
Abstract
Background
Rice is the second-largest food crop in the world and vulnerable to bacterial leaf streak disease. A thorough comprehension of the genetic foundation of agronomic traits was essential for effective implementation of molecular marker-assisted selection.
Results
Our study aimed to evaluate the vulnerability of rice to bacterial leaf streak disease (BLS) induced by the gram-negative bacterium Xanthomonas oryzae pv. oryzicola (Xoc). In order to accomplish this, we first analyzed the population structure of 747 accessions and subsequently assessed their phenotypes 20 days after inoculation with a strain of Xoc, GX01. We conducted genome-wide association studies (GWAS) on a population of 747 rice accessions, consisting of both indica and japonica subpopulations, utilizing phenotypic data on resistance to bacterial leaf streak (RBLS) and sequence data. We identified a total of 20 QTLs associated with RBLS in our analysis. Through the integration of linkage map**, sequence analysis, haplotype analysis, and transcriptome analysis, we were able to identify five potential candidate genes (OsRBLS1—OsRBLS5) that possess the potential to regulate RBLS in rice. In order to gain a more comprehensive understanding of the genetic mechanism behind resistance to bacterial leaf streak, we conducted tests on these genes in both the indica and japonica subpopulations, ultimately identifying superior haplotypes that suggest the potential utilization of these genes in breeding disease-resistant rice varieties.
Conclusions
The findings of our study broaden our comprehension of the genetic mechanisms underlying RBLS in rice and offer significant insights that can be applied towards genetic improvement and breeding of disease-resistant rice in rapidly evolving environmental conditions.
Similar content being viewed by others


Background
Rice, which is considered as the representative species of the Oryza genus, is a cereal crop that originated in China and India. It has been cultivated for over 7,000 years in the Yangtze River basin of China and has played a fundamental role in the development of human civilization [10]. Few studies had explored BLS resistance genes, and the molecular mechanisms underlying rice resistance to Xoc and the pathogenicity of this bacterium were not well understood. BC2 generation using the varieties H359 and Jiannong 8 revealed that resistance to BLS was a quantitative trait controlled by multiple genes [11]. Identified quantitative traits controlled by multiple genes were shown to explain the genetic mode of BLS resistance [12]. Using composite interval map**, five QTLs with significant contributions were identified in the resistant population [13]. Using the BLS-resistant near-isogenic line H359R, the genomic composition of resistance QTLs was analyzed by genotype display analysis. The results showed that the BLS-resistant parent Acc8558 contained three resistance QTL regions, while the susceptible parent H359 contained only one resistance QTL region [14]. One QTL, qBlsr5a, was subsequently mapped to 30 kb using a sub-chromosome segment substitution line, suggesting that the most likely candidate gene was LOC_Os05g01710 [15]. A map** population was constructed by crossing wild rice Oryza rufipogon Griff resistant source 'DY19' with indica rice variety 9311. The results showed that 'DY19' was controlled by a pair of new major recessive genes, bls2, located between SL03 and SL04 on chromosome 2, within a range of about 4 cM [16]. A map** and separation population was constructed using the highly resistant international rice variety BJ1 and the highly susceptible local high-quality rice variety Youzhan 8 as parents. A recessive BLS-resistant gene was identified in BJ1 and this gene mapped to chromosome 10 at about 48.8 cM and was closely linked to RM158 [17]. However, the precise roles of these previously identified genes in disease resistance and susceptibility had not been well characterized. Therefore, there was a need for the identification of additional QTLs/genes that confer resistance to BLS in rice to more fully characterize disease mechanisms. Additionally, molecular marker-assisted selection (MAS) required an explicit understanding of the genetic architecture of agronomic traits [18], so more complete identification of QTLs was required for breeding applications.
In recent years, advances in genomic and transcriptomic technologies had provided powerful tools to investigate the molecular basis of plant-pathogen interactions. Microarray experiments, RNA sequencing (RNA-seq), and other high-throughput methods had enabled researchers to analyze the global gene expression patterns of both the host plant and the pathogen during infection, revealing key genes and pathways involved in the disease process. A microarray experiment was conducted to analyze changes in genome-wide gene expression in response to Xoc at the early stage of infection in both the rice transgenic line and its wild type [19]. One of the differentially expressed pathogenesis-related genes (DEPGs), NRRB, was found to play a role in rice-Xoc interactions [20]. A novel ankyrin-like protein, AnkB, was identified in Xoc and can invade host leaves via stomata and wounds. The type three secretion system (T3SS) of Xoc was found to be pivotal to its pathogenic lifestyle [21]. The effector protein AvrRxo1, an ATP-dependent protease, can enhance the virulence of Xoc and inhibit stomatal immunity in rice by targeting and degrading the rice OsPDX1, which was involved in pyridoxal phosphate synthesis, thereby reducing vitamin B6 levels in rice [22]. A rice multi-parent advanced generation inter-cross (MAGIC) population was used to map QTLs conferring resistance to BLS and another major rice disease, bacterial blight (BB) [23]. A genome-wide association study (GWAS) analysis was conducted on a collection of 236 diverse rice accessions, mainly indica varieties, allowing identification of 12 quantitative trait loci (QTLs) on chromosomes 1, 2, 3, 4, 5, 8, 9 and 11, that conferred resistance to five representative isolates of Thai Xoc [24]. GWAS map** was also conducted to study BLS resistance in rice, and resources were identified with strong resistance and significant SNPs that can potentially be used for breeding BLS-resistant rice [25]. Despite these advances additional work was required to identify new QTLs/genes for BLS resistance in rice and confirm the elite alleles for their utilization in modern molecular breeding.
In this study, we conducted a GWAS to identify resistance to bacterial leaf streak (RBLS) using 747 cultivated rice accessions from the 3,000 Rice Genome (3 K-RG) project [2, 26]. We performed integrated gene annotation and genetic variation, homology, haplotype, and transcriptome analysis to identify candidate functional genes and possible causal polymorphisms for BLS in rice. Our results provided insights into the genetic architecture of BLS, and markers derived from the newly identified genes will be useful in improving resistance to BLS for modern molecular rice breeding.
