
Abstract
Background
Long Terminal Repeat retrotransposons (LTR retrotransposons) are mobile genetic elements composed of a few genes between terminal repeats and, in some cases, can comprise over half of a genome’s content. Available data on LTR retrotransposons have facilitated comparative studies and provided insight on genome evolution. However, data are biased to model systems and marine organisms, including annelids, have been underrepresented in transposable elements studies. Here, we focus on genome of Lamellibrachia luymesi, a vestimentiferan tubeworm from deep-sea hydrocarbon seeps, to gain knowledge of LTR retrotransposons in a deep-sea annelid.
Results
We characterized LTR retrotransposons present in the genome of L. luymesi using bioinformatic approaches and found that intact LTR retrotransposons makes up about 0.1% of L. luymesi genome. Previous characterization of the genome has shown that this tubeworm hosts several known LTR-retrotransposons. Here we describe and classify LTR retrotransposons in L. luymesi as within the Gypsy, Copia and Bel-pao superfamilies. Although, many elements fell within already recognized families (e.g., Mag, CSRN1), others formed clades distinct from previously recognized families within these superfamilies. However, approximately 19% (41) of recovered elements could not be classified. Gypsy elements were the most abundant while only 2 Copia and 2 Bel-pao elements were present. In addition, analysis of insertion times indicated that several LTR-retrotransposons were recently transposed into the genome of L. luymesi, these elements had identical LTR’s raising possibility of recent or ongoing retrotransposon activity.
Conclusions
Our analysis contributes to knowledge on diversity of LTR-retrotransposons in marine settings and also serves as an important step to assist our understanding of the potential role of retroelements in marine organisms. We find that many LTR retrotransposons, which have been inserted in the last few million years, are similar to those found in terrestrial model species. However, several new groups of LTR retrotransposons were discovered suggesting that the representation of LTR retrotransposons may be different in marine settings. Further study would improve understanding of the diversity of retrotransposons across animal groups and environments.
Similar content being viewed by others

Introduction
Retrotransposons are transposable elements that replicate via an RNA intermediate [1]. They often make up a substantial fraction of the host genome in which they reside, occupying more than 40% of the human genome [2] and more than 50% of the maize genome [3]. Retrotransposons play a role in genome evolution [4] and can ultimately impact gene expression. However, our understanding of phylogenetic diversity of retrotransposons and their role in genome evolution is largely based on model organisms such as Drosophila melanogaster, Caenorhabditis elegans, Danio rerio, Mus musculus, Bombyx mori, etc. Animals living in marine environments and the deep-sea have been particularly underrepresented in transposable elements studies. For this reason, we explored the genome of the deep-sea tubeworm Lamellibrachia luymesi (Siboglinidae, Annelida) [5] which employs chemoautotrophic endosymbionts to inhabit hydrocarbon seeps in the Gulf of Mexico.
Retrotransposons are usually classified into two categories: LTR retrotransposons and non-LTR retrotransposons. Long terminal repeat retrotransposons (LTR retrotransposons) are transposable elements that are characterized by having long terminal repeats (LTRs) flanking an internal coding region. LTR retrotransposons usually serve as a model for the study of retroviruses [6], because both are structurally similar and phylogenetically related [7]. The main distinguishing characteristic is the presence of an envelope (env) gene in retroviruses which is absent in LTR retrotransposons. LTR retrotransposons are classified into three super families (Copia, Gypsy and Bel-pao), which differ in the arrangement of the protein domains encoded within the pol gene [8]. The two most common LTR retrotransposon super-families – Copia and Gypsy, are found in almost all eukaryotic lineages sampled to date [9]. These superfamilies display different distribution, abundance and diversity based on the element type and the host taxon been considered [10].
