
Abstract
Background
Up to 80% of cases of prostate cancer present with multifocal independent tumour lesions leading to the concept of a field effect present in the normal prostate predisposing to cancer development. In the present study we applied Whole Genome DNA Sequencing (WGS) to a group of morphologically normal tissue (n = 51), including benign prostatic hyperplasia (BPH) and non-BPH samples, from men with and men without prostate cancer. We assess whether the observed genetic changes in morphologically normal tissue are linked to the development of cancer in the prostate.
Results
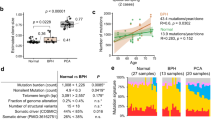
Single nucleotide variants (P = 7.0 × 10–03, Wilcoxon rank sum test) and small insertions and deletions (indels, P = 8.7 × 10–06) were significantly higher in morphologically normal samples, including BPH, from men with prostate cancer compared to those without. The presence of subclonal expansions under selective pressure, supported by a high level of mutations, were significantly associated with samples from men with prostate cancer (P = 0.035, Fisher exact test). The clonal cell fraction of normal clones was always higher than the proportion of the prostate estimated as epithelial (P = 5.94 × 10–05, paired Wilcoxon signed rank test) which, along with analysis of primary fibroblasts prepared from BPH specimens, suggests a stromal origin. Constructed phylogenies revealed lineages associated with benign tissue that were completely distinct from adjacent tumour clones, but a common lineage between BPH and non-BPH morphologically normal tissues was often observed. Compared to tumours, normal samples have significantly less single nucleotide variants (P = 3.72 × 10–09, paired Wilcoxon signed rank test), have very few rearrangements and a complete lack of copy number alterations.
Conclusions
Cells within regions of morphologically normal tissue (both BPH and non-BPH) can expand under selective pressure by mechanisms that are distinct from those occurring in adjacent cancer, but that are allied to the presence of cancer. Expansions, which are probably stromal in origin, are characterised by lack of recurrent driver mutations, by almost complete absence of structural variants/copy number alterations, and mutational processes similar to malignant tissue. Our findings have implications for treatment (focal therapy) and early detection approaches.
Similar content being viewed by others

Background
Prostate cancer is a multifocal, highly heterogeneous disease [1, 2] that is the most common cancer diagnosed in men in the world, with an estimated 50% of men over 60 having cancer present in the prostate [3]. The phenomenon of field cancerization was first described by Slaughter et al. [4] after observing the presence of multiple independent tumours in 11% of patients with oral squamous cell carcinomas. It was proposed that the areas surrounding these lesions were acting as a “field”, a preconditioned epithelium that could lead to cancer development. This theory suggests that tissue with a histomorphologically normal appearance can harbour a significant burden of mutations, early clonal expansions, distinct expression profiles and methylation changes that could potentially lead to tumour development. Numerous reports of somatic mutations and clonal expansions in aging individuals are in agreement with this theory [5,6,7,8]: there is clear evidence that somatic mutations are present in morphologically normal skin [5, 9], brain [10], liver [11], oesophagus [6, 12], and colorectum [13] – in some cases affecting cancer-associated driver genes. Comparable findings have been reported in blood, where the detection of clonal expansions in healthy patients over 65 has been associated with a significant increase in the risk of leukemia [14,15,3A). We observe that mutation clusters in normal tissue are all subclonal (Additional file 3), with a shared N1/N2 subclone in case 0007, two subclones (N1 and N3) in case 0006, and one clone in N2 in case 0008. These results show that multiple clonal expansions of morphologically normal cells are present in the prostate of some men with prostate cancer. There is no shared trunk between tumour clones and normal clones, indicating that they arise independently.

Phylogenies of patients with multiple samples. Phylogenies revealing the relationships between clones for each case. A patients where we have collected multiple tumours and normal. B patients where there was data from a tumour, non-BPH normal tissue, and BPH normal tissue. Each coloured line represents a clone/subclone detected in a particular sample. When two or more coloured lines are together, they represent a clone that is found in all the samples represented. The length of the line is proportional to the weighted number of single nucleotide variants present in each clone; the thickness represents the clonal cell fraction associated with that clone (more detail in Additional file 3). For example, case 0077 contains a shared subclone with 8% N, 33% BPH and 2% T (Tb) supported by 113 SNVs and 4 indels. Dotted lines are associated with samples that have no evidence of a unique sample specific clone. The very low fraction tumour subclone (< 4%) shared with normal and BPH tissue in case 0077 and between normal and tumour in case 0072 suggests cancer targeted tissue contained some of the N/BPH cells. Additional phylogenies can be found in Supplementary Fig. 3
BPH and non-BPH morphologically normal tissue taken from the same prostate shared a subclone in all three cases examined (0065, 0073 and 0077, Fig. 3B). Generally, mutations present in morphologically normal tissue (BPH or non-BPH) and cancer were distinct but in case 0077 a subclone was observed with 2% contribution in the tumour sample, 8% in the morphologically normal sample and 33% in the BPH sample, consistent with a model in which the tumour sample contains a small proportion of the non-BPH/BPH subclone.
In the remaining 11 patients, where morphologically normal (either BPH or non-BPH) and tumour samples were taken, two patterns were present. The first pattern (Cases 0066, 0074, 0115, 0149 Supplementary Fig. 3) was characterised by separate cancer and non-BPH morphologically normal lineage. In the second pattern (Cases 0072, 0076, 0120, 0146, 0156, 0159, 0162) there was evidence of a subclone found in the normal cells also being present in the cancer sample at a low CCF (< 13%, median of 3, IQR of 2; Additional file 3). The minimum distance between cancer and normal samples for the prostates with independent lineages (median of 19 mm; IQR = 9) was on average larger than prostates where the cancer samples had a normal clone present (median 7.1 mm; IQR = 5) (Additional file 1), but this was not statistically significant (P = 0.18, Wilcoxon rank sum test).
In patients with at least one clonal expansion under selective pressure the association between the number of clones and the minimum proximity of the normal samples to the matched tumour was not statistically significant (P = 0.307, Wilcoxon rank sum test). Similarly, there was no evidence of an association between the matched tumour being multifocal and the presence of at least one clonal expansion (P = 0.79, Wilcoxon rank sum test).
Mutational signatures
Mutational signatures were inferred for each sample using SigProfiler [41] using the set of signatures defined by Alexandrov et al. [47] (Additional file 6). The cosine similarity between the reference signatures and the reconstructed profiles was high for all samples but higher in tumour compared to normal samples (median of 0.97 for tumour vs 0.88 normal), likely the result of a lower number of SNVs in normal tissues. Mutational signatures 1, 5, 8, 18 and 40 were detected both in tumour and in morphologically normal tissue/BPH samples (Fig. 4). All of these signatures have been previously been identified in prostate cancer samples [47]. Signature 1 was overrepresented in tumour samples (P = 4.89 × 10–03, Fisher’s exact test). This signature is thought to result from an endogenous mutational process started by the deamination of 5-methylcytosine and has been associated with ageing. Because of this we would expect a similar representation of this signature in both normal and tumour samples. The aetiologies of signatures 5, 8 and signature 40 are unknown [47]. Three signatures (3, 4, and 28) were unique to morphologically normal tissue. Signatures 4 and 28 were present in only one sample, whereas signature 3 is present in 10 samples. Signature 3 has been linked with defective homologous recombination-based repair, Signature 4 has been associated with tobacco smoking and the aetiology of signature 28 is unknown. There were no differences between non-BPH morphologically normal tissue and BPH.

