues demonstrated a progressive accumulation of long-chain fatty acids (LCFAs), which, instead of providing an energy source, hamper the mitochondrial function and rewire the lipid metabolism pathways. In addition, intrapancreatic CD8 + T cells inhibit the very long-chain acyl-CoA dehydrogenase (VLCAD) enzyme, which worsens the accumulation of LCFAs and very-long-chain fatty acids (VLCFA), subsequently inducing lipotoxicity. In fact, recently obesity has also been described as a booster of antitumor pharmacotherapy in some cancers [60], but the mechanism remains unknown.
MDSCs in T-cell lipid (de)regulation
Myeloid-derived suppressive cells (MDSCs) manifest negative regulatory activity by promoting immunosuppression in immune-related diseases [61, 62]. In tumors, MDSCs accelerate tumor proliferation, tumor expansion, and immune escape, thereby further exacerbating the TME [63, 64]. MDSCs reshape TME by inhibiting T cells and natural killer (NKT) cells while inducing regulatory T cells (Tregs) and regulatory B cells (Bregs) [65, 66]. From recent studies, it is evident that lipid metabolism in tumor-infiltrating MDSCs (T-MDSCs) is rewired for raised fatty acid uptake, FAO upgrade, oxygen consumption rate (OCR), mitochondrial mass, and expression of core FAO enzymes [67]. It is interesting to note that only T-MDSCs, but not splenic MDSCs, raise lipid uptake [68], which implies that only infiltrating MDSCs undergo lipid metabolic reprogramming. This differential scheme of pro-tumor metabolic features sheds light on the complexity of TME.
In mammals, Liver X receptors (LXRs) are involved in lipid homeostasis. Previous studies have revealed that administration of LXR agonists initiates MDSC apoptosis and reduces tumor volume [69, 70]. In addition, lectin-type oxidized LDL receptor 1 (LOX-1) is present in PMN-MDSCs of cancer patients but is absent in healthy individuals [71]. Similarly, Caijun Wu et al. noticed the enhanced immunosuppressive role of monocytic MDSCs after administration of a multidose clinical regimen of gemcitabine (GEM). These authors have implicated that the deregulation of lipid metabolism in residual tumor cells is partially responsible for promoting immunosuppression [72]. It is plausible to conclude that tumor-derived MDSCs are forced to rewire lipid metabolism primarily because of robust lipid storage and related signaling activation.
CD36 and T-cell regulation in TME
CD36 is a scavenger receptor of oxidized lipids, and is expressed in multiple cell types, including T cells [73, 74]. Previous studies have highlighted that tumor-associated immune cells undergo CD36-oriented lipid metabolic reprogramming, which leads to immune evasion and cancer progression [75]. Shihao Xu and coworkers have reported that CD8+ tumor-infiltrating lymphocytes (TILs) are responsive to lipids in the TME, mediated by CD36, which is associated with progressive T-cell dysfunction. It has been explained that T-cell dysfunction occurs in a CD36-dependent manner, which leads to a raise in oxidized low-density lipoproteins (OxLDL) in T cells, promotion of lipid peroxidation downstream, and occurrence of ferroptosis. Interestingly, overexpression of glutathione peroxidase 4 reverses lipid peroxidation to improve the effector capacity of T cells [76]. Similar evidence has been provided by other research groups, associating overexpression of CD36 with shorter survival of melanoma patients with tumor-infiltrating CD8+ T cells, while CD36-depleted CD8+ T cells showed greater antitumor potential and survival compared with wild-type CD8+ T cells [77,78,79,80].
Importantly, there are very few identified metabolic drug targets that work in the same direction in both Tregs and Teffs. As presented above, metabolic drug targets are often context- and T-cell subtype–dependent, in which Tregs and Teffs promote or inhibit tumor, respectively. However, CD36 offers a rare opportunity because its deletion on both Tregs and Teffs results in enhanced antitumor activities [81]. For instance, Wang et al. stated that genetic knockdown of CD36 in Treg cells reduced tumor growth and intratumoral Treg cells, promoting the antitumor function of tumor-infiltrating lymphocytes [82]. Although only few studies have been reported in this direction, CD36 presents a viable common drug target that requires future research.
Effects of lipid metabolism on Tregs
The relative ratio of cytotoxic T cells and Tregs in TME plays a pivotal role in tumor progression and immune evasion [83]. Tregs contribute to immune evasion in TME [84,85,86]. Systematic ablation of Tregs in several cancer types has resulted in tumor suppression and cellular alterations within the TME [87, 88]. It is suspected that cytotoxic Teffs and Tregs follow different activation and proliferation pathways as Tregs are abundant even in the unfavorable metabolic states in TME [89]. Indeed, Weinberg et al. have stressed the necessity of mitochondrial metabolism in Tregs to maintain their immunosuppressive function [90]. A recent study has shown raised production of FFAs by RHOA Y42-mutated gastric cancer, modulated via the PI3K pathway, which favors the accumulation of Tregs in a low-glucose TME. Similarly, the expression levels of FAS, CPT-1, PPARα, and PPARɣ were also higher in gastric cancer with RHOA Y42 mutation [91]. It remains unknown what metabolic switch enables Tregs to expand and proliferate differently from Teffs in the same TME. However, one study confirmed that intratumoral Tregs indirectly promote M2-like TAMs by boosting SREBP1-dependent lipid metabolism and then limiting the CD8+ T-produced interferon-gamma (IFNγ), thereby leading to tumor progression and orchestrating tumor-associated immunosuppression [92]. In addition, inhibition of FABP5 on Tregs causes mitochondrial alterations characterized by impaired lipid metabolism, reduced OXPHOS, and loss of cristae structure. The authors concluded that FABP5 is a gatekeeper of mitochondrial integrity, which is necessary for normal functioning of Tregs [93]. An interesting observation regarding the complicity of Tregs and tumor cells to suppress the T-cell functioning has recently been highlighted. ** T-cell senescence in cancer models in vivo and in vitro [94].