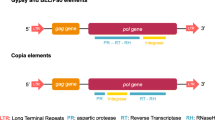
LTR retrotransposons (Fig. 1) includes long terminal repeats flanking elements that range from a few hundred bases to more than 5kb and usually start with 5’TG-3’ and ends with 5’-CA3’, a target site duplication (TSD) of 4-6bp, a polypurine tract (PPT), a primer binding site (PBS) and also gag and pol genes between the two LTRs [11, 12]. The gag gene encodes a structural protein that is essential for assembly of viral-like particles while the pol gene encodes four proteins domains including a protease (PR) which cleaves the Pol polyprotein, a ribonuclease H (RH) which cleaves the RNA in the DNA-RNA hybrid, a reverse transcriptase (RT) that copies retrotransposons RNA into cDNA and an integrase (INT) which integrates the cDNA into the genome. Occasionally, an additional open reading frame (aORF) may be downstream or upstream of the gag-pol gene, in sense or antisense orientation [13, 14]. Those located in the sense orientation encode proteins with certain structural and functional similarities to the env domain of retroviruses, and hence are sometimes called env-like domains [15, 16]. The env domain encodes for protein that is responsible for binding the cellular receptor and facilitates the early steps in the virus-cell interaction, and drives the fusion of viral and host cellular membrane [17]. In contrast, function of the aORF located in the antisense orientation is not clearly known, however , studies carried out so far suggests that they may be playing a regulatory role in retrotransposition [16, 18, 19].

Structure of a LTR retrotransposon. Gag - group-specific antigen gene; TSD- target site duplication; PR - aspartic protease gene; RT - reverse transcriptase gene; RH - ribonuclease-H gene; INT- integrase gene; PBS - primer binding site; PPT - polypurine tract. LTR retrotransposon structure was generated using Adobe Illustrator.
In previous reports, retroelements have been identified in marine organisms including sea urchins [20], corals endosymbionts [21] and crustaceans [22]. However, to the best of our knowledge, there has been minimal effort to characterize the LTR retrotransposons present in deep-sea (>200m) animals or in annelids. Available studies [5, 23, 24] tend to only consider transposable elements in context of their role in genome composition rather than detailed assessment of the elements and their evolution. Of particular interest, Li et al. assessed Lamellibrachia luymesi van der Land & Norrevang 1975; a deep-sea annelid. L. luymesi is a vestimentiferan tubeworm that forms bush-like aggregations at hydrocarbon seeps in the Gulf of Mexico. These animals lack a digestive tract and hosts sulfide-oxidizing, horizontally-transmitted bacterial symbionts for nutrition and growth [5, 25,26,27]. Their result showed that 2.52% of the genome consisted of LTR retroelements. However, the goal of the analysis was to see how much of the genome’s DNA was derived from repetitive elements using RepeatModeler [28] and RepeatMasker [29]. Their approach included altered copies such as truncated elements or solo LTR’s to gain a comprehensive view of L. luymesi’s genome composition rather than an exploration of the LTR retroelements biology. In the current study, we further characterized and classified LTR retrotransposons present in the genome of Lamellibrachia luymesi to shed light on the representation of LTR retrotransposon superfamilies, as well as augment understanding of the potential function and structure of intact elements. In addition, we also estimated insertion times of these elements to understand if they are due to recent or ancient events.
We hypothesized the possible presence of unknown LTR-retrotransposon families in marine organisms or unsampled animal lineages. This work represents an important step towards the characterization of LTR retrotransposons in marine systems (70% of the biosphere) and in unexplored animal lineages (e.g., annelids).
Results
Identification and classification of LTR-retrotransposon
A total of 223 intact LTR retrotransposons (Supplementary Table 1, 2) were identified in the 688 Mb L. luymesi genome, by screening and adjustment of LTR candidates from LTRharvest and LTR_Finder using modules employed in LTR_retriever (Fig. 2). Of the 223 intact LTR-retrotransposon identified by LTR_retriever, 51 were classified as unknown, 1 was classified as Copia while 171 were classified as Gypsy.

Bioinformatics pipeline for annotation of LTR retrotransposon in L. luymesi.