Mutational spectra. Mutational signatures detected in tumour and matched morphologically normal tissue from prostate cancer patients and normal tissue from men without prostate cancer. The mutational spectra of each sample, as defined by the triplets of nucleotides around each SNV, were deconvoluted into mutational signatures (SigProfiler [41]) using the set of signatures defined by Alexandrov et al. [47]. The colour of the first row indicates patient when there is more than a normal-tumour (N-T) pair analysed. Six patients had more than two samples analysed and one patient had only a morphologically normal sample without a matched tumour
Gene mutations with functional impact
In morphologically normal, fibroblasts and BPH samples a total of 281 SNVs and indel mutations were observed in coding regions of 165 genes. 110 of the 281 mutations show a potential functional impact according to wANNOVAR [48] and eight of these occurred in known cancer-related genes (PPARG, BRCA1, GATA1, ACR, WHSC1, FAT1, POLE and HOXD11) as reported in the cancer gene census [49] (Additional file 7). Of these, mutations in GATA1, WHSC1, ACR, and POLE were observed in at least one sample from a primary prostate fibroblast culture (WHSC1 and ACR occurring in the same sample). Mutations with predicted functional impact were observed in 11 genes that are designated prognostic markers of poor outcome in The Cancer Genome Atlas Research Network (TCGA) RNAseq dataset [50, 51]: FAT1, SOBP, CTHRC1, IQGAP1, FOXJ3, ATP1A3, PHF12, BCAT1, GMPR2, ADAM28, DHX32, DSG3, DDX19A, KIAA1217, PPARG, PTK2B, RPL18, DONSON, CHPF2 and XKRX. All apart from 4 of the 110 mutations were detected in a single sample: mutations affecting genes GYPA and NACAD were present in multiple samples from different patients, and mutations in genes BCAT1 and FAT2 were present in two samples from the same patient (Additional file 7). Of all the genes identified, only BRCA2 and ADAM28 have been previously classified as recurrently mutated drivers in prostate cancer [52, 53]. A previously described dN/dS driver detection method [5] was performed but no significant hits were found, possible due to the limited number of mutations and samples. From the 110 genes with a predicted functional impact, 13 were also observed to be mutated in at least one tumour sample (Additional file 7). However, there was only one instance where a potentially functionally important mutation occurred in both a normal sample and the matched tumour from the same patient (gene ACOT1 in patient 0122).
We conclude that some of the observed mutations had the potential to generate driver genes but there was an absence of evidence for recurrent mutations in cancer driver genes.
Comparison with tumours
When comparing the morphologically normal samples to their respective tumours, both the number of SNVs (median 421 vs 2560.5) and structural rearrangements (median 0 vs 40) was significantly higher in tumours (P = 3.73 × 10–09, P = 2.70 × 10–06, respectively, paired Wilcoxon signed rank test; Fig. 5A; Additional file 1). In total 17,370 indels (median of 445) were identified in morphologically normal samples whereas tumour samples harboured 11,087 indels (median of 265). The absence of copy number alterations is a notable characteristic of the normal samples, and the number is significantly less than in cancer tissue (median of 42 for cancer vs 0 for morphologically normal, P = 2.68 × 10–06, paired Wilcoxon signed rank test).

Tumours show a distinct mutation profile to normal tissue. A The difference between the number of single nucleotide variants (SNVs) detected in normal tissue compared to tumour tissue. Where multiple samples of either type were present the median number was used. B The distribution of the number of SNVs detected in morphologically normal tissue, tumour tissue with low CNAs (percentage genome altered (PGA) < 6%) and tumour tissue with high CNAs (PGA > 6%). Data from these last two categories came from Wedge et al. [52]
We analysed a total of 91 of the 112 tumours examined by Wedge et al. [52] (removing the metastatic samples; Additional file 8). A group of 23 samples with less than 6% of the genome affected by copy number alterations were identified as “quiet tumours” (Supplementary Table 3). The numbers of SNVs (median = 2250 vs 2796) and structural rearrangements (median = 32 vs 56) were significantly lower in the quiet tumours than their high CNAs counterparts (P = 7.59 × 10–04 and P = 5.27 × 10–03, respectively, Wilcoxon rank sum test). The number of SNVs was significantly higher in “quiet tumours” when compared to samples from morphologically normal tissue (P = 1.88 × 10–10, Wilcoxon rank sum test, median = 421 vs 2250; Fig. 5B).
Discussion
Our study demonstrates several critical and recurrent features of the mutations present in non-neoplastic (BPH and non-BPH) tissue taken from cancerous prostates. Primarily, morphologically normal tissue from patients with prostate cancer had a high number of single nucleotide variants (SNVs) and indels and generally a clonal expansion under selective pressure was present. This contrasted with samples from prostates lacking cancer which had a significantly lower number of mutations and a lack of clonal expansions under selective pressure. Our results indicate that the presence of the clonal expansions in non-neoplastic tissue is a feature associated with development of cancer, a finding previously reported in leukemia [14,15,16,17].
We also show that there is evidence that clonal expansions from non-neoplastic tissue originates from stromal cells. This is highlighted by the finding that the clonal cell fraction of clonal expansions of morphologically normal tissue was always higher than the proportion of the prostate estimated as epithelial. This is supported by the relationship we observe between non-BPH and BPH normal tissue, with BPH in some cases thought to be associated with hyper-proliferation of stromal tissue [54] (although we found no evidence of an association between stromal content and mutation burden). Firstly, our constructed phylogenies reveal non-BPH morphologically normal and BPH samples within the same prostate can have a shared lineage. Secondly, high mutation rates were observed in five primary cell cultures of stromal cells prepared from BPH specimens; four of the cultures exhibiting evidence of selective clonal expansion; and three samples containing potential driver genes. Thirdly, higher mutation rates were observed in stroma-dominated BPH compared to non-BPH morphologically normal tissue. Finally, the cystoprostatectomy sample 0239 – which exhibited BPH – had the highest number of SNVs observed in non-cancer patients and had evidence of a clonal expansion under selection. The importance of stroma in prostate differentiation was established in mouse studies by Cunha et al. [55]. These studies have been extended into human cells [56,57,58] and Maitland et al. have studied prostate stromal influences for more than 20 years, exploiting primary clinical material and cultured cells [59, 60]. Foster et al. have reported clonal expansions in cancer-associated fibroblasts (CAFs) [61] and shown that stromal cells from BPH, unlike stromal cells from normal prostatic tissue, have capability of inducing growth of prostatic epithelia in vivo. Taken together, these findings indicate a model for cancer development wherein the presence of clonal expansions of stromal cells supports cancer development and contributes to the field effect. This theory is in agreement with previous reports of an association between BPH and prostate cancer [62,63,64], although a causal link has not previously been established. If this model is correct, it cannot exclude a role for stroma in non-BPH normal tissue since prostates without BPH also exhibit multifocal disease. Examining the estimated cellular composition of the stroma, derived from single cell sequencing data, in both PC and non-PC donors would further elucidate the differences we observe.