Unconventional T cells in TME
γδ T cells are capable of differentiating into various subtypes of immune cells depending on the TME conditions [95]. Although the scientific knowledge on γδ T cells is underdeveloped and their proclivity as pro-tumorigenic or anticancer immune cells is still unclear [96,97,98], γδ T cells are potential agents against cancer cells [99]. Various studies have reported the chameleon-like nature of γδ T cells, pointing at the flexibility they exhibit in TME [100,101,102]. The phenomenon has been successfully explained in squamous cell carcinoma [103] and colorectal cancer [104], where it has been suggested that TME conditions can affect the proliferation and functional nature of γδ T-cells. For example, a recent study has highlighted two distinct subtypes of γδ T cells, namely, antitumoral IFN-γ–producing γδ T cells (γδIFN cells) and IL17-producing γδ T cells (γδ17 cells) [105]. Interestingly, it has been shown that Vδ2 cells are activated, independent of MHC, by small lipid molecules, phosphoantigens (pAgs), which are derived from the mevalonate pathway [106,107,108]. Furthermore, Emmanuel Scotet et al. identified two different lipid-related ligands of Vɣ9Vδ2 TCR in tumor cells, namely apolipoprotein A1 (Apo-A1) and ATP synthase/F1-ATPase (high-affinity apo A-I receptor). These authors revealed that Apo-A1, which is abundant in high-density lipoproteins (HDL), is needed for the activation of Vɣ9Vδ2 T cells by tumors expressing F1-ATPase [109]. Similarly, a related study by Rodrigues et al. showed that Vδ2 T cells express low-density lipoprotein (LDL) receptors when they are activated and their functions can be modified once LDL attaches to their activated receptors. It has also been demonstrated that expression levels of IFN, NKG2D, and DNAM-1 are downregulated when Vɣ9Vδ2 T cells are treated with LDL-cholesterol [110]. Furthermore, host-derived lipids from lung-infiltrating CD1d + B-1a cells are able to induce γδ T cells for the induction of IL-17 A [111].
As a specialized type of T lymphocytes, Natural killer T cells (NKT cells) recognize lipid antigens presented through CD1d [112, 113]. NKT cells are divided into two distinct types, including I and II NKT cells, which regulate the immune response in the development and progression of tumor [114,115,116]. Both type I and type II NKT cells show intermodulation, but type I NKT cells are known to increase antitumor responses, while type II NKT cells are inclined towards pro-cancer activities [117], with some contextual exceptions where type I can also suppress tumor immunity [115, 118, 119]. However, tumor growth in TME is bound to consume more lipids to support its rapid proliferation and meet excessive energy needs. De novo lipid synthesis, greater and preferential fatty acid uptake from surrounding tissues in TME, and altered equilibrium of polyunsaturated fatty acids (PUFAs) and saturated fatty acids (SFAs) change the lipid repertoire of tumor cells, which can affect membrane fluidity, cell–cell interaction, and membrane protein landscape, subsequently affecting the downstream signaling cascade [120, 121] in cancers [122, 123]. The changes in lipid repertoire are also linked to the altered structure of bio-in cancer cells [124]. In this context, HFD rich in SFA can negatively affect the capability of DCs to activate naïve T cells [125], which is critical for antitumor responses. The availability of lipids is necessary for the NKT cells development [126] as mice deficient in lysosomal lipid transfer enzyme Niemann Pick C (NPC) 2 have a decreased number of type I NKT cells [127]. There is no doubt that excess lipid states lead to the activation of type I NKT cells, which generates a proinflammatory environment in obese patients [128], while CD1d−/−mice show reduced inflammation under similar conditions [129]. Furthermore, the antitumor potential of NKT cells in obesity is reduced and does not inhibit tumor growth [130]. However, human studies were unable to show any changes in the number of NKT cells in a hepatocellular carcinoma (HCC) model [131]. It is interesting to note that higher lipids increase the NKT cell proliferation, leading to proinflammatory responses, but an obese state reduces NKT cells, causing hindrance in tumor immunity. Further research in this direction is vital and has the potential to unveil anticancer drug targets.
Conclusion
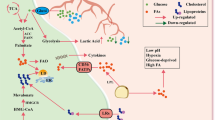
Lipid metabolic features differ in their ability to initiate antitumor or pro-tumor responses in glucose-diminished TME infiltrated by cytotoxic T cells, Tregs, Tmem, and NKT cells (Fig. 1). It may be oversimplified to argue that lipids proliferate Teffs and regress Tregs/Tmem in complicated TMEs where diversified forms of lipids exist. The predisposition of TME to attract and utilize excessive lipids leaves little energy for Teffs to expand. In a complex TME, the identification of lipid-based T-cell drug targets is context-dependent as the same genes or enzymes responsible for attracting lipids to T cells preferentially contribute to cancer cell fat intake. The type of lipid intake peculiarly affects different subsets of T cells. Formulation of a definitive hypothesis in this regard is too early. It is necessary to explore the expression difference of the same gene in different subtypes of T cells under TME to mark it as a drug target. Although CD36, SREBPs, PD-L1/2, FABP5, CPT-1, ACC1, GLUT1, and FAS has shown promising prospects to be potential metabolic drug targets of cancer, their context-dependence and varied implications in T-cell subtypes urge for more research. Identifying the differential expression of lipid-related genes, gatekeepers, and enzymes on T-cell subsets and cancer cells that can be manipulated to draw clinical gains presents an opportunity for future research.