To further classify these elements, TEsorter was used to search their internal regions against Gypsy database (GYDB). Those matching at least one domain profile in GYDB were classified. All the 171 Gypsy and 1 Copia elements classified by LTR-retriever were also classified as Gypsy and Copia respectively in TEsorter. In addition, out of the 51 classified by LTR_retriever as unknown, 7 were classified as Gypsy, 2 were classified as Bel-pao while 1 was classified as Copia in TEsorter. The rest were not classified at all. Hence, in total, TEsorter classified 182 of the 223 intact LTR retrotransposons identified by LTR-retriever (Supplementary Table 2).
Further analyses were carried out on the remaining 41 elements not classified by TEsorter. This was accomplished by manually searching the internal region of these unclassified elements against PFAM [30] and Conserved Domains Database (CDD) [31] to identify domains present within their internal region. Results showed that 24 of the elements lacked domains matching any known profiles in the databases, 10 had domains that were unrelated to LTR retrotransposons (e.g., a transmembrane receptor, coagulation-inhibition site etc.), while the remaining 8 had only RT domains (Supplementary Table 1). To further verify and classify these elements, we used REXdb-metazoan database option of TEsorter. We also performed a manual hmmscan search using GYDB hmm profiles. The REXdb- metazoan option classified these elements as LINEs (Long interspersed nuclear elements) while no match was found in the GYDB hmm profile scan. Due to the inability to accurately classify these 41 elements, they were excluded from further analysis.
Summary details of the 182 LTR retrotransposons used for downstream analysis, which includes 178 Gypsy, 2 Bel-pao and 2 Copia elements are shown in Table 1.
Structural characterization
Of the 182 identified LTR retrotransposons, 32 elements had all domains (Gag and Pol – RT, INT, RH, PR) present with the remainder having at least one domain present. For Gypsy elements, 30 out the 178 had a complete set of domains, both the Bel-pao elements had a complete set of domains and both Copia elements lacked a complete set of domains. Further analysis to describe the position of these elements in relation to coding elements showed that 26.4% of them overlapped with coding elements, 46.2% were located > 5 kb of coding elements, 10.4% were located within 5-10 kb and the remaining 17% were more than 10 kb away from coding elements.
The target site duplication flanking ends of identified LTR retrotransposons ranged from 3 to 5 bp in length, with majority of them being 5 bp in length. Palindromic motifs detected in the elements includes TGCA, TACA, TATA, TCGT, TGAA, TGAC, TGAT and TTAT, with 89% of the LTR-retrotransposons having TGCA motif. In addition, differences in length of identified LTR-retrotransposons were substantial, ranging from 1389 bp-8866 bp while the length of the LTRs ranged from 103 to 1468 bp (Supplementary Table 2).
Estimation of insertion time
Insertion times of LTR retrotransposon elements in L. luymesi genome suggests that most elements were inserted around 1.0 million years ago (MYA; Fig. 3). The oldest observed and complete inserted retrotransposon was a Gypsy element, inserted around 2MYA. Interestingly, 50 Gypsy elements showed a 100% LTR identity, suggesting that they very recently inserted into the genome. However, calculations of insertion times used a substitution rate of 1.3 × 10− 8 substitution per bp per year, the LTR_retriever default based on the rice genome. Although these insertion time estimates for L. luymesi should be viewed with caution, decreasing the rate by two- or three-fold still suggests insertion times within the last few million years.

Insertion time distribution of intact LTR-RT in L. luymesi genome. Chart was generated using GraphPad Prism.
Phylogenetic analysis of LTR-retrotransposons
Phylogenetic analysis corroborates assignments made by TEsorter. However, weak internodal support limited inferences about evolutionary relationships. Final family assignment was done by considering placements of elements with strong nodal support indicating monophyletic lineage representing gene families (Fig. 4 for RT domain, Fig. 5 for RH domain, and Fig. 6 for INT domain). Due to issues of non-concordant evolutionary histories, domains were not combined into a single phylogenetic analysis. Naming conventions based on phylogenetic analyses are described in the Methods section.