We found only very limited evidence that in normal tissue known genetic drivers were affected by mutations with potential functional impact – only PPARG, BRCA1, GATA1, HOXD11, WHSC1, FAT1 and POLE were identified. These genes have been associated with tumour suppression (BRCA1 and FAT1) [65, 66], DNA repair (POLE) [67], morphogenesis (HOXD11), epigenetic regulation (WHSC1) [68], lipid metabolism (PPARG) [69] and red blood cell development (GATA1) [70]. They have been previously linked with leukemia [71, 72], breast [73,74,75,76], bladder [73, 77] colon [78, 79], kidney [80], endometrial [81], head and neck carcinoma [82,83,84], pancreatic [73] and prostate [85,86,87] cancers. The low detection of mutations in potential driver genes agrees with a cross tissue study performed by Moore et al. in participants without detected cancer [7] and raises the possible importance of epigenetic alterations in driving clonal expansion. This involvement of epigenetic changes is supported by the reported high hypermethylation levels in genes such as APC, GTSP1 and RASSF1 in morphologically normal tissue in the prostate [88,89,90,91], that have also been shown to be good predictors of cancer development [88,89,90]. For example, hypermethylation in genes APC and GTSP1 was reported in 95% and 43% respectively in patients with an initial negative biopsy that later developed prostate cancer [88].
Clonal expansions identified in non-neoplastic tissue have a distinct unrelated pattern to those in malignant tissue but are driven by the same processes. Known prostate cancer associated mutational signatures [47, 92] were present in both morphologically normal and tumour tissue, suggesting that the same mutational processes are driving the clonal expansions in both cases. This is consistent with our own study in a smaller dataset [22] and studies at other cancer sites [93, 94]. Despite this, our constructed phylogenies reveal that clones in morphologically normal samples are of a distinct lineage from those in the tumour and their mutational characteristics are different: normal samples have significantly fewer SNVs, have very few rearrangements and a complete lack of copy number alterations. We observed this difference both with samples from the same prostate in this study and in comparison with “quiet” tumours studied by Wedge et al. [52]. Copy number alterations are an important driving feature of prostate cancer and copy number burden has been associated with a poor prognosis [95,96,97]. Homologous recombination, non-allelic homologous recombination, non-homologous end joining and microhomology-mediated break-induced replication are double stranded break (DSB) repair mechanisms that could result in CNAs, rearrangements and hypermutation [98]. The absence of these three types of genetic alterations in normal samples suggest that this type of DNA damage by DSB and errors in the repairing mechanisms (or both) occur at a lower rate in normal samples and supports the potential increase of replication errors and non-DSB DNA damage produced by endogenous or exogenous environmental factors.
In summary, these results provide further evidence that the whole prostate environment, in particular stromal cells, are involved in the development of prostate cancer and insights into potential genomic evolution mechanisms at very early stages of development. Our findings have implications for treatment (focal therapy) and early detection approaches.
Availability of data and materials
The datasets generated during the current study are available in the European Genome-Phenome Archive repository, https://ega-archive.org/datasets/EGAD00001000689 and https://ega-archive.org/datasets/EGAD00001004125. The variant calls generated are available from the corresponding author on reasonable request.
Abbreviations
- BPH:
-
Benign prostatic hyperplasia
- BWA-SW:
-
Burrows-Wheeler Aligner’s Smith-Waterman Alignment
- CAFs:
-
Cancer-associated fibroblasts
- CCF:
-
Clonal Cell Fraction
- CNAs:
-
Copy number alterations
- IQR:
-
Inter-quartile range
- Indels:
-
Insertions and deletions
- SNVs:
-
Single nucleotide variants
- TCGA:
-
The Cancer Genome Atlas Research Network
- WGS:
-
Whole Genome DNA Sequencing
References
Andreoiu M, Cheng L. Multifocal prostate cancer: biologic, prognostic, and therapeutic implications. Hum Pathol. 2010;41:781–93. Available from: http://linkinghub.elsevier.com/retrieve/pii/S0046817710001085.
Nonn L, Ananthanarayanan V, Gann PH. Evidence for field cancerization of the prostate. Prostate. 2009;69:1470–9. Available from: https://doi.org/10.1002/pros.20983 Wiley-Blackwell[cited 2015 Aug 31].
Zlotta AR, Egawa S, Pushkar D, Govorov A, Kimura T, Kido M, et al. Prevalence of prostate cancer on autopsy: cross-sectional study on unscreened Caucasian and Asian men. J Natl Cancer Inst. 2013;105:1050–8. Available from: http://www.ncbi.nlm.nih.gov/pubmed/23847245.
Slaughter DP, Southwick HW, Smejak W. Field cancerization in oral stratified squamous epithelium; clinical implications of multicentric origin. Cancer. 1953;6:963–8. Available from: http://www.ncbi.nlm.nih.gov/pubmed/13094644 [cited 2015 Jan 16].
Martincorena I, Roshan A, Gerstung M, Ellis P, Van Loo P, McLaren S, et al. High burden and pervasive positive selection of somatic mutations in normal human skin. Science (80-). 2015;348:880–6.
Martincorena I, Fowler JC, Wabik A, Lawson ARJ, Abascal F, Hall MWJ, et al. Somatic mutant clones colonize the human esophagus with age. Science (80-). 2018;3879:e3879. Available from: https://doi.org/10.1126/science.aau3879.
Moore L, Cagan A, Coorens THH, Neville MDC, Sanghvi R, Sanders MA, et al. The mutational landscape of human somatic and germline cells. Nature. 2021;597(7876):381–6.
Li R, Di L, Li J, Fan W, Liu Y, Guo W, et al. A body map of somatic mutagenesis in morphologically normal human tissues. Nature. Springer US; 2021; Available from: https://doi.org/10.1038/s41586-021-03836-1
Tang J, Fewings E, Chang D, Zeng H, Liu S, Jorapur A, et al. The genomic landscapes of individual melanocytes from human skin. Nature. 2020;586:600–5. Available from: https://www.nature.com/articles/s41586-020-2785-8 5867830. Nature Publishing Group; 2020 [cited 2021 Nov 30].
Lodato MA, Rodin RE, Bohrson CL, Coulter ME, Barton AR, Kwon M, et al. Aging and neurodegeneration are associated with increased mutations in single human neurons. Science (80-). 2018;359:555–9. Available fromhttps://doi.org/10.1126/science.aao4426 American Association for the Advancement of Science; [cited 2021 Nov 30].
Brunner SF, Roberts ND, Wylie LA, Moore L, Aitken SJ, Davies SE, et al. Somatic mutations and clonal dynamics in healthy and cirrhotic human liver. Nature. 2019;574:538–42. Available from: https://www.nature.com/articles/s41586-019-1670-9 5747779. Nature Publishing Group; 2019 [cited 2021 Nov 30].
Yokoyama A, Kakiuchi N, Yoshizato T, Nannya Y, Suzuki H, Takeuchi Y, et al. Age-related remodelling of oesophageal epithelia by mutated cancer drivers. Nature. 2019;565:312–7. Available from: https://www.nature.com/articles/s41586-018-0811-x 5657739. Nature Publishing Group; 2019 [cited 2021 Nov 30].
Lee-Six H, Olafsson S, Ellis P, Osborne RJ, Sanders MA, Moore L, et al. The landscape of somatic mutation in normal colorectal epithelial cells. Nature. 2019;574:532–7.
Jaiswal S, Fontanillas P, Flannick J, Manning A, Grauman PV, Mar BG, et al. Age-Related Clonal Hematopoiesis Associated with Adverse Outcomes. N Engl J Med. 2014;371:2488–98. Available from: https://doi.org/10.1056/NEJMoa1408617 .