Differential T-cell responses after metabolic rewiring in response to lipids within TME. Raised expression of CD36 leads to T-cell dysregulation and immune evasion in TME. Excess lipids generate differential responses in T-MDSCs and S-MDSCs, leading to the activation of Tregs and causing T-cell dysregulation. Similarly, PD-1/PD-L1 complex converts glycolysis to FAO, thereby affecting T-cell exhaustion and playing a role in tumor progression. Under the state of metabolic competition within TME, T cells may opt to consume more lipids to compensate for depleted glucose and in-process lead to T cells activation.TME: Tumor microenvironment; FAO: Fatty acid oxidation; T-MDSCs: Tumor MDSCs; S-MDSCs: Splenic MDSCs; oxLDL: Oxidized low-density lipoproteins; Tregs: T regulatory cells; SREBPs: Sterol-responsive element-binding proteins. Raised OxLDL intake: Raised intake of oxidized low-density lipoproteins (OxLDL) increases lipid peroxidation downstream and causes ferroptosis in T cells, leading to suppression of antitumor immunity. Lipids induce metabolic reprogramming: Myeloid-derived suppressive cells (MDSCs) rewiring lipid metabolism have a direct role in remodeling TME by repressing T cells as well as natural killer (NK) cells and generating regulatory T cells (Tregs) and regulatory B cells (Bregs), supporting immune escape. Metabolic reprogramming: As glycolysis is necessary for T cells to obtain the required amount of energy for proliferation, PD-1/PD-L1–involved metabolic reprogramming from glycolysis to fatty acid oxidation (FAO) causes T-cell exhaustion, which is likely to sabotage antitumor immunity in TME. Antitumor metabolic reprogramming: Sterol-responsive element-binding proteins (SREBPs) increase activated T cells with greater metabolic demands by enhancing fatty acid synthesis (FAS), therefore driving the antitumor immunity.
Data Availability
Not applicable.
Abbreviations
- TME:
-
tumormicroenvironment
- FAS:
-
fatty acid synthesis
- SREBPs:
-
sterolresponsive element-binding proteins
- FAO:
-
fatty acid oxidation
- MDSCs:
-
myeloid-derived suppressive cells
- LXRs:
-
liver X receptors
References
Howie D, Ten Bokum A, Necula AS, Cobbold SP, Waldmann H. The Role of Lipid Metabolism in T Lymphocyte Differentiation and Survival. Front Immunol 2017, 8:1949.
Geltink RIK, Kyle RL, Pearce EL. Unraveling the Complex Interplay Between T Cell Metabolism and Function. Annu Rev Immunol. 2018;36:461–88.
Patsoukis N, Bardhan K, Weaver J, Herbel C, Seth P, Li L, Boussiotis VA. The role of metabolic reprogramming in T cell fate and function. Curr Trends Immunol. 2016;17:1–12.
Kaymak I, Williams KS, Cantor JR, Jones RG. Immunometabolic Interplay in the Tumor Microenvironment. Cancer Cell. 2021;39:28–37.
Bader JE, Voss K, Rathmell JC. Targeting Metabolism to Improve the Tumor Microenvironment for Cancer Immunotherapy. Mol Cell. 2020;78:1019–33.
Wegiel B, Vuerich M, Daneshmandi S, Seth P. Metabolic Switch in the Tumor Microenvironment Determines Immune Responses to Anti-cancer Therapy. Front Oncol. 2018;8:284.
**a L, Oyang L, Lin J, Tan S, Han Y, Wu N, Yi P, Tang L, Pan Q, Rao S, et al. The cancer metabolic reprogramming and immune response. Mol Cancer. 2021;20:28.
Cantor JR, Sabatini DM. Cancer cell metabolism: one hallmark, many faces. Cancer Discov. 2012;2:881–98.
Kong H, Chandel NS. Regulation of redox balance in cancer and T cells. J Biol Chem. 2018;293:7499–507.
Sugiura A, Rathmell JC. Metabolic Barriers to T Cell Function in Tumors. J Immunol. 2018;200:400–7.
Ho PC, Bihuniak JD, Macintyre AN, Staron M, Liu X, Amezquita R, Tsui YC, Cui G, Micevic G, Perales JC, et al. Phosphoenolpyruvate Is a Metabolic Checkpoint of Anti-tumor T Cell Responses. Cell. 2015;162:1217–28.
Chang CH, Curtis JD, Maggi LB Jr, Faubert B, Villarino AV, O’Sullivan D, Huang SC, van der Windt GJ, Blagih J, Qiu J, et al. Posttranscriptional control of T cell effector function by aerobic glycolysis. Cell. 2013;153:1239–51.
Corn KC, Windham MA, Rafat M. Lipids in the tumor microenvironment: From cancer progression to treatment. Prog Lipid Res. 2020;80:101055.
Balaban S, Shearer RF, Lee LS, van Geldermalsen M, Schreuder M, Shtein HC, Cairns R, Thomas KC, Fazakerley DJ, Grewal T, et al. Adipocyte lipolysis links obesity to breast cancer growth: adipocyte-derived fatty acids drive breast cancer cell proliferation and migration. Cancer Metab. 2017;5:1.
Chen WJ, Ho CC, Chang YL, Chen HY, Lin CA, Ling TY, Yu SL, Yuan SS, Chen YJ, Lin CY, et al. Cancer-associated fibroblasts regulate the plasticity of lung cancer stemness via paracrine signalling. Nat Commun. 2014;5:3472.
Yu Y, **ao CH, Tan LD, Wang QS, Li XQ, Feng YM. Cancer-associated fibroblasts induce epithelial-mesenchymal transition of breast cancer cells through paracrine TGF-β signalling. Br J Cancer. 2014;110:724–32.
Kidani Y, Elsaesser H, Hock MB, Vergnes L, Williams KJ, Argus JP, Marbois BN, Komisopoulou E, Wilson EB, Osborne TF, et al. Sterol regulatory element-binding proteins are essential for the metabolic programming of effector T cells and adaptive immunity. Nat Immunol. 2013;14:489–99.
Hukelmann JL, Anderson KE, Sinclair LV, Grzes KM, Murillo AB, Hawkins PT, Stephens LR, Lamond AI, Cantrell DA. The cytotoxic T cell proteome and its sha** by the kinase mTOR. Nat Immunol. 2016;17:104–12.
Düvel K, Yecies JL, Menon S, Raman P, Lipovsky AI, Souza AL, Triantafellow E, Ma Q, Gorski R, Cleaver S, et al. Activation of a metabolic gene regulatory network downstream of mTOR complex 1. Mol Cell. 2010;39:171–83.
Macintyre AN, Finlay D, Preston G, Sinclair LV, Waugh CM, Tamas P, Feijoo C, Okkenhaug K, Cantrell DA. Protein kinase B controls transcriptional programs that direct cytotoxic T cell fate but is dispensable for T cell metabolism. Immunity. 2011;34:224–36.