RT domain phylogenetic tree. RT phylogenetic tree was generated in IQtree with the LG + F + R6 model. Tree lines are color-coded according to the superfamily above it. Elements in red are elements identified in the genome of L. luymesi.

RnaseH domain phylogenetic tree. RnaseH phylogenetic tree was generated in IQtree with the LG + R7 model. Tree lines are color-coded according to the superfamily above it. Elements in red are elements identified in the genome of L. luymesi.

INT domain phylogenetic tree. INT phylogenetic tree was generated in IQtree with the LG + R7 model. Tree lines are color-coded according to the superfamily name above it. Elements in red are elements identified in the genome of L. luymesi.
For Gypsy elements, phylogenetic analysis of the RT, RH and INT sequences showed that some elements fall into recognized families such as CSRN1 [32], Gmr1 [33] and Mag [Classification of discovered LTR retrotransposons Classification of LTR retrotransposons is dependent upon the presence and order of protein domains within the pol gene [11] (Fig 1). LTR_retriever based the classification of LTR retrotransposons on identification of conserved protein domains of each LTR retrotransposon candidate using profile Hidden Markov Models (pHMMs) of LTR retrotransposon domains from Pfam database [30]. Elements returning ambiguous pHMMs matches were classified as unknown. To refine classification, we employed the program TEsorter v1.2.5 [59] which translated nucleotide sequence of LTR retrotransposon candidates in all six frames and searched these sequences against HMM profiles obtained from existing mobile elements protein databases – specifically , REXdb [14] and Gyspsy database of mobile genetic elements [60]. For each domain of a sequence, only the best hit with highest score is retained. Classification into superfamilies and families were based on hits of the pol and gag genes to curated database. Elements lacking at least one domain were not classified. To do this step, fasta sequences of LTR retrotransposon candidates were first extracted using the call_by_seq_list.pl script from LTR_retriever package. Obtained sequences were then input into TEsorter (parameters = ‘-db gydb, -st nucl and -p 10’) for further classification. To facilitate communication, naming conventions for LTR retrotransposons families and elements identified in this study were created. Gypsy families were designated as LGF (Lamellibrachia Gypsy Family), followed by a unique number (e.g., LGF1, LGF2 etc.), Copia families were designated as LCF (Lamellibrachia Copia Family), followed by a unique number (e.g., LCF1) while Bel-pao families were designated as LBF (Lamellibrachia Bel-pao Family), followed by a unique number (e.g., LBF1). For individual elements, identified LTR retrotransposons were designated as LLXY#, where LL denotes 2 letters representing L. luymesi, XY denotes the first two letters of the superfamily it belongs to and # denotes the element number (e.g., LLGY1 represents a Gypsy element). Phylogenetic analysis was used to further validate family-level assignment of these elements and to access the evolutionary position of L. luymesi LTR retrotransposon candidates. For this purpose, amino acid sequences of INT, RT and RH domains were extracted from the LTR retrotransposon candidates following the guideline from TEsorter package. Gag and Protease (PR) sequences were excluded from analyses as they are known for their variability which prevents reliable alignments [61, 62]. To infer phylogenetic trees, amino acid sequence of INT,RH and RT from other known organisms were obtained from the GYDB database and recent studies [47, 53, 63], and aligned using MAFFT v7.407 [64] to amino acid sequence of INT, RT and RH from LTR retrotransposons