Genovese G, Kähler AK, Handsaker RE, Lindberg J, Rose SA, Bakhoum SF, et al. Clonal Hematopoiesis and Blood-Cancer Risk Inferred from Blood DNA Sequence. N Engl J Med. 2014;371:2477–87 (Massachusetts Medical Society).
**e M, Lu C, Wang J, McLellan MD, Johnson KJ, Wendl MC, et al. Age-related mutations associated with clonal hematopoietic expansion and malignancies. Nat Med. 2014;20:1472–8 (2014/10/19).
Zink F, Stacey SN, Norddahl GL, Frigge ML, Magnusson OT, Jonsdottir I, et al. Clonal hematopoiesis, with and without candidate driver mutations, is common in the elderly. Blood. 2017;130:742–52 (American Society of Hematology).
Yizhak K, Aguet F, Kim J, Hess J, Kubler K, Grimsby J, et al. A comprehensive analysis of RNA sequences reveals macroscopic somatic clonal expansion across normal tissues. 2018;416339. Available from: https://www.biorxiv.org/content/early/2018/09/13/416339.
Colom B, Herms A, Dentro S, King C, Sood R, Alcolea M, et al. Precancer: Mutant clones in normal epithelium outcompete and eliminate esophageal micro-tumors. bioRxiv. 2021;2021.06.25.449880. Available from: http://biorxiv.org/content/early/2021/06/25/2021.06.25.449880.abstract
Shoag J, Barbieri CE. Clinical variability and molecular heterogeneity in prostate cancer. Asian J Androl. 2016;18:543–8.
Svensson M a, LaFargue CJ, MacDonald TY, Pflueger D, Kitabayashi N, Santa-Cruz AM, et al. Testing mutual exclusivity of ETS rearranged prostate cancer. Lab Invest. 2011;91:404–12 (Nature Publishing Group).
Cooper CS, Eeles R, Wedge DC, Van Loo P, Gundem G, Alexandrov LB, et al. Analysis of the genetic phylogeny of multifocal prostate cancer identifies multiple independent clonal expansions in neoplastic and morphologically normal prostate tissue. Nat Genet. 2015;47:367–72. https://doi.org/10.1038/ng.3221 (Nature Publishing Group, a division of Macmillan Publishers Limited. All Rights Reserved).
Parr RL, Dakubo GD, Crandall KA, Maki J, Reguly B, Aguirre A, et al. Somatic mitochondrial DNA mutations in prostate cancer and normal appearing adjacent glands in comparison to age-matched prostate samples without malignant histology. J Mol Diagn. 2006;8:312–9 (American Society for Investigative Pathology).
Grossmann S, Hooks Y, Wilson L, Moore L, O’Neill L, Martincorena I, et al. Development, maturation, and maintenance of human prostate inferred from somatic mutations. Cell Stem Cell. 2021;28:1262-1274.e5.
Chandran UR, Dhir R, Ma C, Michalopoulos G, Becich M, Gilbertson J. Differences in gene expression in prostate cancer, normal appearing prostate tissue adjacent to cancer and prostate tissue from cancer free organ donors. BMC Cancer. 2005;5:45 (BioMed Central).
Yu YP, Landsittel D, **g L, Nelson J, Ren B, Liu L, et al. Gene Expression Alterations in Prostate Cancer Predicting Tumor Aggression and Preceding Development of Malignancy. J Clin Oncol. 2004;22:2790–9 (American Society of Clinical Oncology).
Luo JH, Ding Y, Chen R, Michalopoulos G, Nelson J, Tseng G, et al. Genome-wide methylation analysis of prostate tissues reveals global methylation patterns of prostate cancer. Am J Pathol. 2013;182:2028–36 (American Society for Investigative Pathology).
Yang B, Bhusari S, Kueck J, Weeratunga P, Wagner J, Leverson G, et al. Methylation profiling defines an extensive field defect in histologically normal prostate tissues associated with prostate cancer. Neoplasia (United States). 2013;15:399–408.
Warren AY, Whitaker HC, Haynes B, Sangan T, Mcduffus L, Kay JD, et al. Method for SamplingTissue for ResearchWhich Preserves Pathological Data in Radical Prostatectomy. Prostate. 2013;202:194–202.
Cussenot O, Berthon P, Cochand-Priollet B, Maitland NJ, Le Duc A. Immunocytochemical comparison of cultured normal epithelial prostatic cells with prostatic tissue sections. Exp Cell Res. 1994;214:83–92. Available from: http://www.ncbi.nlm.nih.gov/pubmed/8082751.
Pellacani D, Droop AP, Frame FM, Simms MS, Mann VM, Collins AT, et al. Phenotype-independent DNA methylation changes in prostate cancer. Br J Cancer. 2018;119:1133–43. Available from: http://www.ncbi.nlm.nih.gov/pubmed/30318509.
Li H, Durbin R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics. 2010;26:589–95. Available from: http://www.ncbi.nlm.nih.gov/pubmed/20080505.
Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–60 (Oxford University Press).
Jones D, Raine KM, Davies H, Tarpey PS, Butler AP, Teague JW, et al. cgpCaVEManWrapper: Simple Execution of CaVEMan in Order to Detect Somatic Single Nucleotide Variants in NGS Data. Curr Protoc Bioinformatics. 2016;56:15.10.1-15.10.18. Available from: https://doi.org/10.1002/cpbi.20 (Hoboken, NJ, USA: John Wiley & Sons, Inc. [cited 2018 May 29]).
Nik-Zainal S, Alexandrov LB, Wedge DC, Van Loo P, Greenman CD, Raine K, et al. Mutational Processes Molding the Genomes of 21 Breast Cancers. Cell. 2012;149:979–93. Available from: http://www.ncbi.nlm.nih.gov/pubmed/22608084 [cited 2012 Jul 13].
Alioto TS, Buchhalter I, Derdak S, Hutter B, Eldridge MD, Hovig E, et al. A comprehensive assessment of somatic mutation detection in cancer using whole-genome sequencing. Nat Commun. 2015;6:10001. Available from: https://doi.org/10.1038/ncomms10001 Nature Publishing Group.
Raine KM, Hinton J, Butler AP, Teague JW, Davies H, Tarpey P, et al. cgpPindel: Identifying Somatically Acquired Insertion and Deletion Events from Paired End Sequencing. Curr Protoc Bioinformatics. 2015;52:15.7.1-12. Available from: https://doi.org/10.1002/0471250953.bi1507s52 (Hoboken, NJ, USA: John Wiley & Sons, Inc. [cited 2018 May 29]).
Zerbino DR, Birney E. Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 2008;18:821–9. Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=2336801&tool=pmcentrez&rendertype=abstract [cited 2011 Jul 16].
Nik-Zainal S, Van Loo P, Wedge DC, Alexandrov LB, Greenman CD, Lau KW, et al. The life history of 21 breast cancers. Cell. 2012;149:994–1007. Available from: http://linkinghub.elsevier.com/retrieve/pii/S0092867412005272 Elsevier; [cited 2013 Oct 12].
Alexandrov LB, Kim J, Haradhvala NJ, Huang MN, Tian Ng AW, Wu Y, et al. The repertoire of mutational signatures in human cancer. Nature. Nature Publishing Group; 2020;578:94–101. [cited 2022 Sep 8]. Available from: https://www.nature.com/articles/s41586-020-1943-3.
Bergstrom EN, Huang MN, Mahto U, Barnes M, Stratton MR, Rozen SG, et al. SigProfilerMatrixGenerator: a tool for visualizing and exploring patterns of small mutational events. BMC Genomics. 2019;20:1–12. Available from: https://doi.org/10.1186/s12864-019-6041-2 (201 . BioMed Central; 2019 [cited 2021 Aug 13]).