Lim SA, Wei J, Nguyen TM, Shi H, Su W, Palacios G, Dhungana Y, Chapman NM, Long L, Saravia J, et al. Lipid signalling enforces functional specialization of T(reg) cells in tumours. Nature. 2021;591:306–11.
Wen YA, **ong X, Zaytseva YY, Napier DL, Vallee E, Li AT, Wang C, Weiss HL, Evers BM, Gao T. Downregulation of SREBP inhibits tumor growth and initiation by altering cellular metabolism in colon cancer. Cell Death Dis. 2018;9:265.
Berod L, Friedrich C, Nandan A, Freitag J, Hagemann S, Harmrolfs K, Sandouk A, Hesse C, Castro CN, Bähre H, et al. De novo fatty acid synthesis controls the fate between regulatory T and T helper 17 cells. Nat Med. 2014;20:1327–33.
Lee J, Walsh MC, Hoehn KL, James DE, Wherry EJ, Choi Y. Regulator of fatty acid metabolism, acetyl coenzyme a carboxylase 1, controls T cell immunity. J Immunol. 2014;192:3190–9.
Berod L, Friedrich C, Nandan A, Freitag J, Hagemann S, Harmrolfs K, Sandouk A, Hesse C, Castro CN, Bähre H. De novo fatty acid synthesis controls the fate between regulatory T and T helper 17 cells. Nat Med. 2014;20:1327–33.
Raha S, Raud B, Oberdörfer L, Castro CN, Schreder A, Freitag J, Longerich T, Lochner M, Sparwasser T, Berod L, et al. Disruption of de novo fatty acid synthesis via acetyl-CoA carboxylase 1 inhibition prevents acute graft-versus-host disease. Eur J Immunol. 2016;46:2233–8.
Ma Y, Wang W, Devarakonda T, Zhou H, Wang XY, Salloum FN, Spiegel S, Fang X. Functional analysis of molecular and pharmacological modulators of mitochondrial fatty acid oxidation. Sci Rep. 2020;10:1450.
Malinarich F, Duan K, Hamid RA, Bi** A, Lin WX, Poidinger M, Fairhurst AM, Connolly JE. High mitochondrial respiration and glycolytic capacity represent a metabolic phenotype of human tolerogenic dendritic cells. J Immunol. 2015;194:5174–86.
Cubillos-Ruiz JR, Silberman PC, Rutkowski MR, Chopra S, Perales-Puchalt A, Song M, Zhang S, Bettigole SE, Gupta D, Holcomb K, et al. ER Stress Sensor XBP1 Controls Anti-tumor Immunity by Disrupting Dendritic Cell Homeostasis. Cell. 2015;161:1527–38.
O’Neill LA, Pearce EJ. Immunometabolism governs dendritic cell and macrophage function. J Exp Med. 2016;213:15–23.
Huang SC, Everts B, Ivanova Y, O’Sullivan D, Nascimento M, Smith AM, Beatty W, Love-Gregory L, Lam WY, O’Neill CM, et al. Cell-intrinsic lysosomal lipolysis is essential for alternative activation of macrophages. Nat Immunol. 2014;15:846–55.
Raud B, McGuire PJ, Jones RG, Sparwasser T, Berod L. Fatty acid metabolism in CD8(+) T cell memory: Challenging current concepts. Immunol Rev. 2018;283:213–31.
Horlad H, Ma C, Yano H, Pan C, Ohnishi K, Fujiwara Y, Endo S, Kikukawa Y, Okuno Y, Matsuoka M, et al. An IL-27/Stat3 axis induces expression of programmed cell death 1 ligands (PD-L1/2) on infiltrating macrophages in lymphoma. Cancer Sci. 2016;107:1696–704.
Schildberg FA, Klein SR, Freeman GJ, Sharpe AH. Coinhibitory Pathways in the B7-CD28 Ligand-Receptor Family. Immunity. 2016;44:955–72.
Zhang L, Gajewski TF, Kline J. PD-1/PD-L1 interactions inhibit antitumor immune responses in a murine acute myeloid leukemia model. Blood. 2009;114:1545–52.
Kuang DM, Zhao Q, Peng C, Xu J, Zhang JP, Wu C, Zheng L. Activated monocytes in peritumoral stroma of hepatocellular carcinoma foster immune privilege and disease progression through PD-L1. J Exp Med. 2009;206:1327–37.
Patsoukis N, Bardhan K, Chatterjee P, Sari D, Liu B, Bell LN, Karoly ED, Freeman GJ, Petkova V, Seth P, et al. PD-1 alters T-cell metabolic reprogramming by inhibiting glycolysis and promoting lipolysis and fatty acid oxidation. Nat Commun. 2015;6:6692.
Chang CH, Qiu J, O’Sullivan D, Buck MD, Noguchi T, Curtis JD, Chen Q, Gindin M, Gubin MM, van der Windt GJ, et al. Metabolic Competition in the Tumor Microenvironment Is a Driver of Cancer Progression. Cell. 2015;162:1229–41.
Staron MM, Gray SM, Marshall HD, Parish IA, Chen JH, Perry CJ, Cui G, Li MO, Kaech SM. The transcription factor FoxO1 sustains expression of the inhibitory receptor PD-1 and survival of antiviral CD8(+) T cells during chronic infection. Immunity. 2014;41:802–14.
van der Windt GJ, Everts B, Chang CH, Curtis JD, Freitas TC, Amiel E, Pearce EJ, Pearce EL. Mitochondrial respiratory capacity is a critical regulator of CD8 + T cell memory development. Immunity. 2012;36:68–78.
Ahn E, Araki K, Hashimoto M, Li W, Riley JL, Cheung J, Sharpe AH, Freeman GJ, Irving BA, Ahmed R. Role of PD-1 during effector CD8 T cell differentiation. Proc Natl Acad Sci U S A. 2018;115:4749–54.
Yi J, Zhu J, Wu J, Thompson CB, Jiang X. Oncogenic activation of PI3K-AKT-mTOR signaling suppresses ferroptosis via SREBP-mediated lipogenesis. Proc Natl Acad Sci U S A. 2020;117:31189–97.