Results
Population structure and phenotypic variation in RBLS traits among 747 cultivated rice accessions
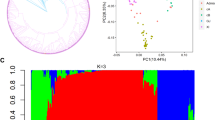
The 747 accessions were collected and a map was plotted using their latitude and longitude based on their clear geographic distribution (Fig. 1a, Additional file 2: Table S1). The Population structure and phenotypic variation in RBLS traits among 747 cultivated rice accessions were analyzed (Fig. 1b-d, Additional file 2: Table S1). We conducted principal component analysis (PCA) using 2.86 million SNPs to assess the population structure of 747 accessions (Fig. 1d). Our analysis revealed a distinct and profound subpopulation structure within this germplasm collection. The PCA indicated that the accessions were classified into two distinct subspecies groups: Oryza sativa ssp. indica (comprising 458 accessions) and japonica (comprising 289 accessions). The first two principal components (PCs) accounted for almost half of the genetic variation, with PC1 and PC2 accounting for 38% and 7% of the total genetic variation, respectively. Furthermore, the neighbor-joining tree also demonstrated a clear subpopulation structure within the 747 accessions (Fig. 1c).

Population genomic analyses and phenotype statistics for RBLS in rice. a. The geographical distribution of 747 cultivated rice accessions using the R package “leaflet”, and the map was based on “Esri.WorldStreetMap” provided by OpenStreetMap. Dark red and light blue dots on the world map represented japonica and indica, respectively. b. Distribution of resistance to bacterial leaf streak (RBLS) in the full population. c. Phylogenetic tree of all accessions inferred from whole-genome SNPs. Major clades were indicated, and dark red and light blue lines represented japonica and indica, respectively. d. Principal component analysis (PCA) for different subpopulations
We utilized ADMIXTURE software [27] to investigate the population structure and genotype structure of the 747 accessions. The analysis was based on the maximum likelihood estimation model and cross-validated for the number of subpopulations (K) to determine the optimal number of ancestral components (K = 1–10). Our results from the structural simulation analysis revealed that the cross-validation error (CV) was minimized when K = 2 (Additional file 1: Fig. S1). Therefore, we selected a k value of 2 to evaluate the genetic structure of the 747 rice genotypes. The distinct levels of K illustrated a clear separation between the indica and japonica subpopulations when K = 2 (Additional file 1: Fig. S1). As a result, we utilized the full population, as well as the indica and japonica subpopulations, to conduct further phenotypic analysis and GWAS.
The resistance to rice bacterial leaf streak displayed a variation that ranged from 0.416 to 2.042 in the full population and indica varieties, and from 0.421 to 1.816 in japonica varieties. Our phenotypic data exhibited a normal distribution and were deemed suitable for association analysis (Fig. 1b).
GWAS and QTL identification of resistance to rice bacterial leaf streak
We conducted GWAS for the full population and the indica and japonica subpopulations, using the phenotypic data of resistance to rice bacterial leaf streak and sequence data from the 747 rice accessions. We employed the general linear model (GLM), mixed linear model (MLM), and fixed and random model circulating probability unification (FarmCPU) implemented in rMVP software [28]. To minimize false positives arising from population structure, we compared the quantile–quantile plots from the three models for BLS resistance for each population and determined that FarmCPU was more suitable than GLM and MLM. The first three principal components were used as covariates within the GWAS model to account for subpopulation structure. We established the suggestive significant threshold at -log (P) = 5 to detect significant association signals. For each GWAS result, we identified SNPs with P-values lower than the threshold (Fig. 2, Additional file 1: Figs. S2-S6).

GWAS results of RBLS in different populations. Manhattan plots and Quantile–quantile plots for GWAS in full (a), indica (b), and japonica (c) using FarmCPU. The red and black genes indicated known genes and candidate genes involved in the RBLS and described in the text, respectively. A dotted horizontal line for each figure indicates the significance threshold (P = 10−5)
Based on this criterion, we identified a total of 89 signals for RBLS across the full, indica, and japonica populations, with 28, 21, and 40 signals detected in the full, indica, and japonica populations, respectively (Fig. 2, Table 1, Additional file 3: Table S2). The QTLs detected by GWAS offered valuable insights into the genetic architecture of the observed phenotypic variations. To further investigate these associations for RBLS, we compared the QTLs identified by GWAS with previous linkage map** results. Our analysis revealed that nine QTLs associated with RBLS had been previously identified in genetic linkage map** studies [16, 31] and Osaba1 (Zeaxanthin epoxidase) [32] (Fig. 2, Additional file 3: Table S2).
OsPSKR1 had been shown to rescue root growth and influence susceptibility to Pseudomonas syringae pv. DC3000 in Arabidopsis pskr1-3 mutants. Moreover, the expression of OsPSKR1 was found to be up-regulated following inoculation with RS105, a strain of Xoc that causes bacterial leaf streak in rice [31]. Based on our sequence data, we found four SNPs (P ≤ 10–2) located in the promoter region of OsPSKR1 (Fig. 3a). We conducted haplotype analysis for OsPSKR1 using the genotypes of these four SNPs for the 747 accessions, and we identified four haplotypes (hap1, hap2, hap3, and hap4) (Fig. 3a). In the japonica subpopulation, we observed a significant difference in the mean value of RBLS between hap2 and hap4; the mean RBLS for hap4 (1.089, 9 accessions) was higher than that for hap2 (0.835, 163 accessions). However, no significant difference was found between the four haplotypes in the indica subpopulation. Therefore, we concluded that OsPSKR1 was a functional gene that regulates RBLS, and hap2 of OsPSKR1 was identified as the superior genotype. Increasing the frequency of hap2 in the japonica subpopulation could enhance resistance to rice bacterial leaf streak. In summary, hap2 of OsPSKR1 could be utilized to improve resistance to RBLS in rice (Fig. 3a).