Rosenthal R, McGranahan N, Herrero J, Taylor BS, Swanton C. deconstructSigs: delineating mutational processes in single tumors distinguishes DNA repair deficiencies and patterns of carcinoma evolution. Genome Biol. 2016;17:31 (London: BioMed Central).
Bolli N, Avet-Loiseau H, Wedge DC, Van Loo P, Alexandrov LB, Martincorena I, et al. Heterogeneity of genomic evolution and mutational profiles in multiple myeloma. Nat Commun. 2014;5:2997. Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3905727&tool=pmcentrez&rendertype=abstract Nature Publishing Group; [cited 2014 Jul 10].
Gundem G, Van Loo P, Kremeyer B, Alexandrov LB, Tubio JMC, Papaemmanuil E, et al. The evolutionary history of lethal metastatic prostate cancer. Nature. 2015;520:353–7. Available from: http://www.nature.com/doifinder/10.1038/nature14347.
Williams MJ, Werner B, Barnes CP, Graham TA, Sottoriva A. Identification of neutral tumor evolution across cancer types. Nat Genet. 2016;48:238–44. Available from: https://doi.org/10.1038/ng.3489 (Nature Publishing Group).
Jiao W, Vembu S, Deshwar AG, Stein L, Morris Q. Inferring clonal evolution of tumors from single nucleotide somatic mutations. BMC Bioinformatics. 2014;15:35 (BioMed Central).
Alexandrov LB, Kim J, Haradhvala NJ, Huang MN, Ng AW, Boot A, et al. The Repertoire of Mutational Signatures in Human Cancer. bioRxiv. Cold Spring Harbor Laboratory; 2018 [cited 2019 Sep 30];322859. Available from: https://doi.org/10.1101/322859v1
Chang X, Wang K. wANNOVAR: annotating genetic variants for personal genomes via the web. J Med Genet. 2012;49:433–6.
Sondka Z, Bamford S, Cole CG, Ward SA, Dunham I, Forbes SA. The COSMIC Cancer Gene Census: describing genetic dysfunction across all human cancers. Nat Rev Cancer. 2018;18:696–705.
Network CGAR, Weinstein JN, Collisson EA, Mills GB, Shaw KRM, Ozenberger BA, et al. The Cancer Genome Atlas Pan-Cancer analysis project. Nat Genet. 2013;45:1113–20. Available from: https://doi.org/10.1038/ng.2764 (Nature Research).
Uhlén M, Fagerberg L, Hallström BM, Lindskog C, Oksvold P, Mardinoglu A, et al. Tissue-based map of the human proteome. Science (80- ). American Association for the Advancement of Science; 2015 [cited 2022 Feb 1];347. Available from: https://doi.org/10.1126/science.1260419
Wedge DC, Gundem G, Mitchell T, Woodcock DJ, Martincorena I, Ghori M, et al. Sequencing of prostate cancers identifies new cancer genes, routes of progression and drug targets. Nat Genet. 2018;50:682–92. Available from: http://www.nature.com/articles/s41588-018-0086-z Nature Publishing Group.
Armenia J, Wankowicz SAMM, Liu D, Gao J, Kundra R, Reznik E, et al. The long tail of oncogenic drivers in prostate cancer. Nat Genet. 2018;50:645–51. Available from: http://www.nature.com/articles/s41588-018-0078-z Nature Publishing Group.
Barclay WW, Woodruff RD, Hall MC, Cramer SD. A system for studying epithelial-stromal interactions reveals distinct inductive abilities of stromal cells from benign prostatic hyperplasia and prostate cancer. Endocrinology. 2005;146:13–8. Available from: http://www.ncbi.nlm.nih.gov/pubmed/15471963.
Cunha GR, Lung B. The importance of stroma in morphogenesis and functional activity of urogenital epithelium. In Vitro. 1979;15:50–71. Available from: http://www.ncbi.nlm.nih.gov/pubmed/437808.
Chung LW, Davies R. Prostate epithelial differentiation is dictated by its surrounding stroma. Mol Biol Rep. 1996;23:13–9. Available from: http://www.ncbi.nlm.nih.gov/pubmed/8983015.
Cunha GR, Hayward SW, Wang YZ, Ricke WA. Role of the stromal microenvironment in carcinogenesis of the prostate. Int J Cancer. 2003;107:1–10. Available from: https://doi.org/10.1002/ijc.11335.
Zhao H, Peehl DM. Tumor-promoting phenotype of CD90 hi prostate cancer-associated fibroblasts. Prostate. 2009;69:991–1000. Available from: https://doi.org/10.1002/pros.20946.
Lang SH, Stark M, Collins A, Paul AB, Stower MJ, Maitland NJ. Experimental prostate epithelial morphogenesis in response to stroma and three-dimensional matrigel culture. Cell Growth Differ. 2001;12:631–40. Available from: http://www.ncbi.nlm.nih.gov/pubmed/1175145.
Hall JA, Maitland NJ, Stower M, Lang SH. Primary prostate stromal cells modulate the morphology and migration of primary prostate epithelial cells in type 1 collagen gels. Cancer Res. 2002;62:58–62. Available from: http://www.ncbi.nlm.nih.gov/pubmed/11782359.
Foster DS, Ransom RC, Jones RE, Salhotra A, Hu MS, Longaker MT. Abstract 10: Characterizing the Clonal Nature of Cancer Associated Fibroblasts. Plast Reconstr Surg Glob Open. 2018;6:8 (Wolters Kluwer Health).
Ørsted DD, Bojesen SE. The link between benign prostatic hyperplasia and prostate cancer. Nat Rev Urol. 2013;10:49–54 (Nature Publishing Group).
Miah S, Catto J. BPH and prostate cancer risk. Indian J Urol. 2014;30:214–8 (India: Medknow Publications & Media Pvt Ltd).
Chokkalingam AP, Gridley G, Mclaughlin JK, Adami H, et al. Prostate Carcinoma Risk Subsequent to Diagnosis of Benign Prostatic Hyperplasia A Population-Based Cohort Study in Sweden. 2003.
Silver DP, Livingston DM. Mechanisms of BRCA1 tumor suppression. Cancer Discov. 2012;2:679–84 (2012/07/27).
Katoh M. Function and cancer genomics of FAT family genes (review). Int J Oncol. 2012;41:1913–8 (D.A. Spandidos).
Ogi T, Limsirichaikul S, Overmeer R, Volker M, Takenaka K, Cloney R, et al. Three DNA Polymerases, Recruited by Different Mechanisms, Carry Out NER Repair Synthesis in Human Cells. Mol Cell. 2010;37:714–27.
Campos-Sanchez E, Deleyto-Seldas N, Dominguez V, Carrillo-de-Santa-Pau E, Ura K, Rocha PP, et al. Wolf-Hirschhorn Syndrome Candidate 1 Is Necessary for Correct Hematopoietic and B Cell Development. Cell Rep. 2017;19:1586–601.
Lefterova MI, Zhang Y, Steger DJ, Schupp M, Schug J, Cristancho A, et al. PPARgamma and C/EBP factors orchestrate adipocyte biology via adjacent binding on a genome-wide scale. Genes Dev. 2008;22:2941–52 (Cold Spring Harbor Laboratory Press).