Acúrcio RC, Pozzi S, Carreira B, Pojo M, Gómez-Cebrián N, Casimiro S, Fernandes A, Barateiro A, Farricha V, Brito J, et al: Therapeutic targeting of PD-1/PD-L1 blockade by novel small-molecule inhibitors recruits cytotoxic T cells into solid tumor microenvironment. J Immunother Cancer 2022, 10.
Brahmer JR, Tykodi SS, Chow LQ, Hwu WJ, Topalian SL, Hwu P, Drake CG, Camacho LH, Kauh J, Odunsi K, et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med. 2012;366:2455–65.
Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, Powderly JD, Carvajal RD, Sosman JA, Atkins MB, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. 2012;366:2443–54.
Hashimoto M, Kamphorst AO, Im SJ, Kissick HT, Pillai RN, Ramalingam SS, Araki K, Ahmed R. CD8 T Cell Exhaustion in Chronic Infection and Cancer: Opportunities for Interventions. Annu Rev Med. 2018;69:301–18.
Barber DL, Wherry EJ, Masopust D, Zhu B, Allison JP, Sharpe AH, Freeman GJ, Ahmed R. Restoring function in exhausted CD8 T cells during chronic viral infection. Nature. 2006;439:682–7.
Freeman GJ, Long AJ, Iwai Y, Bourque K, Chernova T, Nishimura H, Fitz LJ, Malenkovich N, Okazaki T, Byrne MC, et al. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J Exp Med. 2000;192:1027–34.
Iwai Y, Ishida M, Tanaka Y, Okazaki T, Honjo T, Minato N. Involvement of PD-L1 on tumor cells in the escape from host immune system and tumor immunotherapy by PD-L1 blockade. Proc Natl Acad Sci U S A. 2002;99:12293–7.
Dong H, Strome SE, Salomao DR, Tamura H, Hirano F, Flies DB, Roche PC, Lu J, Zhu G, Tamada K, et al. Tumor-associated B7-H1 promotes T-cell apoptosis: a potential mechanism of immune evasion. Nat Med. 2002;8:793–800.
Lerrer S, Tocheva AS, Bukhari S, Adam K, Mor A. PD-1-stimulated T cell subsets are transcriptionally and functionally distinct. iScience. 2021;24:103020.
Lu L, Koo S, McPherson S, Hull MA, Rees CJ, Sharp L. Systematic review and meta-analysis: Associations between metabolic syndrome and colorectal neoplasia outcomes. Colorectal Dis 2022.
Chrysavgis L, Giannakodimos I, Diamantopoulou P, Cholongitas E. Non-alcoholic fatty liver disease and hepatocellular carcinoma: Clinical challenges of an intriguing link. World J Gastroenterol. 2022;28:310–31.
Nishina S, Hino K. CD26/DPP4 as a Therapeutic Target in Nonalcoholic Steatohepatitis Associated Hepatocellular Carcinoma. Cancers (Basel) 2022, 14.
Nieman KM, Kenny HA, Penicka CV, Ladanyi A, Buell-Gutbrod R, Zillhardt MR, Romero IL, Carey MS, Mills GB, Hotamisligil GS, et al. Adipocytes promote ovarian cancer metastasis and provide energy for rapid tumor growth. Nat Med. 2011;17:1498–503.
Ringel AE, Drijvers JM, Baker GJ, Catozzi A, García-Cañaveras JC, Gassaway BM, Miller BC, Juneja VR, Nguyen TH, Joshi S, et al. Obesity Shapes Metabolism in the Tumor Microenvironment to Suppress Anti-Tumor Immunity. Cell. 2020;183:1848–66.e1826.
Corrado M, Edwards-Hicks J, Villa M, Flachsmann LJ, Sanin DE, Jacobs M, Baixauli F, Stanczak M, Anderson E, Azuma M, et al. Dynamic Cardiolipin Synthesis Is Required for CD8(+) T Cell Immunity. Cell Metab. 2020;32:981–95.e987.
Kienzl M, Hasenoehrl C, Maitz K, Sarsembayeva A, Taschler U, Valadez-Cosmes P, Kindler O, Ristic D, Raftopoulou S, Santiso A, et al. Monoacylglycerol lipase deficiency in the tumor microenvironment slows tumor growth in non-small cell lung cancer. Oncoimmunology. 2021;10:1965319.
Shang C, Huang J, Guo H. Identification of an Metabolic Related Risk Signature Predicts Prognosis in Cervical Cancer and Correlates With Immune Infiltration. Front Cell Dev Biol. 2021;9:677831.
Assumpção JAF, Pasquarelli-do-Nascimento G, Duarte MSV, Bonamino MH, Magalhães KG. The ambiguous role of obesity in oncology by promoting cancer but boosting antitumor immunotherapy. J Biomed Sci. 2022;29:12.
Schmidt K, Zilio S, Schmollinger JC, Bronte V, Blankenstein T, Willimsky G. Differently immunogenic cancers in mice induce immature myeloid cells that suppress CTL in vitro but not in vivo following transfer. Blood. 2013;121:1740–8.
Grover A, Sanseviero E, Timosenko E, Gabrilovich DI. Myeloid-Derived Suppressor Cells: A Propitious Road to Clinic. Cancer Discov. 2021;11:2693–706.
Pergamo M, Miller G. Myeloid-derived suppressor cells and their role in pancreatic cancer. Cancer Gene Ther. 2017;24:100–5.
Malek E, de Lima M, Letterio JJ, Kim BG, Finke JH, Driscoll JJ, Giralt SA. Myeloid-derived suppressor cells: The green light for myeloma immune escape. Blood Rev. 2016;30:341–8.
Trovato R, Canè S, Petrova V, Sartoris S, Ugel S, De Sanctis F. The Engagement Between MDSCs and Metastases: Partners in Crime. Front Oncol. 2020;10:165.
Pastaki Khoshbin A, Eskian M, Keshavarz-Fathi M, Rezaei N. Roles of Myeloid-Derived Suppressor Cells in Cancer Metastasis: Immunosuppression and Beyond. Arch Immunol Ther Exp (Warsz). 2019;67:89–102.