Exploration of two cloned genes for RBLS. a The cloned genes for RBLS and heat map of the ratio of RPKM. Different colors show ratio of RPKM in the Oryza sativa L. ssp. japonica cv. Nipponbare leaves at 48 h after inoculation with 10 geographically diverse strains of Xanthomonas oryzae pv. oryzicola and mock inoculated. Rows of the heat map correspond to the two cloned genes for RBLS listed on the left of the table. Haplotype analysis of OsPSKR1 (b) and Osaba1 (c). Different haplotypes and a comparison of RBLS among haplotypes of OsPSKR1 and Osaba1 in the indica and japonica subgroups. The green violins represent indica, the red violins represent japonica rice, and different letters indicate significant differences (P < 0.05) detected by one-way ANOVA
The Osaba1 mutant has been shown to be resistant to Xoc [32]. We identified five significant SNPs located in the promoter region of Osaba1. Haplotype analysis was conducted for Osaba1 using genotypic data from the 747 accessions, which revealed a total of three haplotypes (hap1, hap2, and hap3) in both the indica and japonica subpopulations (Fig. 3b). In the indica subpopulation, hap1 and hap2 of Osaba1 exhibited a significant difference in RBLS compared to hap3, with the mean RBLS of hap3 (1.049, 67 accessions) being higher than that for hap1 (0.896, 332 accessions) and hap2 (0.759, 8 accessions). Therefore, we tentatively identified this gene as a functional gene that controls bacterial leaf streak in rice, with hap1 and hap2 of Osaba1 being identified as the superior genotypes. However, we also observed a higher proportion of superior haplotypes for this gene in both indica and japonica rice, indicating that these superior haplotypes have already been widely utilized in modern breeding processes (Fig. 3b).
Transcriptome analysis of candidate genes
To ensure the reliability of our results, we conducted transcriptome analysis on Nipponbare leaves 48 h after inoculation with 10 geographically diverse strains of Xoc. We utilized three biological replicates for each condition, and three replicates of mock-inoculated O. sativa were used as controls. We compared the reads per kilobase per million mapped reads between the samples inoculated with the Xoc strains and the mock-inoculated samples, denoted as RPKMXoc and RPKMMock, respectively. The genes detected in the GWAS for RBLS were classified as up-regulated or down-regulated genes based on their RPKMXoc/RPKMMock values. Genes with values greater than 1.5 were classified as up-regulated, while those with values lower than 0.67 were classified as down-regulated, between the inoculation with 10 geographically diverse strains of Xoc and the mock-inoculated samples (Figs. 3c and 4). As previously described, our GWAS results identified two cloned genes that may be functional genes for RBLS in the japonica population. OsPSKR1 was identified as an up-regulated gene, with the ratios of its RPKM values between the 10 geographically diverse strains of Xoc and the mock-inoculated samples being greater than 1.5. The ratio of RPKM in B8-12 to the mock sample for OsPSKR1 was 2.32, and the lowest ratio of RPKM in RS105 to mock for OsPSKR1 was 1.53. Osaba1 was identified as a down-regulated gene, with the ratios of its RPKM values between the 10 geographically diverse strains of Xoc and the mock-inoculated samples being all lower than 0.67. The lowest ratio for Osaba1 was 0.42, which was the ratio of RPKM in CFBP7341 to the mock sample. The largest ratio of RPKM in CFBP2286 to mock for Osaba1 was 0.65 (Fig. 3c). These results were in agreement with previous reports, indicating that the transcriptomic analyses were trustworthy and could be employed to further identify potential candidate genes for the QTLs detected by GWAS.

Candidate genes for RBLS and heat map of the ratio of RPKM. Different colors show the ratio of RPKM in leaves of Nipponbare 48 h after inoculation with 10 geographically diverse strains of Xoc and mock-inoculated. The rows of the heat map correspond to the 20 candidate genes for RBLS listed on the left of the table
With the reliability of the transcriptomic analyses established, we proceeded to identify potential candidate genes within the QTL for RBLS by comparing the RPKM values of genes within each QTL detected by GWAS between the samples inoculated with 10 geographically diverse strains of Xoc and the mock-inoculated samples. We utilized the transcriptomic data to identify 20 candidate genes that corresponded to the eight QTLs detected by GWAS. Among these, seven genes were up-regulated, while 13 genes were found to be down-regulated (Fig. 4). Based on the transcriptomic analysis, these genes were the most likely candidate genes for RBLS among the 20 candidates we identified.
To investigate the function of these candidate genes, the same approach of KEGG pathway enrichment analysis for cloned and candidate genes were applied. The cloned and candidate genes were enriched for KEGG pathway (Additional file 1: Figs. S7-S8). The terms “MAPK signaling pathway—plant”, “Plant hormone signal transduction” and “Biosynthesis of secondary metabolites” from cloned genes (Additional file 1: Fig. S7), and “MAPK signaling pathway—plant”, “Biosynthesis of secondary metabolites” and “Metabolic pathways” from candidate genes (Additional file 1: Fig. S8) were both related to development and resistance. Taken together, these candidate genes were most likely the genes that control RBLS, and could provide theoretical guidance for subsequent research on disease resistance in rice.
Candidate gene analysis in QTLs
To gain a deeper understanding of the potential candidate genes, we conducted haplotype analysis for these genes. Our analysis revealed significant differences in the mean RBLS values between haplotypes for five of the genes (Fig. 5).

Investigation of five candidate genes for RBLS. Different haplotypes and a comparison of RBLS between the haplotypes of OsRBLS1 (a), OsRBLS2 (b), OsRBLS3 (c), OsRBLS4 (d) and OsRBLS5 (e) in the indica and japonica subgroups. The green violins represent indica, the red violins represent japonica rice, and different letters indicate significant differences (P < 0.05) detected by one-way ANOVA
LOC_Os08g10260 was located in qRBLS8-1 and encoded a NBS-LRR disease resistance protein. We found four SNPs (P ≤ 10–2) within the promoter region of the gene. Haplotype analysis was conducted for LOC_Os08g10260 using the genotypes of these four SNPs for all 747 accessions, ultimately leading to the identification of seven haplotypes (Fig. 5a). In the indica subpopulation, we observed a significant difference in RBLS between hap2 (1.092, 30 accessions) and hap3 (1.079, 73 accessions) compared to hap5 (0.867, 77 accessions), hap6 (0.826, 32 accessions), and hap7 (0.838, 13 accessions). Hap2 and hap3 exhibited higher mean RBLS values than hap5, hap6, and hap7. In japonica, we observed a significant difference in RBLS between hap1 (1.066, 33 accessions), and hap4 (0.761, 99 accessions), hap7 (0.686, 12 accessions), with the mean RBLS value for hap1 greater than that of hap4 and hap7. Based on our findings, we concluded that LOC_Os08g10260, which we named OsRBLS1, is a functional gene that controls RBLS. Hap4, hap5, hap6, and hap7 of OsRBLS1 were identified as the superior genotypes. Thus, increasing their frequency in both the indica and japonica subpopulations could enhance resistance to rice bacterial leaf streak (Fig. 5a).