Ferreira R, Ohneda K, Yamamoto M, Philipsen S. GATA1 function, a paradigm for transcription factors in hematopoiesis. Mol Cell Biol. 2005;25:1215–27 (American Society for Microbiology).
Shimizu R, Engel JD, Yamamoto M. GATA1-related leukaemias. Nat Rev Cancer. 2008;8:279–87.
Swaroop A, Oyer JA, Will CM, Huang X, Yu W, Troche C, et al. An activating mutation of the NSD2 histone methyltransferase drives oncogenic reprogramming in acute lymphocytic leukemia. Oncogene. 2019;38:671–86.
Consortium APG. AACR Project GENIE: Powering Precision Medicine through an International Consortium. Cancer Discov. 2017;7:818–31 (2017/06/01).
Rosen EM, Fan S, Pestell RG, Goldberg ID. BRCA1 gene in breast cancer. J Cell Physiol. 2003;196:19–41.
Cao XH, Lv JX, Wei XY, Ltuamba EDGL, Hu HL, Zhang YN. FAT1 expression in different breast lesions and its down-regulation in breast cancer development. Int J Clin Exp Pathol. 2017;10:7242–8.
Yu S, Jiang X, Li J, Li C, Guo M, Ye F, et al. Comprehensive analysis of the GATA transcription factor gene family in breast carcinoma using gene microarrays, online databases and integrated bioinformatics. Sci Rep. 2019;9:4467.
Cazier J-B, Rao SR, McLean CM, Walker AK, Walker AL, Wright BJ, et al. Whole-genome sequencing of bladder cancers reveals somatic CDKN1A mutations and clinicopathological associations with mutation burden. Nat Commun. 2014;5:3756.
Guerra J, Pinto C, Pinto D, Pinheiro M, Silva R, Peixoto A, et al. POLE somatic mutations in advanced colorectal cancer. Cancer Med. 2017;6:2966–71 (2017/10/26. John Wiley and Sons Inc.).
Yu J, Liu M, Liu H, Zhou L. GATA1 promotes colorectal cancer cell proliferation, migration and invasion via activating AKT signaling pathway. Mol Cell Biochem. 2019;457:191–9 (Springer US).
Peters I, Dubrowinskaja N, Tezval H, Kramer MW. Decreased mRNA expression of GATA1 and GATA2 is associated with tumor aggressiveness and poor outcome in clear cell renal cell carcinoma. 2015. p. 267–75.
Imboden S, Nastic D, Ghaderi M, Rydberg F, Rau TT, Mueller MD, et al. Phenotype of POLE-mutated endometrial cancer. PLoS ONE. 2019;14:1–15.
Saloura V, Cho H-S, Kiyotani K, Alachkar H, Zuo Z, Nakakido M, et al. WHSC1 Promotes Oncogenesis through Regulation of NIMA-Related Kinase-7 in Squamous Cell Carcinoma of the Head and Neck. Mol Cancer Res. 2015;13:293 LP – 304.
Lin SC, Lin LH, Yu SY, Kao SY, Chang KW, Cheng HW, et al. FAT1 somatic mutations in head and neck carcinoma are associated with tumor progression and survival. Carcinogenesis. 2018;39:1320–30.
de Barros e Lima Bueno R, Ramão A, Pinheiro DG, Alves CP, Kannen V, Jungbluth AA, et al. HOX genes: potential candidates for the progression of laryngeal squamous cell carcinoma. Tumor Biol. 2016;37:15087–96.
Castro E, Eeles R. The role of BRCA1 and BRCA2 in prostate cancer. Asian J Androl. 2012;14:409–14 (2012/04/23. Nature Publishing Group).
Li N, Xue W, Yuan H, Dong B, Ding Y, Liu Y, et al. AKT-mediated stabilization of histone methyltransferase WHSC1 promotes prostate cancer metastasis. J Clin Invest. 2017;127:1284–302 (2017/03/20. American Society for Clinical Investigation).
Elix C, Pal SK, Jones JO. The role of peroxisome proliferator-activated receptor gamma in prostate cancer. Asian J Androl. 2018;20:238–43 (Medknow Publications & Media Pvt Ltd).
Trock BJ, Brotzman MJ, Mangold LA, Bigley JW, Epstein JI, McLeod D, et al. Evaluation of GSTP1 and APC methylation as indicators for repeat biopsy in a high-risk cohort of men with negative initial prostate biopsies. BJU Int. 2012;110:56–62 (2011/11/11).
Partin AW, Van Neste L, Klein EA, Marks LS, Gee JR, Troyer DA, et al. Clinical validation of an epigenetic assay to predict negative histopathological results in repeat prostate biopsies. J Urol. 2014;192:1081–7 (2014/04/18).
Stewart GD, Leander VN, Philippe D, Paul D, Agnès D, Alan MS, et al. Clinical Utility of an Epigenetic Assay to Detect Occult Prostate Cancer in Histopathologically Negative Biopsies: Results of the MATLOC Study. J Urol. 2013;189:1110–6 (AUA Elsevier (Legacy Content)).
Henrique R, Jerónimo C, Teixeira MR, Hoque MO, Carvalho AL, Pais I, et al. Epigenetic Heterogeneity of High-Grade Prostatic Intraepithelial Neoplasia: Clues for Clonal Progression in Prostate Carcinogenesis. Mol Cancer Res. 2006;4:1 LP – 8.
Alexandrov LB, Kim J, Haradhvala NJ, Huang MN, Tian Ng AW, Wu Y, et al. The repertoire of mutational signatures in human cancer. Nature. 2020;578:94–101.
Blokzijl F, de Ligt J, Jager M, Sasselli V, Roerink S, Sasaki N, et al. Tissue-specific mutation accumulation in human adult stem cells during life. Nature. 2016;538:260 (Macmillan Publishers Limited, part of Springer Nature. All rights reserved).
Ju YS, Martincorena I, Gerstung M, Petljak M, Alexandrov LB, Rahbari R, et al. dynamics in the early human embryo. Nat Publ Gr. 2017;543:714–8 (Nature Publishing Group).
Camacho N, Van Loo P, Edwards S, Kay JD, Matthews L, Haase K, et al. Appraising the relevance of DNA copy number loss and gain in prostate cancer using whole genome DNA sequence data. PLoS Genet. 2017;13:e1007001. Available from: https://doi.org/10.1371/journal.pgen.1007001 (Beroukhim R, editor. Public Library of Science).
Taylor BS, Schultz N, Hieronymus H, Gopalan A, **ao Y, Carver BS, et al. Integrative Genomic Profiling of Human Prostate Cancer. Cancer Cell. 2010;18:11–22. Available from: http://www.ncbi.nlm.nih.gov/pubmed/20579941 Elsevier Ltd; [cited 2010 Jul 7].
Hieronymus H, Schultz N, Gopalan A, Carver BS, Chang MT, **ao Y, et al. Copy number alteration burden predicts prostate cancer relapse. Proc Natl Acad Sci U S A. 2014;111:11139–44. Available from: http://www.ncbi.nlm.nih.gov/pubmed/25024180.
Ponder RG, Fonville NC, Rosenberg SM. A switch from high-fidelity to error-prone DNA double-strand break repair underlies stress-induced mutation. Mol Cell. 2005;19:791–804.