Hossain F, Al-Khami AA, Wyczechowska D, Hernandez C, Zheng L, Reiss K, Valle LD, Trillo-Tinoco J, Maj T, Zou W, et al. Inhibition of Fatty Acid Oxidation Modulates Immunosuppressive Functions of Myeloid-Derived Suppressor Cells and Enhances Cancer Therapies. Cancer Immunol Res. 2015;3:1236–47.
Al-Khami AA, Zheng L, Del Valle L, Hossain F, Wyczechowska D, Zabaleta J, Sanchez MD, Dean MJ, Rodriguez PC, Ochoa AC. Exogenous lipid uptake induces metabolic and functional reprogramming of tumor-associated myeloid-derived suppressor cells. Oncoimmunology. 2017;6:e1344804.
Tavazoie MF, Pollack I, Tanqueco R, Ostendorf BN, Reis BS, Gonsalves FC, Kurth I, Andreu-Agullo C, Derbyshire ML, Posada J, et al. LXR/ApoE Activation Restricts Innate Immune Suppression in Cancer. Cell. 2018;172:825–40.e818.
LXR Agonism Depletes. MDSCs to Promote Antitumor Immunity. Cancer Discov. 2018;8:263.
Condamine T, Dominguez GA, Youn JI, Kossenkov AV, Mony S, Alicea-Torres K, Tcyganov E, Hashimoto A, Nefedova Y, Lin C, et al: Lectin-type oxidized LDL receptor-1 distinguishes population of human polymorphonuclear myeloid-derived suppressor cells in cancer patients. Sci Immunol 2016, 1.
Wu C, Tan X, Hu X, Zhou M, Yan J, Ding C. Tumor Microenvironment following Gemcitabine Treatment Favors Differentiation of Immunosuppressive Ly6C(high) Myeloid Cells. J Immunol. 2020;204:212–23.
Subramanian M, Marelli-Berg FM. CD36 pumps fat to defang killer T cells in tumors. Cell Metab. 2021;33:1509–11.
Torres-Hernandez A, Wang W, Nikiforov Y, Tejada K, Torres L, Kalabin A, Adam S, Wu J, Lu L, Chen R, et al. γδ T Cells Promote Steatohepatitis by Orchestrating Innate and Adaptive Immune Programming. Hepatology. 2020;71:477–94.
Wang J, Li Y. CD36 tango in cancer: signaling pathways and functions. Theranostics. 2019;9:4893–908.
Xu S, Chaudhary O, Rodríguez-Morales P, Sun X, Chen D, Zappasodi R, Xu Z, Pinto AFM, Williams A, Schulze I, et al. Uptake of oxidized lipids by the scavenger receptor CD36 promotes lipid peroxidation and dysfunction in CD8(+) T cells in tumors. Immunity. 2021;54:1561–77.e1567.
Mallick R, Basak S, Duttaroy AK. Fatty acids and evolving roles of their proteins in neurological, cardiovascular disorders and cancers. Prog Lipid Res. 2021;83:101116.
Ma X, **ao L, Liu L, Ye L, Su P, Bi E, Wang Q, Yang M, Qian J, Yi Q. CD36-mediated ferroptosis dampens intratumoral CD8(+) T cell effector function and impairs their antitumor ability. Cell Metab. 2021;33:1001–12.e1005.
CD36 Activity Causes. Ferroptosis in Tumor-Infiltrating CD8(+) T Cells. Cancer Discov. 2021;11:Of24.
Kolonin MG: Bad Cholesterol Uptake by CD36 in T-Cells Cripples Anti-Tumor Immune Response. Immunometabolism 2021, 3.
Horton BL, Spranger S. CD36 - the Achilles’ heel of T(reg) cells. Nat Immunol. 2020;21:251–3.
Wang H, Franco F, Tsui YC, **e X, Trefny MP, Zappasodi R, Mohmood SR, Fernández-García J, Tsai CH, Schulze I, et al. CD36-mediated metabolic adaptation supports regulatory T cell survival and function in tumors. Nat Immunol. 2020;21:298–308.
Ayers M, Lunceford J, Nebozhyn M, Murphy E, Loboda A, Kaufman DR, Albright A, Cheng JD, Kang SP, Shankaran V, et al. IFN-γ-related mRNA profile predicts clinical response to PD-1 blockade. J Clin Invest. 2017;127:2930–40.
Sasidharan Nair V, Elkord E. Immune checkpoint inhibitors in cancer therapy: a focus on T-regulatory cells. Immunol Cell Biol. 2018;96:21–33.
Kumar P, Bhattacharya P, Prabhakar BS. A comprehensive review on the role of co-signaling receptors and Treg homeostasis in autoimmunity and tumor immunity. J Autoimmun. 2018;95:77–99.
Grzywa TM, Justyniarska M, Nowis D, Golab J. Tumor Immune Evasion Induced by Dysregulation of Erythroid Progenitor Cells Development. Cancers (Basel) 2021, 13.
Bos PD, Plitas G, Rudra D, Lee SY, Rudensky AY. Transient regulatory T cell ablation deters oncogene-driven breast cancer and enhances radiotherapy. J Exp Med. 2013;210:2435–66.
Delgoffe GM, Woo SR, Turnis ME, Gravano DM, Guy C, Overacre AE, Bettini ML, Vogel P, Finkelstein D, Bonnevier J, et al. Stability and function of regulatory T cells is maintained by a neuropilin-1-semaphorin-4a axis. Nature. 2013;501:252–6.
Wang H, Franco F, Ho PC. Metabolic Regulation of Tregs in Cancer: Opportunities for Immunotherapy. Trends Cancer. 2017;3:583–92.
Weinberg SE, Singer BD, Steinert EM, Martinez CA, Mehta MM, Martínez-Reyes I, Gao P, Helmin KA, Abdala-Valencia H, Sena LA, et al. Mitochondrial complex III is essential for suppressive function of regulatory T cells. Nature. 2019;565:495–9.
Kumagai S, Togashi Y, Sakai C, Kawazoe A, Kawazu M, Ueno T, Sato E, Kuwata T, Kinoshita T, Yamamoto M, et al. An Oncogenic Alteration Creates a Microenvironment that Promotes Tumor Progression by Conferring a Metabolic Advantage to Regulatory T Cells. Immunity. 2020;53:187–203.e188.