ASR1 (LOC_Os02g33820), located in qRBLS2-7, encoded an abscisic stress-ripening gene. We found four SNPs (P ≤ 10–2) within the promoter region of ASR1. Prior research had demonstrated that overexpression of ASR1 in transgenic rice plants led to increased tolerance to both drought and cold stress. Furthermore, ASR1 and ASR5 had been shown to function complementarily in the regulation of gene expression in response to aluminium (Al) toxicity [33, 34]. We conducted haplotype analysis for ASR1 using the genotypes of these four SNPs for all 747 accessions, ultimately leading to the identification of three haplotypes (Fig. 5b). Within the indica subspecies, we noted a significant difference in RBLS between hap1, hap3, and hap2. The mean RBLS value of hap2 (0.758, 46 accessions) was lower than those for hap1 (0.96, 79 accessions) and hap3 (0.879, 139 accessions). In the japonica subspecies, a significant difference in RBLS was observed between hap1 and hap2, with hap1 exhibiting a higher mean RBLS value than hap2. Based on our results, we concluded that ASR1 (LOC_Os02g33820), which we named OsRBLS2, was a functional gene that controls RBLS. Regarding OsRBLS2, hap2 was identified as the superior genotype. Thus, increasing the frequency of hap2 in the indica subpopulation could enhance resistance to rice bacterial leaf streak (Fig. 5b).
We observed a significant difference in RBLS between hap1 and hap2 of LOC_Os03g47160, LOC_Os10g32990, and LOC_Os10g33210 within both the indica and japonica subpopulations. LOC_Os03g47160 was located within qRBLS3-1 and encoded an expressed protein that we named OsRBLS3. LOC_Os10g32990 and LOC_Os10g33210 were both located in qRBLS10-2. LOC_Os10g32990 encoded a precursor of receptor-like protein kinase 2, named OsRBLS4. LOC_Os10g33210, on the other hand, encoded a peptide transporter PTR3-A that we named OsRBLS5. In both the indica and japonica subpopulations, the mean RBLS values of hap2 for OsRBLS3 and OsRBLS5 were significantly lower than those for hap1. While, the mean RBLS value of hap2 of OsRBLS4 was significantly higher than that for hap1 in both indica and japonica. Therefore, we concluded that OsRBLS3, OsRBLS4, and OsRBLS5 were functional genes that control RBLS. The hap2 of OsRBLS3 and OsRBLS5, and hap1 of OsRBLS4 were superior genotypes. Increasing the frequency of these genotypes in either the indica or japonica subpopulations could improve resistance to rice bacterial leaf streak (Fig. 5c-e). By employing GWAS and linkage map** in conjunction with sequence analysis, haplotype analysis, and transcriptome analysis, we identified 20 QTLs and five candidate genes that control resistance to bacterial leaf streak in rice. We also determined the superior haplotypes of these genes and assessed their breeding potential in both the indica and japonica subspecies. These genes could be useful in further investigations aimed at elucidating the genetic mechanisms underlying resistance to bacterial leaf streak in rice.
Discussion
Genetic factors contributing to resistance to bacterial leaf streak in rice
Efforts to develop BLS-resistant rice varieties necessitated the exploration and utilization of genes associated with RBLS. To date, several genes related to bacterial leaf streak disease in rice have been cloned [31, 32, 35,36,37,42]. The resistance of hybrid rice to the tested pathogen was dominant, but the susceptibility was recessive, as determined by spray inoculation during the seedling stage, indicating that the resistance of the restorer lines determined the resistance of the hybrid material [43]. The resistance of Duar and IR36 to the tested pathogen was identified by needle inoculation, and was suggested to be controlled by two pairs of recessive genes [44]. Eight materials were randomly selected from 57 resistant wild rice materials for hybridization and backcrossing with 9311 and resistance was found to be inherited recessively [45]. BC2 generations were constructed using H359 and Jiannong 8, and resistance to bacterial leaf streak disease was found to be controlled by multiple genes as a quantitative trait [11]. Similarly, another study found that inheritance of resistance to bacterial leaf streak disease was controlled by multiple genes as a quantitative trait [12]. Four resistant rice varieties, Dular, IR1545-339, IR26, and IR36, as well as a susceptible variety, **gang 30, were used for hybridization and backcrossing and the resistance gene in IR1545-339 was found to be non-allelic [46]. A highly pathogenic bacterial strain, S-103, was used to inoculate resistant 90IRBBN44, and genetic analysis revealed that the resistance in 90IRBBN44 was controlled by a pair of recessive genes [14]. The BLS resistance quantitative trait locus (QTL) qBlsr3d in rice was verified to be controlled by quantitative inheritance by constructing a H359-BLSR3D single-chromosome segment substitution line [16]. The BLS resistance gene in the international rice variety BJ1 was located using the BSA method. Resistance was found to be controlled by a pair of recessive genes located on the chromosome 10 at a distance of approximately 48.8 cM and tightly linked to marker RM158 [17]. A doubled haploid (DH) population was constructed using the parents Taichung Native 1 and Chunjiang 06 and four effective quantitative trait loci (QTL) were identified through needle inoculation during the seedling stage on the 2, 4, 5, and 8 chromosomes of rice [50, To summarize, we analyzed the population structure of 747 rice accessions and evaluated their phenotypes 20 days after inoculation with Xoc strain GX01. We then conducted GWAS on the full population and the indica and japonica subpopulations based on both the phenotypic resistance to rice bacterial leaf streak and the sequence data of the 747 rice accessions. Our analysis led to the identification of 20 QTLs associated with RBLS in rice. Subsequently, we used a combination of linkage map**, sequence analysis, haplotype analysis, and transcriptome analysis to identify five candidate genes that control resistance to BLS in rice. We also determined their superior haplotypes and breeding potential in both indica and japonica rice varieties. These findings suggested that these genes could be employed in future studies aimed at elucidating the genetic mechanisms underlying resistance to bacterial leaf streak in rice. The rice diversity panel comprised 747 cultivated rice accessions sourced from the 3 K Rice Genome (3 K-RG) [2, 26]. This panel included 290 genotypes from the mini-core collection, which was initially selected from a core set of 4,310 accessions [53], and 457 lines from the International Rice Molecular Breeding Network [54]. The two collections consisted of accessions from 45 countries, encompassing major rice-growing regions worldwide (Table S1). A map for 747 accessions