Acknowledgements
The authors would like to thank those men with prostate cancer and the subjects who have donated their time and samples for this study. We thank the funders: Cancer Research UK (C5047/A29626/A22530/A17528), the Dallaglio Foundation, and a Prostate Cancer UK Movember Training, Leadership & Development Award (TLD-S15-003). We acknowledge additional support from Cancer Research UK (C309/A11566, C368/A6743, A368/A7990, C14303/A17197). We acknowledge the National Cancer Research Institute (NIHR) Collaborative Study: “Prostate Cancer: Mechanisms of Progression and Treatment (PROMPT)” (grant G0500966/75466). We thank the National Institute for Health Research, Hutchison Whampoa Limited, University of Cambridge, the Human Research Tissue Bank (Addenbrooke’s Hospital) which is supported by the NIHR Cambridge Biomedical Research Centre, and The Core Facilities at the Cancer Research UK Cambridge Institute. We also acknowledge support of the research staff in S4 who so carefully curated the samples and the follow-up data (J. Burge, M. Corcoran, A. George and S. Stearn). The Cambridge Human Research Tissue Bank and A.Y.W. are supported by the NIHR Cambridge Biomedical Research Centre. A.J.W. acknowledges The Cambridge Urological Malignancies Programme, part of the CRUK Cambridge Centre, funded by Cancer Research UK Major Centre Award C9685/A25117. This project used the UPMC Hillman Cancer Center and Tissue and Research Pathology/Pitt Biospecimen Core shared resource which is supported in part by award P30CA047904. We acknowledge and thank support received from the Prostate Cancer Research, Big C, Bob Champion Cancer Trust, The Masonic Charitable Foundation successor to The Grand Charity, The Alan Boswell Group, The King Family and the Stephen Hargrave Trust. C.E.M. was supported by a CRUK Major Centre Award through the CRUK Cambridge Centre Early Detection Programme and Urological Malignancies Programme. We acknowledge support from the NIHR to the Biomedical Research Centre at The Institute of Cancer Research and Royal Marsden NHS Foundation Trust. A.G.L. acknowledges the support of the University of St Andrews and the Cambridge Cancer Research Fund. G.S.B. was supported by the Academy of Finland; Cancer Society of Finland, and Sigrid Juselius Foundation. N.J.M. acknowledges the support of Prostate Cancer UK (RIA15-ST2-022), Charity Soul, and York Against Cancer. Some of the research presented in this paper was carried out on the High Performance Computing Cluster supported by the Research and Specialist Computing Support service at the University of East Anglia. We thank D. Holland from the Infrastructure Management Team, and P. Clapham from the Informatics Systems Group at the Wellcome Trust Sanger Institute. We thank M. Stratton for discussions when setting up the CR-UK Prostate Cancer ICGC Project. We thank R. Rahbari for useful comments during the PhD examination of C.B.
CRUK-ICGC Prostate Group Members
Abraham Gihawi13, Adam Butler1, Adam Lambert2, Alan Thompson3, Andrew Futreal1, Andrew Menzies1, Andy G Lynch4,5, Anne Baddage6, Anne Y Warren7, Anthony Ng8, Atef Sahil9, Barbara Kremeyer1,10, Bissan Al-Lazikani11, Charlie E Massie6,12, Christopher Greenman13, Christopher Ogden3, Christopher S Foster14,15, Clare Verrill16,17, Claudia Buhigas13, Colin S Cooper18,13, Cyril Fisher3, Dan Berney19, Dan Burns18, Dan J Woodcock9, Daniel Leongamornlert18,1, Daniel S Brewer13,20, David E. Neal21,22, David Jones1, David Nicol3, David C Wedge23,9, Declan Cahill3, Douglas Easton24, Edward Rowe25, Ekaterina Riabchenko26, Elizabeth Bancroft18,3, Erik Mayer3, Ezequiel Anokian18, Freddie Hamdy2, G. Steven Bova26, Gahee Park6, Gill Pelvender27, Gregory Leeman1, Gunes Gundem1,28, Hayley J Luxton21, Hayley C Whitaker29, Hongwei Zhang30, Ian G Mills31, **g**g Zhang13, Jon Teague1, Jonathan Kay21, Jorge Zamora1, Katalin Karaszi2, Kieran Raine1, Lucy Matthews27, Lucy Stebbings1, Ludmil B Alexandrov1, Luke Marsden2, Mahbubl Ahmed18, Matti Nykter26, Mohammed Ghori1, Naomi Livni3, Nening Dennis3, Nicholas Van As3, Niedzica Camacho28, Nimish Shah32, Pardeep Kumar3, Peter Campbell1, Peter Van Loo33,34, Radoslaw Lach6, Rosalind A Eeles18,3, Sandra Edwards35, Sara Pita6, Sarah J Field36, Sarah Thomas3, Simon Tavaré37, Stefania Scalabrino22, Steve Hawkins21,38, Steven Hazell3, Stuart McLaren1, Sue Merson18, Tapio Visakorpi26, Thomas J Mitchell1,39,40, Tim Dudderidge41, Tokhir Dadaev18, Ultan McDermott1, Valeria Bo37, Valeriia Haberland13, Vincent Gnanapragasam32,42, Vincent Khoo3, William Howat43,44, Wing-Kit Leung5, Yaobo Xu1, Yong Jie-Lu45,46, Yongwei Yu30, Zsofia Kote-Jarai18
1 The Cancer, Ageing and Somatic Mutation Programme, Wellcome Trust Sanger Institute, Hinxton, CB10 1SA, UK.
2 The University of Oxford, Oxford, OX1 2JD, UK.
3 Royal Marsden NHS Foundation Trust, London and Sutton, SM2 5PT, UK.
4 School of Mathematics and Statistics/School of Medicine, University of St Andrews, St Andrews, Fife, KY16 9SS, UK.
5 Cancer Research UK Cambridge Institute, University of Cambridge, Li Ka Shing Centre, Robinson Way, Cambridge CB2 0RE, UK.
6 Department of Oncology, Hutchison/MRC Research Centre, University of Cambridge, Cambridge, CB2 0XZ, UK.
7 Department of Histopathology, Cambridge University Hospitals NHS Foundation Trust, Cambridge, CB2 0QQ, UK.
8 The Chinese University of Hong Kong, Hong Kong, China.
9 Oxford Big Data Institute, University of Oxford, Old Road Campus, Oxford, OX3 7LF, UK.
10 Carcassonne, Languedoc-Roussillon, France.
11 Cancer Research UK Cancer Therapeutics Unit, The Institute Of Cancer Research, London, SW7 3RP, UK.
12 Early Detection Programme, CRUK Cambridge Centre, Cancer Research UK Cambridge Institute, Robinson Way, Cambridge, CB2 0RE, UK.
13 Norwich Medical School, University of East Anglia, Norwich, NR4 7TJ, UK.
14 HCA Laboratories, London, WC1E 6JA, UK.
15 University of Liverpool, Liverpool, UK.
16 Nuffield Department of Surgical Sciences, University of Oxford, Oxford, UK.
17 Oxford NIHR Biomedical Research Centre, Oxford, UK.
18 The Institute Of Cancer Research, London, SW7 3RP, UK.
19 Department of Molecular Oncology, Barts Cancer Centre, Barts and the London School of Medicine and Dentistry, London, E1 2AD, UK.