found in L. luymesi genome. Each of the 3 domains was analyzed separately and a combined analysis was not done due to difference in taxon sampling and the fact that the domains may have distinct evolutionary histories. Maximum likelihood with bootstrap analysis was employed to construct phylogenetic trees using IQtree v1.6.12 [65] with the following parameters ‘-bb 100000, -nt AUTO, --runs 5’. The substitution model employed by IQtree for the INT domain tree was LG+R7, the RT domain tree was LG+F+R6 while the RH domain tree was LG+R7. Phylogenetic trees were mid-point rooted, visualized and edited using Figtree v1.4.2 [66]. Time since initial insertion of LTR retrotransposon candidates was estimated using scripts implemented in the LTR_retriever package. Insertion time were calculated as T = K/2 μ, where K is the divergence rate measured by the Jukes-Cantor model with K = − 3/4*ln (1-d*4/3) [67] and μ is the neutral mutation which is set at 1.3 × 10− 8 mutations per bp per year [68].Naming conventions
Phylogenetic analysis
Estimation of insertion time
Availability of data and materials
Assembled whole genomic sequence of Lamellibrachia luymesi generated by Li et al. was accessed from NCBI repository (https://www.ncbi.nlm.nih.gov/bioproject/PRJNA516467/) with WGS project - SDWI01, Bio project number - PRJNA516467 and Bio sample number -SAMN10789628. All data generated or analyzed during this study are included in this published article (and its supplementary information files). Scripts for bioinformatic analyses generated herein are available at https://github.com/clavia96/LTR_retrotransposon.git.
Abbreviations
- LTR:
-
Long Terminal Repeat
- RT:
-
Reverse Transcriptase
- RH:
-
Ribonuclease H
- INT:
-
Integrase
- HMM:
-
Hidden Markov Models
- BSV:
-
Bootstrap Value
- GYDB:
-
Gypsy Database of Mobile Genetic Elements
- MYA:
-
Million Years Ago
References
Boeke J, Stoye J. Retrotransposons, endogenous retroviruses, and the evolution of retroelements. Retroviruses. Cold Spring Harbor Laboratory Press. 1997.
Smit AF. Interspersed repeats and other mementos of transposable elements in mammalian genomes. Curr Opin Genet Dev. 1999;9(6):657–63.
SanMiguel P, Gaut BS, Tikhonov A, Nakajima Y, Bennetzen JL. The paleontology of intergene retrotransposons of maize. Nat Genet. 1998;20(1):43–5.
Kumar A, Bennetzen JL. Plant Retrotransposons. Annu Rev Genet. 1999;33(1):479–532.
Li Y, Tassia MG, Waits DS, Bogantes VE, David KT, Halanych KM. Genomic adaptations to chemosymbiosis in the deep-sea seep-dwelling tubeworm Lamellibrachia luymesi. BMC Biol. 2019;17(1):1–14.
Bowen NJ, McDonald JF. Drosophila euchromatic LTR retrotransposons are much younger than the host species in which they reside. Genome Res. 2001;11(9):1527–40.
**ong Y, Eickbush TH. Similarity of reverse transcriptase-like sequences of viruses, transposable elements, and mitochondrial introns. Mol Biol Evol. 1988;5(6):675–90.
Alzohairy AM, Sabir JSM, Gyulai G, Younis RAA, Jansen RK, Bahieldin A. Environmental stress activation of plant long-terminal repeat retrotransposons. Funct Plant Biol. 2014;41(6):557–67.
Muszewska A, Hoffman-Sommer M, Grynberg M. LTR Retrotransposons in Fungi. PLoS One. 2011;6(12):e29425 Redfield RJ, editor.
Thomas-Bulle C, Piednoël M, Donnart T, Filée J, Jollivet D, Bonnivard É. Mollusc genomes reveal variability in patterns of LTR-retrotransposons dynamics. BMC Genomics. 2018;19(1):821.
Wicker T, Sabot F, Hua-Van A, Bennetzen JL, Capy P, Chalhoub B, et al. A unified classification system for eukaryotic transposable elements. Nat Rev Genet. 2007;8(12):973–82.
Zhang L, Yan L, Jiang J, Wang Y, Jiang Y, Yan T, et al. The structure and retrotransposition mechanism of LTR-retrotransposons in the asexual yeast Candida albicans. Virulence. 2014;5(6):655–64.