was plotted using the R package “leaflet” (https://rstudio.github.io/leaflet/), and the map was based on “Esri.WorldStreetMap” provided by OpenStreetMap, with latitude and longitude of based on their clear geographic distribution. Transcriptomic data were obtained from the NCBI (https://www.ncbi.nlm.nih.gov/gds) with series accession ID: PRJNA280380 [55]. A total of 747 varieties were grown under field conditions in Nanning, Guangxi, southern China, with a plant spacing of 20 cm x 30 cm, and each variety was replicated twice. The bacterial strains of rice bacterial leaf streak were obtained from GX01, which was isolated at the Guangxi Academy of Agricultural Sciences. At the maximum tillering stage, the 747 varieties were inoculated with Xoc using the acupuncture method [10]. Phenotypic assessments were conducted 20 days after inoculation (DAI), and the diseased leaf areas were measured using the Standard Evaluation System for Rice (5th Edition, 2014). The genetic variation data for the 747 accessions, in the form of single nucleotide polymorphisms (SNPs), were obtained from the publicly available 3 K-RG database. The database comprised approximately 17 million highly credible SNPs and 2.4 million indels, which were aligned to the Nipponbare IRGSP 1.0 reference genome [2, 26]. After removing SNPs with missing rates > 30% and minor allele frequencies < 5% in the full, indica, and japonica populations, we identified 2,860,820 SNPs as a credible SNP set. To account for population structure, we performed principal component (PC) and kinship matrix analyses for the 747 accessions. To infer the basal group of rice, we used a total of 2,860,820 SNPs with a missing rate of ≤ 50% to construct a phylogenetic tree using the unweighted neighbor-joining (NJ) method with the 'phangorn' R package. For constructing the PC matrix, we used the first three principal components (PCs). To conduct PCA, we used R (version 4.0.3) and plotted the first three eigenvectors. We further called independent SNPs using Plink v1.90b4 with the parameter '–indep-pairwise 50 5 0.3'. This resulted in the effective numbers of independent SNPs being 113,223, 95,164, and 45,752 for the full, indica, and japonica populations, respectively [56]. To analyze the population structure of the rice accessions, we used the 113,223 independent SNPs to perform a maximum likelihood clustering analysis with ADMIXTURE (version 1.3) [27]. To estimate the genetic ancestry of each sample, we ran the cross-validation error (CV) procedure with varying levels of K (K = 1–10). Based on the cross-validation error, we determined that a value of K = 2 was optimal. We plotted the ADMIXTURE result using an R script. We obtained a total of 2,860,820 SNPs with a missing rate of ≤ 50% and minor allele frequencies of ≥ 5%. To account for population structure, we performed principal component (PC) and kinship matrix analyses, using the first three PCs to construct the PC matrix. We performed a genome-wide association study (GWAS) to identify genetic variants associated with resistance to rice bacterial leaf streak (BLS) in the 747 accessions. And we used three different models: general linear model (GLM), mixed linear model (MLM), and Fixed and random model Circulating Probability Unification (FarmCPU). The GWAS was conducted using the SNP set and default settings in the rMVP software [28]. The strong linkage disequilibrium (LD) among SNPs resulted in non-independence, which caused the thresholds derived from the total number of markers to be too stringent for detecting significant associations. To address this issue, we calculated suggestive thresholds using the formula "-log10(1 / effective number of independent SNPs)", as previously described [50, 52, 57, 58]. And we determined the effective numbers of independent SNPs using PLINK [56], with a window size of 50, step size of 50, and r2 ≥ 0.3. The effective numbers of independent SNPs were found to be 113,223, 95,164, and 45,752 in the full population, and the indica and japonica subpopulations, respectively. To identify significant associations, we set the threshold at -log (P) = 5. We used genome structure annotation information from MSU-RGAP 7.0 to annotate nonsynonymous SNPs using SnpEff [59]. An in-house Perl script was used to separate these SNPs from all the identified SNPs in the 747 accessions. For haplotype analysis, we used SNPs with a significance level of P ≤ 10–2 in the 2 k promoter and exons of the gene. Haplotypes that contained at least five varieties were used for statistical testing. We calculated the differences in phenotypic value between haplotypes of each gene using one-way ANOVA or Student's t-tests with an R script [52, 60]. Genes with RPKMXoc/RPKMmock > 1.5 were defined as up-regulated genes, and those with RPKMXoc/RPKMmock < 0.67 were defined as down-regulated genes.Conclusion
Methods
Materials and sequencing data
Phenotypic data for RBLS
Phylogenetic and population structure analysis
Genome-wide association study
Haplotype analysis
Availability of data and materials
The genomic data for 747 rice accessions from the 3 K-RG can be downloaded from http://snp-seek.irri.org. The sequencing data used in our analyses can be accessed through project accession PRJEB6180 on NCBI (https://www.ncbi.nlm.nih.gov/sra/?term=PRJEB6180). The public transcriptome data includes data for Nipponbare leaves at 48 h after inoculation with 10 geographically diverse strains of Xoc, with three biological replicates for each compared to three replicates of mock-inoculated O. sativa as the control (PRJNA280380). All data generated or analyzed during this study are included in this published article or its supplementary information files or are available from the corresponding authors on reasonable request.
Abbreviations
- BLS:
-
Bacterial leaf streak
- Xoc:
-
Xanthomonas oryzae Pv. oryzicola
- T3SS:
-
Type three secretion system
- VB6:
-
Vitamin B6
- GWAS:
-
Genome-wide association study
- QTL:
-
Quantitative trait loci
- MAS:
-
Marker-assisted selection
- 3 K-RG:
-
3,000 Rice Genome
- RBLS:
-
Resistance to bacterial leaf streak
- PCA:
-
Principle component analysis
- CV:
-
Cross-validation error
- GLM:
-
General linear model
- MLM:
-
Mixed linear model
- FarmCPU:
-
Fixed and random model Circulating Probability Unification
- SA:
-
Salicylic acid
- RPKM:
-
Reads Per Kilobase per Million mapped reads
- RPKMXoc :
-
RPKM of inoculation with 10 geographically diverse strains of Xanthomonas oryzae pv. oryzicola
- RPKMmock :
-
RPKM of inoculation with mock
References
Wang X, Wang W, Tai S, Li M, Gao Q, Hu Z, Hu W, Wu Z, Zhu X, **e J, et al. Selective and comparative genome architecture of Asian cultivated rice (Oryza sativa L.) attributed to domestication and modern breeding. J Adv Res. 2022;42:1–16. https://doi.org/10.1016/j.jare.2022.08.004.