20 The Earlham Institute, Norwich, NR4 7UH, UK.
21 Urological Research Laboratory, Cancer Research UK, Cambridge Institute, Cambridge, CB2 0RE, UK.
22 Department of Surgical Oncology, University of Cambridge, Addenbrooke’s Hospital, Cambridge, CB2 0QQ, UK.
23 The University of Manchester, Oxford Rd, Manchester, M13 9PL, UK.
24 Centre for Cancer Genetic Epidemiology, Department of Oncology, University of Cambridge, Cambridge, CB1 8RN, UK.
25 North Bristol NHS Trust, Bristol.
26 Institute of Biosciences and Medical Technology, BioMediTech, University of Tampere and Fimlab Laboratories, Tampere University Hospital, Tampere, FI-33520, Finland.
27 Oxford University Hospitals NHS Trust, John Radcliffe Hospital, Oxford, OX3 9DU, UK.
28 Memorial Sloan-Kettering Cancer Center, NY 10,065, New York, USA.
29 University College London Charles Bell House 43–45 Foley Street London W1W 7 T.
30 Second Military Medical University, Shanghai, China 200,433.
31 Nuffield Department of Surgical Sciences, University of Oxford, UK.
32 Department of Urology, Addenbrooke’s Hospital, Cambridge, CB2 0QQ, UK.
33 The Francis Crick Institute, London NW1 1AT.
34 Dept of Human Genetics, University of Leuven, 3000 Leuven, Belgium.
35 Division of Genetics & Epidemiology, The Institute of Cancer Research, London SW7 3RP.
36 Cancer Research UK Cambridge Institute, University of Cambridge, Li Ka Shing Centre, Robinson Way, Cambridge CB2 0RE, UK.
37 Statistics and Computational Biology Laboratory, Cancer Research UK Cambridge Institute, University of Cambridge, Li Ka Shing Centre, Robinson Way, Cambridge CB2 0RE, UK.
38 Now at CRUK—CRICK, London.
Funding
This project was funded by Cancer Research UK (C5047/A29626/A22530/A17528), the Dallaglio Foundation, and a Prostate Cancer UK Movember Training, Leadership & Development Award (TLD-S15-003). The funders played no role in the design of the study, collection, analysis, or interpretation of data.
Author information
Authors and Affiliations
Consortia
Contributions
CSC, RAE, DEN, DSB, CSF, GSB, AGL, CEM, DCW designed the study and funding acquisition. CB, AYW, WL, HCW, HJL, SH, SM, FMF, NJM, CSF, CEM, AGL, RAE, CSC, DSB coordinated and were involved with sample collection, pathology review and processing. CB, AB, YX, DJW, IM, AS, FA, PC, CEM, AGL, DCW, DSB supported, directed, and performed the bioinformatics and statistical analyses. CB, CSC, DJW, IM, DCW, CEM, AGL, DSB interpretated the data. CEM, AGL, RAE, CSC, DCW, DSB jointly supervised this work. CB, DSB, and CSC wrote the paper, and all other authors contributed to revisions. All authors read and approved the manuscript. All authors critiqued the manuscript for important intellectual content. The full CRUK-ICGC Prostate Group created and maintains overall study direction. AGL, CEM, GSB, DCW, DSB, CSC, and RAE, are joint PIs of the CR-UK Prostate Cancer ICGC Project.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Ethical approval was obtained from the NHS East of England-Cambridge REC [03/018] and from the NHS Hull and East Yorkshire (REC ref/07/H1304/121) for the morphologically normal samples (including BPH) and fibroblasts, respectively. Samples were collected subject to ICGC standards of ethical consent (https://icgc.org/).
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1.
Sample summary.
Additional file 2:
Supplementary Figure 1. Age vs number of SNVs detected for normal tissue from Prostate cancer patients and non-prostate cancer donors. For the non-cancer donors, the number of SNVs detected is remarkably consistent (range: 104 to 159), apart from one outlier from a cystoprostatectomy with an exceptionally high number of mutations (1202) that is uniquely classified as BPH. There is no significant correlation in the non-cancer donors between age and SNVs (ρ = -0.015, P = 0.98, Spearman’s correlation; this is retained even when the outlier is included: ρ = 0.49, P = 0.27). The number of SNVs detected for non-cancer patients is over 100 SNVs lower than all prostate cancer patients except for one prostate cancer outlier, even in the three samples which are of similar age to the prostate cancer cohort. Looking at only samples in the range 50-73 there is a statistically significant difference in the number of SNVs in prostate cancer vs non-prostate cancer donors (P = 0.0093; Wilcoxon rank sum test; excluding the normal outlier). Supplementary Figure 2. Example density plots of cell cultured fibroblasts and morphologically normal samples from patients where phylogenies could not be reconstructed due to only having one sample per patient or no detected clonal expansions under positive selection. They show the posterior distribution of the fraction of cells bearing a mutation, modelled by a one-dimensional Bayesian Dirichlet process [43]. The median density is indicated by the purple line and 95% confidence intervals by the blue region. The grey histogram shows the observed frequency density of mutations as a function of the fraction of cells bearing the mutation. Supplementary Figure 3. Subclonal architecture of patients with morphologically normal and matched tumour (N-T). Phylogenies revealing the relationships between clones for each case. Each coloured line represents a clone/subclone detected in a particular sample. When two or more coloured lines are together, they represent a clone that is found in all the samples represented. The length of the line is proportional to the weighted number of single nucleotide variants present in each clone; the thickness represents the clonal cell fraction associated with that clone (more detail in Additional file 3). Dotted lines are associated with samples that have no evidence of a unique sample specific clone. Supplementary Figure 4. The relationship between the clonal cell fraction (CCF) and the type of normal samples. Boxplots showing the distribution of estimated CCF for each clone detected and the type of normal sample (non-BPH normal tissue, normal tissue with BPH and BPH fibroblasts). Supplementary Table 1. Summary of patients with multiple normal samples. Patients 0006, 0007 and 0008 have multiple samples from non-BPH normal and tumour tissue and patients 0065, 0073 and 0077 have a sample from non-BPH and BPH normal tissue. Supplementary Table 2. The number of mutations in common between normal samples from the same donor. Supplementary Table 3. The mutation characteristics of three groups of samples defined by the proportion of genome affected by copy number alterations. Group 1: Tumour samples examined by Wedge et al. [30] with less than 6% of the genome affected by CNAs; Group 2: Tumour samples examined by Wedge et al. [30] with more than 6% of the genome affected by CNAs; and Group 3: normal samples examined in our study where no CNAs were detected. The median number of SNVs and indels are shown for each group.
Additional file 3.
Subclonal analysis summary in multiple samples: Worksheet 1 summarises the number of clusters and clonal cell fraction for each patient after applying a multidimensional Bayesian Dirichlet process. Worksheet 2 reports the total number of patients included in the subclonal analysis and the location of normal samples in relation to the tumour sample.
Additional file 4.
Sample summary of samples with clonal expansions under selection pressure.
Additional file 5.
Proportion of epithelial and stromal contents for each morphologically normal sample.
Additional file 6.
Mutational signatures in each patient: Results of mutational signature analyses before and after bootstrap.
Additional file 7.
Mutations in coding regions with functional significance: Functional impact was assessed using wANNOWAR [48].
Additional file 8.
Sample summary for the comparison of the distribution of the number of SNVs detected in morphologically normal tissue, tumour tissue with low CNAs (percentage genome altered (PGA) <6 %) and tumour tissue with high CNAs (PGA >6 %).
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Buhigas, C., Warren, A.Y., Leung, WK. et al. The architecture of clonal expansions in morphologically normal tissue from cancerous and non-cancerous prostates. Mol Cancer 21, 183 (2022). https://doi.org/10.1186/s12943-022-01644-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12943-022-01644-3