Liu C, Chikina M, Deshpande R, Menk AV, Wang T, Tabib T, Brunazzi EA, Vignali KM, Sun M, Stolz DB, et al. Treg Cells Promote the SREBP1-Dependent Metabolic Fitness of Tumor-Promoting Macrophages via Repression of CD8(+) T Cell-Derived Interferon-γ. Immunity. 2019;51:381–97.e386.
Field CS, Baixauli F, Kyle RL, Puleston DJ, Cameron AM, Sanin DE, Hippen KL, Loschi M, Thangavelu G, Corrado M, et al. Mitochondrial Integrity Regulated by Lipid Metabolism Is a Cell-Intrinsic Checkpoint for Treg Suppressive Function. Cell Metab. 2020;31:422–37.e425.
Liu X, Hartman CL, Li L, Albert CJ, Si F, Gao A, Huang L, Zhao Y, Lin W, Hsueh EC, et al: Reprogramming lipid metabolism prevents effector T cell senescence and enhances tumor immunotherapy. Sci Transl Med 2021, 13.
Corsale AM, Di Simone M, Lo Presti E, Picone C, Dieli F, Meraviglia S: Metabolic Changes in Tumor Microenvironment: How Could They Affect γδ T Cells Functions? Cells 2021, 10.
Chitadze G, Oberg HH, Wesch D, Kabelitz D. The Ambiguous Role of γδ T Lymphocytes in Antitumor Immunity. Trends Immunol. 2017;38:668–78.
Wu P, Wu D, Ni C, Ye J, Chen W, Hu G, Wang Z, Wang C, Zhang Z, **a W, et al. γδT17 cells promote the accumulation and expansion of myeloid-derived suppressor cells in human colorectal cancer. Immunity. 2014;40:785–800.
Ma S, Cheng Q, Cai Y, Gong H, Wu Y, Yu X, Shi L, Wu D, Dong C, Liu H. IL-17A produced by γδ T cells promotes tumor growth in hepatocellular carcinoma. Cancer Res. 2014;74:1969–82.
Lo Presti E, Di Mitri R, Pizzolato G, Mocciaro F, Dieli F, Meraviglia S. γδ cells and tumor microenvironment: A helpful or a dangerous liason? J Leukoc Biol. 2018;103:485–92.
Wesch D, Glatzel A, Kabelitz D. Differentiation of resting human peripheral blood gamma delta T cells toward Th1- or Th2-phenotype. Cell Immunol. 2001;212:110–7.
Ness-Schwickerath KJ, ** C, Morita CT. Cytokine requirements for the differentiation and expansion of IL-17A- and IL-22-producing human Vgamma2Vdelta2 T cells. J Immunol. 2010;184:7268–80.
Fenoglio D, Poggi A, Catellani S, Battaglia F, Ferrera A, Setti M, Murdaca G, Zocchi MR. Vdelta1 T lymphocytes producing IFN-gamma and IL-17 are expanded in HIV-1-infected patients and respond to Candida albicans. Blood. 2009;113:6611–8.
Meraviglia S, Lo Presti E, Tosolini M, La Mendola C, Orlando V, Todaro M, Catalano V, Stassi G, Cicero G, Vieni S, et al. Distinctive features of tumor-infiltrating γδ T lymphocytes in human colorectal cancer. Oncoimmunology. 2017;6:e1347742.
Lo Presti E, Toia F, Oieni S, Buccheri S, Turdo A, Mangiapane LR, Campisi G, Caputo V, Todaro M, Stassi G, et al. Squamous Cell Tumors Recruit γδ T Cells Producing either IL17 or IFNγ Depending on the Tumor Stage. Cancer Immunol Res. 2017;5:397–407.
Lopes N, McIntyre C, Martin S, Raverdeau M, Sumaria N, Kohlgruber AC, Fiala GJ, Agudelo LZ, Dyck L, Kane H, et al. Distinct metabolic programs established in the thymus control effector functions of γδ T cell subsets in tumor microenvironments. Nat Immunol. 2021;22:179–92.
Roelofs AJ, Jauhiainen M, Mönkkönen H, Rogers MJ, Mönkkönen J, Thompson K. Peripheral blood monocytes are responsible for gammadelta T cell activation induced by zoledronic acid through accumulation of IPP/DMAPP. Br J Haematol. 2009;144:245–50.
Constant P, Davodeau F, Peyrat MA, Poquet Y, Puzo G, Bonneville M, Fournié JJ. Stimulation of human gamma delta T cells by nonpeptidic mycobacterial ligands. Science. 1994;264:267–70.
Tanaka Y, Morita CT, Tanaka Y, Nieves E, Brenner MB, Bloom BR. Natural and synthetic non-peptide antigens recognized by human gamma delta T cells. Nature. 1995;375:155–8.
Scotet E, Martinez LO, Grant E, Barbaras R, Jenö P, Guiraud M, Monsarrat B, Saulquin X, Maillet S, Estève JP, et al. Tumor recognition following Vgamma9Vdelta2 T cell receptor interactions with a surface F1-ATPase-related structure and apolipoprotein A-I. Immunity. 2005;22:71–80.
Rodrigues NV, Correia DV, Mensurado S, Nóbrega-Pereira S, deBarros A, Kyle-Cezar F, Tutt A, Hayday AC, Norell H, Silva-Santos B, Dias S. Low-Density Lipoprotein Uptake Inhibits the Activation and Antitumor Functions of Human Vγ9Vδ2 T Cells. Cancer Immunol Res. 2018;6:448–57.
Wang X, Lin X, Zheng Z, Lu B, Wang J, Tan AH, Zhao M, Loh JT, Ng SW, Chen Q, et al: Host-derived lipids orchestrate pulmonary γδ T cell response to provide early protection against influenza virus infection. Nat Commun 2021, 12:1914.
Godfrey DI, MacDonald HR, Kronenberg M, Smyth MJ, Van Kaer L. NKT cells: what’s in a name? Nat Rev Immunol. 2004;4:231–7.