Wang W, Mauleon R, Hu Z, Chebotarov D, Tai S, Wu Z, Li M, Zheng T-Q, Fuentes RR, Zhang F, et al. Genomic variation in 3,010 diverse accessions of Asian cultivated rice. Nature. 2018;557(7703):43–9. https://doi.org/10.1038/s41586-018-0063-9.
Ke Y, Deng H, Wang S. Advances in understanding broad-spectrum resistance to pathogens in rice. Plant J. 2016;90(4):738–48. https://doi.org/10.1111/tpj.13438.
Niño-Liu D, Ronald P, Bogdanove A. Xanthomonas oryzae pathovars: model pathogens of a model crop. Mol Plant Pathol. 2006;7(5):303–24. https://doi.org/10.1111/j.1364-3703.2006.00344.x.
Wang W, Zhang R, Luo C, Yin X, Liu Y, Lu F, Chen Z. Biological characteristics and genetic diversity of ustilaginoidea virens from rice regions in China. Scientia Agricultura Sinica. 2014;47(14):2762–73. https://doi.org/10.3864/j.issn.0578-1752.2014.14.007.
Ji Z, Zakria M, Zou L, **ong L, Zheng L, Ji G, Chen G. Genetic diversity of transcriptional activator-like effector genes in Chinese isolates of Xanthomonas oryzae pv. oryzicola. Phytopathology. 2014;104(7):672–82. https://doi.org/10.1094/PHYTO-08-13-0232-R.
Huang C. Analysis of the prevalence of bacterial leaf streak disease of rice and quarantine control measures. Anhui Agri Sci Bull. 2008;14(3):88–90. https://doi.org/10.16377/j.cnki.issn1007-7731.2008.03.017.
Jiang F. Identification and control methods of rice bacterial leaf streak. Hortic Seed. 2017;2:36–8. https://doi.org/10.16530/j.cnki.cn21-1574/s.2017.02.014.
Tang S, Yang Y, Pan S, Ling M, Dai L, Li W. Control of rice bacterial leaf streak: research progress. J Agric. 2018;8(11):16–20. https://doi.org/10.1111/j.1364-3703.2006.00344.x.
Zhao Y, Luo D, He S, Liu F. Comparative study on four inoculation methods of rice bacterial leaf streak. Subtropical Agric Res. 2018;14(4):242–6. https://doi.org/10.13321/j.cnki.subtrop.agric.res.2018.04.005.
Tang D, Li W, Wu W. Inheritance of the resistance to rice bacterial leaf streak. Fujian Agri Univ. 1998;27(2):133–7.
Chen C, Zheng W, Huang X, Zhang D, Lin X. Major QTL conferring resistance to rice bacterial leaf streak. Agri Sci in China. 2006;5(3):216–20. https://doi.org/10.1016/S1671-2927(06)60041-2.
Tang D, Wu W, Li W, Lu H, Worland AJ. Map** of QTLs conferring resistance to bacterial leaf streak in rice. Theor Appl Genet. 2000;101(1):286–91. https://doi.org/10.1007/s001220051481.
Chen Z, **g Y, LI X, Zhou Y, Diao Z, Li S, Wu W. Verification and more precise map** of a QTL qBlsr5a underlying resistance to bacterial leaf streak in rice. Fujian Agri For Univ. 2006;35(6):619–22.
** sub-CSSLs in rice. PloS One. 2014;9(4):e95751. https://doi.org/10.1371/journal.pone.0095751.
Shi L, Luo D, Zhao Y, Cen Z, Liu F, Li R. Genetic analysis and map** of bacterial leaf streak resistance genes in Oryzae rufipogon Griff. J South China Agric Univ. 2019;40(2):1–5. https://doi.org/10.7671/j.issn.1001-411X.201805009.
Qin L, ** of major gene conferring resistance to rice bacterial leaf streak. Southwest China J Agric Sci. 2020;33(1):7–12. https://doi.org/10.16213/j.cnki.scjas.2020.1.002.
Zeng D, Tian Z, Rao Y, Dong G, Yang Y, Huang L, Leng Y, Xu J, Sun C, Zhang G, et al. Rational design of high-yield and superior-quality rice. Nat Plants. 2017;3:17031. https://doi.org/10.1038/nplants.2017.31.
Xu M, Vera Cruz CM, Fu B, Zhu L, Zhou Y, Li Z. Different patterns of gene expression in rice varieties undergoing a resistant or susceptible interaction with the bacterial leaf streak pathogen. African J Biotech. 2011;10(65):14419–38. https://doi.org/10.5897/AJB11.1317.
Guo L, Guo C, Li M, Wang W, Luo C, Zhang Y, Chen L. Suppression of expression of the putative receptor-like kinase gene NRRB enhances resistance to bacterial leaf streak in rice. Mol Biol Rep. 2014;41(4):2177–87. https://doi.org/10.1007/s11033-014-3069-x.
Pan X, Xu S, Wu J, Duan Y, Zhen Z, Wang J, Song X, Zhou M. The ankyrin-like protein AnkB interacts with CatB, affects catalase activity, and enhances resistance of Xanthomonas oryzae pv. oryzae and Xanthomonas oryzae pv. oryzicola to phenazine-1-carboxylic acid. Appl Environ Microb. 2017;84(4):e02145–17. https://doi.org/10.1128/AEM.02145-17.
Liu H, Lu C, li Y, Tao W, Zhang B, Liu B, Feng W, Xu Q, Dong H, He S, et al. The bacterial effector AvrRxo1 inhibits vitamin B6 biosynthesis to promote infection in rice. Plant Commun. 2022;3:100324. https://doi.org/10.1016/j.xplc.2022.100324.
Bossa-Castro A, Tekete C, Raghavan C, Delorean E, Dereeper A, Dagno K, Koita O, Mosquera G, Leung H, Verdier V, et al. Allelic variation for broad-spectrum resistance and susceptibility to bacterial pathogens identified in a rice MAGIC population. Plant Biotechnol J. 2018;16(9):1559–68. https://doi.org/10.1111/pbi.12895.
Sattayachiti W, Wanchana S, Arikit S, Nubankoh P, Patarapuwadol S, Vanavichit A, Darwell CT, Too**da T. Genome-wide association analysis identifies resistance loci for bacterial leaf streak resistance in rice (Oryza sativa L.). Plants. 2020;9:1673.
Jiang N, Fu J, Zeng Q, Liang Y, Shi Y, Li Z, ** for resistance to bacterial blight and bacterial leaf streak in rice. Planta. 2021;253:94. https://doi.org/10.1007/s00425-021-03612-5.
Li Z, Fu B-Y, Gao Y-M, Wang W, Xu J, Zhang F, Zhao X-Q, Zheng T-Q, Zhou Y, Zhang G, et al. The 3,000 Rice Genomes Project. GigaScience. 2014;3:7. https://doi.org/10.1186/2047-217X-3-7.
Alexander D, Novembre J, Lange K. Fast Model-based estimation of ancestry in unrelated individuals. Genome Res. 2009;19(9):1655–64. https://doi.org/10.1101/gr.094052.109.
Yin L, Zhang H, Tang Z, Xu J, Yin D, Zhang Z, Yuan X, Zhu M, Zhao S, Li X, et al. rMVP: A memory-efficient, visualization-enhanced, and parallel-accelerated tool for genome-wide association study. Genom Proteom Bioinf. 2021;19(4):619–28. https://doi.org/10.1016/j.gpb.2020.10.007.
**e X, Zheng Y, Lu L, Yuan J, Hu J, Suhong B, Lin Y, Liu Y, Guan H, Wu W. Genome-wide association study of QTLs conferring resistance to bacterial leaf streak in rice. Plants. 2021;10(10):2039. https://doi.org/10.3390/plants10102039.
Wei M, Ma Z, Huang D, Qin Y, Liu C, Lu Y, Luo T, Li Z, Zhang Y, Qin G. QTL-seq analysis for identification of resistance locus to bacterial leaf streak in rice. Chinese J Rice Sci. 2023;37(2):133–41. https://doi.org/10.16819/j.1001-7216.2023.220607.
Yang W, Zhang B, Qi G, Shang L, Liu H, Ding X, Chu Z. Identification of the phytosulfokine receptor 1 (OsPSKR1) confers resistance to bacterial leaf streak in rice. Planta. 2019;250(5):1603–12. https://doi.org/10.1007/s00425-019-03238-8.
Zhang D, Tian C, Yin K, Wang W, Qiu J. Post-invasive bacterial resistance conferred by open stomata in rice. Mol Plant Microbe In. 2019;32(2):255–66. https://doi.org/10.1094/MPMI-06-18-0162-R.
Joo J, Lee Y, Nahm B, Song S. Abiotic stress responsive rice ASR1 and ASR3 exhibit different tissue-dependent sugar and hormone-sensitivities. Mol Cells. 2013;35(5):421–35. https://doi.org/10.1007/s10059-013-0036-7.
Arenhart R, Schunemann M, Bücker-Neto L, Margis R, Wang Z-Y, Margis-Pinheiro M. Rice ASR1 and ASR5 are complementary transcription factors regulating aluminum responsive genes. Plant Cell Environ. 2015;39(3):645–51. https://doi.org/10.1111/pce.12655.
Xu X, Xu Z, Li Z, Zakria M, Zou L, Chen G. Increasing resistance to bacterial leaf streak in rice by editing the promoter of susceptibility gene OsSULRT3;6. Plant Biotechnol J. 2021;19(6):1101–3. https://doi.org/10.1111/pbi.13602.
Tao W, Zhang H, Yuan B, Liu H, Kong L, Chu Z, Ding X. Tal2b targets and activates the expression of OsF3H03g to hijack OsUGT74H4 and synergistically interfere with rice immunity. New Phytol. 2022;233:1864–80. https://doi.org/10.1111/nph.17877.
Ju Y, Tian H, Zhang R, Zuo L, ** G, Xu Q, Ding X, Li X, Chu Z. Overexpression of OsHSP18.0-CI enhances resistance to bacterial leaf streak in rice. Rice. 2017;10(1):12. https://doi.org/10.1186/s12284-017-0153-6.
Li B, Liu Y, Tao W, Wang J, **e G, Chu Z, Ding X. OsBGLU19 and OsBGLU23 regulate disease resistance to bacterial leaf streak in rice. J Integr Agr. 2019;18(6):1199–210. https://doi.org/10.1016/S2095-3119(18)62117-3.
Guo L, Li M, Wang W, Wang L, Hao G, Guo C, Chen L. Over-expression in the nucleotide-binding site-leucine rich repeat gene DEPG1 increases susceptibility to bacterial leaf streak disease in transgenic rice plants. Mol Biol Rep. 2011;39:3491–504. https://doi.org/10.1007/s11033-011-1122-6.
Tong X, Xu H, Zhu C, Xu Y, Xu F. Assessment of yield loss due to the bacterial leaf streak of rice. Zhejiang Agri Univ. 1995;21(4):357–60.
Xu J, Wang H, Lin Y, ** Y. Studies on the inheritance of the resistance of rice to bacterial leaf streak and bacterial leaf blight. Acta Genet Sin. 1997;24(4):330–5.
Nayak P, Reddy R, Misra RN. Pattern of inheritance to bacterial leaf streak resistance in rice. Curr Sci INDIA. 1975;44:600–1.
He Y, Wen Y, Huang R, Zeng X. Inheritance of resistance to bacterial leaf streak in hybrid rice. Acta Agri Univ Jiangxiensis. 1994;16(1):62–5.
Zhou M, Xu Z, Li H, You Y, Zhou Y. Inheritance of resistance to bacterial leaf streak in two indica rice cultivars. Nan**g Agri Univ. 1999;22(4):27–9.
Huang D, Cen Z, Liu C, He W, Chen Y, Ma Z, Yang L, Wei S, Liu Y, Huang S, et al. Identification and genetic analysis of resistance to bacterial leaf streak in wild rice. Plant Genet Resour. 2008;9(1):11–4. https://doi.org/10.13430/j.cnki.jpgr.2008.01.014.
Zhang H, Lu Z, Zhu L. Inheritance of resistance to bacterial leaf streak (Xanthomonas oryzae pv. oryzicola) in four indica rice cultivars. Chinese Journal of Rice Sci. 1996;10(4):193–6. https://doi.org/10.16819/j.1001-7216.1996.04.001.
Zheng J, Li Y, Fang X. Detection of QTL conferring resistance to bacterial leaf streak in rice chromosome 2 (O. sativa L. spp. indica). Scientia Agricultura Sinica. 2005;38(9):1923–25.