Cohen NR, Garg S, Brenner MB. Antigen Presentation by CD1 Lipids, T Cells, and NKT Cells in Microbial Immunity. Adv Immunol. 2009;102:1–94.
Berzofsky JA, Terabe M. NKT cells in tumor immunity: opposing subsets define a new immunoregulatory axis. J Immunol. 2008;180:3627–35.
Sadegh L, Chen PW, Brown JR, Han Z, Niederkorn JY. NKT cells act through third party bone marrow-derived cells to suppress NK cell activity in the liver and exacerbate hepatic melanoma metastases. Int J Cancer. 2015;137:1085–94.
Dhodapkar MV, Kumar V. Type II NKT Cells and Their Emerging Role in Health and Disease. J Immunol. 2017;198:1015–21.
Terabe M, Berzofsky JA. Tissue-Specific Roles of NKT Cells in Tumor Immunity. Front Immunol. 2018;9:1838.
Renukaradhya GJ, Sriram V, Du W, Gervay-Hague J, Van Kaer L, Brutkiewicz RR. Inhibition of antitumor immunity by invariant natural killer T cells in a T-cell lymphoma model in vivo. Int J Cancer. 2006;118:3045–53.
Yang W, Li H, Mayhew E, Mellon J, Chen PW, Niederkorn JY. NKT cell exacerbation of liver metastases arising from melanomas transplanted into either the eyes or spleens of mice. Invest Ophthalmol Vis Sci. 2011;52:3094–102.
Dowhan W, Bogdanov M. Lipid-protein interactions as determinants of membrane protein structure and function. Biochem Soc Trans. 2011;39:767–74.
Escribá PV, González-Ros JM, Goñi FM, Kinnunen PK, Vigh L, Sánchez-Magraner L, Fernández AM, Busquets X, Horváth I, Barceló-Coblijn G. Membranes: a meeting point for lipids, proteins and therapies. J Cell Mol Med. 2008;12:829–75.
Kuhajda FP, Jenner K, Wood FD, Hennigar RA, Jacobs LB, Dick JD, Pasternack GR. Fatty acid synthesis: a potential selective target for antineoplastic therapy. Proc Natl Acad Sci U S A. 1994;91:6379–83.
Medes G, Thomas A, Weinhouse S. Metabolism of neoplastic tissue. IV. A study of lipid synthesis in neoplastic tissue slices in vitro. Cancer Res. 1953;13:27–9.
Tiwary S, Berzofsky JA, Terabe M. Altered Lipid Tumor Environment and Its Potential Effects on NKT Cell Function in Tumor Immunity. Front Immunol. 2019;10:2187.
Zeyda M, Säemann MD, Stuhlmeier KM, Mascher DG, Nowotny PN, Zlabinger GJ, Waldhäusl W, Stulnig TM. Polyunsaturated fatty acids block dendritic cell activation and function independently of NF-kappaB activation. J Biol Chem. 2005;280:14293–301.
Porubsky S, Speak AO, Salio M, Jennemann R, Bonrouhi M, Zafarulla R, Singh Y, Dyson J, Luckow B, Lehuen A, et al. Globosides but not isoglobosides can impact the development of invariant NKT cells and their interaction with dendritic cells. J Immunol. 2012;189:3007–17.
Schümann J, Facciotti F, Panza L, Michieletti M, Compostella F, Collmann A, Mori L, De Libero G. Differential alteration of lipid antigen presentation to NKT cells due to imbalances in lipid metabolism. Eur J Immunol. 2007;37:1431–41.
Wu L, Parekh VV, Gabriel CL, Bracy DP, Marks-Shulman PA, Tamboli RA, Kim S, Mendez-Fernandez YV, Besra GS, Lomenick JP, et al. Activation of invariant natural killer T cells by lipid excess promotes tissue inflammation, insulin resistance, and hepatic steatosis in obese mice. Proc Natl Acad Sci U S A. 2012;109:E1143–52.
Satoh M, Hoshino M, Fujita K, Iizuka M, Fujii S, Clingan CS, Van Kaer L, Iwabuchi K. Adipocyte-specific CD1d-deficiency mitigates diet-induced obesity and insulin resistance in mice. Sci Rep. 2016;6:28473.
Michelet X, Dyck L, Hogan A, Loftus RM, Duquette D, Wei K, Beyaz S, Tavakkoli A, Foley C, Donnelly R, et al. Metabolic reprogramming of natural killer cells in obesity limits antitumor responses. Nat Immunol. 2018;19:1330–40.
Ma C, Kesarwala AH, Eggert T, Medina-Echeverz J, Kleiner DE, ** P, Stroncek DF, Terabe M, Kapoor V, ElGindi M, et al. NAFLD causes selective CD4(+) T lymphocyte loss and promotes hepatocarcinogenesis. Nature. 2016;531:253–7.
Acknowledgements
We thank LetPub (www.letpub.com) for its linguistic assistance during the preparation of this manuscript.
Funding
This work was supported by The Zhejiang Provincial Natural Science Foundation of China (LQ21H160009), National Natural Science Foundation of China (82103334, 81901625), Science and Technology Program of Zhejiang Province (LGF19H290002), Zhejiang Medical and Health Science and Technology Plan (2021KY409) and Youth Medical Science and Technology Innovation Project of Xuzhou Municipal Health Commission, China (XWKYHT20200047).
Author information
Authors and Affiliations
Contributions
Zejun Fang and Huihui Xu contributed to the study conception and design. Wanshuang Lou and Chaoju Gong wrote the main manuscript text and prepared the figure. Zhuoni Ye wrote the revised manuscript. Yanyan Hu and Min**g Zhu provided advice regarding the manuscript. All authors have seen and approved the final version of the manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Wanshuang Lou, Chaoju Gong the authors contributed equally to this work
Electronic supplementary material
Below is the link to the electronic supplementary material.

Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

{kind=link}
Cite this article
Lou, W., Gong, C., Ye, Z. et al. Lipid metabolic features of T cells in the Tumor Microenvironment. Lipids Health Dis 21, 94 (2022). https://doi.org/10.1186/s12944-022-01705-y
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12944-022-01705-y