Cao J, Chen Z, Lin D, Wu W, **e X. Verification of QTL qBlsr3d conferring resistance to bacterial leaf streak in rice by constructing SSSL. Mol Plant Breeding. 2014;12(3):416–20. https://doi.org/10.13271/j.mpb.012.000416.
Ma L, Fang Y, **ao J, ** C, ** Z, Ye W, Rao Y. QTL exploration of bacterial leaf streak and their gene expression in rice. Chinese Bulletin of Botany. 2018;53(4):468–76. https://doi.org/10.11983/CBB17250.
Yuan J, Wang X, Zhao Y, Khan N, Zhao Z, Zhang Y, Wen X, Tang F, Wang F, Li Z. Genetic basis and identification of candidate genes for salt tolerance in rice by GWAS. Sci Rep. 2020;10:9958. https://doi.org/10.1038/s41598-020-66604-7.
Zhang Q, **e J, Wang X, Liu M, **aoyang Z, Yang T, Khan N, Sun C, Li J, Zhang Z, et al. Natural variation of RGN1a regulates grain number per panicle in japonica rice. Front Plant Sci. 2022;13:1097622. https://doi.org/10.3389/fpls.2022.1097622.
Wang X, Zhao Y, Jiang C, Wang L, Chen L, Li F, Zhang Y, Pan Y, Zhang T. Evolution of different rice ecotypes and genetic basis of flooding adaptability in Deepwater rice by GWAS. BMC Plant Biol. 2022;22(1):526. https://doi.org/10.1186/s12870-022-03924-y.
Zhang H, Zhang D, Wang M, Sun J, Qi Y, Li J, Wei X, Han L, Qiu Z, Tang S, et al. A core collection and mini core collection of Oryza sativa L. China Theor Appl Genet. 2011;122(1):49–61. https://doi.org/10.1007/s00122-010-1421-7.
Yu S, Xu WJ, Vijayakumar C, Ali J, Fu B, Xu J, Jiang YZ, Marghirang R, Domingo J, Aquino C, et al. Molecular diversity and multilocus organization of the parental lines used in the International Rice Molecular Breeding Program. Theor Appl Genet. 2004;108(1):131–40. https://doi.org/10.1007/s00122-003-1400-3.
Wilkins K, Booher N, Wang L, Bogdanove A. TAL effectors and activation of predicted host targets distinguish Asian from African strains of the rice pathogen Xanthomonas oryzae pv. oryzicola while strict conservation suggests universal importance of five TAL effectors. Front Plant Sci. 2015;6:536. https://doi.org/10.3389/fpls.2015.00536.
Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira M, Bender D, Maller J, Sklar P, Bakker P, Daly M, et al. Plink: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81(3):559–75. https://doi.org/10.1086/519795.
Lippert C, Listgarten J, Liu Y, Kadie C, Davidson R, Heckerman D. FaST Linear Mixed Models for genome-wide association studies. Nat Methods. 2011;8(10):833–5. https://doi.org/10.1038/nmeth.1681.
Wang X, Wang H, Liu S, Ferjani A, Li J, Yan J, Yang X, Qin F. Genetic variation in ZmVPP1 contributes to drought tolerance in maize seedlings. Nat Genet. 2016;48(10):1233–41. https://doi.org/10.1038/ng.3636.
Cingolani P, Platts A, Wang L, Coon M, Nguyen T, Luan W, Land S, Lu X, Ruden D. A program for annotating and predicting the effects of single nucleotide polymorphisms. SnpEff Fly. 2012;6(2):80–92. https://doi.org/10.4161/fly.19695.
Zhao Y, Zhang H, Xu J, Jiang C, Yin Z, **ong H, **e J, Wang X, Zhu X, Li Y, et al. Loci and natural alleles underlying robust roots and adaptive domestication of upland ecotype rice in aerobic conditions. PLOS Genet. 2018;14(8):e1007521. https://doi.org/10.1371/journal.pgen.1007521.
Funding
The present study was supported by the Project of Guangxi Natural Science Foundation (2023GXNSFDA026060), Hainan Yazhou Bay Seed Laboratory in Hainan Province (B21Y10402, B22C10403), the China Postdoctoral Science Foundation (2022M722808), Hainan Provincial Natural Science Foundation of China (323QN313), High-performance Computing Platform of YZBSTCACC, the National Natural Science Foundation of China (32060452, 32160447, and 32160645), the Open Project of Guangxi Key Laboratory of Rice Genetics and Breeding (2022–36-Z01-KF03, 2022–36-Z01-KF05, and 2022–36-Z01-ZZ07), the Hainan Yazhou Bay Seed Laboratory and China National Seed Group (JBGS-B23YQ1517, JBGS-B23CQ15HP), the Basic Business Project of Guangxi Academy of Agricultural Sciences (GuiNongKe 2022YM01 and 2023YM63), and the project of the base and talent (Guike AD17129064 and Guike AD20297093).
Author information
Authors and Affiliations
Contributions
WX, PY and ZX designed the project; ZX performed SNP calling; WX, ZX and CL performed the data analysis; WX, ZZ, LJ, LZ and ZH interpreted the data and results; WX, ZX and PY wrote the manuscript with input from all authors. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Study complied with local and national regulations for using plants.
Consent for publication
No applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1:
Fig. S1. Population structure analyses of 747 rice accessions. Fig. S2. Quantile–quantile plots for the GWAS in full population using GLM, MLM and FarmCPU. Fig. S3. Quantile–quantile plots for the GWAS in indica population using GLM, MLM and FarmCPU. Fig. S4. Quantile–quantile plots for the GWAS in japonica population using GLM, MLM and FarmCPU. Fig. S5. GWAS results of RBLS in different populations using MLM. Fig. S6. GWAS results of RBLS in different populations using GLM. Fig. S7. Pathway enrichment of the cloned genes for RBLS. Fig. S8. Pathway enrichment of the candidate genes for RBLS.
Additional file 2:
Table S1. Description of 747 rice accessions.
Additional file 3:
Table S2. Significant association signals for RBLS in the indica and japonica populations detected by rMVP using FarmCPU model.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Zhu, X., Chen, L., Zhang, Z. et al. Genetic-based dissection of resistance to bacterial leaf streak in rice by GWAS. BMC Plant Biol 23, 396 (2023). https://doi.org/10.1186/s12870-023-04412-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12870-023-04412-7