Steinbauerová V, Neumann P, Novák P, Macas J. A widespread occurrence of extra open reading frames in plant Ty3/gypsy retrotransposons. Genetica. 2011;139(11-12):1543–55.
Neumann P, Novák P, Hoštáková N, Macas J. Systematic survey of plant LTR-retrotransposons elucidates phylogenetic relationships of their polyprotein domains and provides a reference for element classification. Mob DNA. 2019;10:1.
Carvalho M, Ribeiro T, Viegas W, Morais-Cecilio L, Rocheta M. Presence of env-like sequences in Quercus suber retrotransposons. J Appl Genet. 2010;51(4):461–7.
Gómez-Orte E, Vicient CM, Martínez-Izquierdo JA. Grande retrotransposons contain an accessory gene in the unusually long 3'-internal region that encodes a nuclear protein transcribed from its own promoter. Plant Mol Biol. 2013;81(6):541–51.
Steckbeck JD, Kuhlmann AS, Montelaro RC. Structural and functional comparisons of retroviral envelope protein C-terminal domains: still much to learn. Viruses. 2014;6(1):284–300.
McLane LM, Pulliam KF, Devine SE, Corbett AH. The Ty1 integrase protein can exploit the classical nuclear protein import machinery for entry into the nucleus. Nucleic Acids Res. 2008;36(13):4317–26.
Vicient CM, Casacuberta JM. Additional ORFs in Plant LTR-Retrotransposons. Front Plant Sci. 2020;11:555.
Gonzalez P, Lessios HA. Evolution of sea urchin retroviral-like (SURL) elements: evidence from 40 echinoid species. Mol Biol Evol. 1999;16(7):938–52.
Chen JE, Cui G, Wang X, Liew YJ, Aranda M. Recent expansion of heat-activated retrotransposons in the coral symbiont Symbiodinium microadriaticum. ISME J. 2018;12(2):639–43.
Piednoël M, Donnart T, Esnault C, Graça P, Higuet D, Bonnivard E. LTR-retrotransposons in R. exoculata and other crustaceans: the outstanding success of GalEa-like copia elements. PLoS One. 2013;8(3):e57675.
Wang K, Shen Y, Yang Y, Gan X, Liu G, Hu K, et al. Morphology and genome of a snailfish from the Mariana Trench provide insights into deep-sea adaptation. Nat Ecol Evol. 2019;3(5):823–33.
Sun J, Zhang Y, Xu T, Zhang Y, Mu H, Zhang Y, et al. Adaptation to deep-sea chemosynthetic environments as revealed by mussel genomes. Nat Ecol Evol. 2017;1(5):121.
Schulze A. Phylogeny of Vestimentifera (Siboglinidae, Annelida) inferred from morphology. Zool Scr. 2003;32(4):321–42.
Schulze A, Halanych KM. Siboglinid evolution shaped by habitat preference and sulfide tolerance. In: Hydrobiologia: Springer; 2003. p. 199–205.
Halanych KM. Molecular phylogeny of siboglinid annelids (a.k.a. pogonophorans): a review. In: Morphology, molecules, evolution and phylogeny in polychaeta and related taxa: Springer-Verlag; 2005. p. 297–307.
RepeatModeler Download Page. Available from: http://www.repeatmasker.org/RepeatModeler/. Cited 2021 Feb 20
RepeatMasker Home Page. Available from: http://www.repeatmasker.org/. Cited 2021 Feb 20
Finn RD, Bateman A, Clements J, Coggill P, Eberhardt RY, Eddy SR, et al. Pfam: The protein families database. Nucleic Acids Res. 2014;42(D1):222–30.
Marchler-Bauer A, Bryant SH. CD-Search: Protein domain annotations on the fly. Nucleic Acids Res. 2004;32(Web Server issue):W327–31.
Bae YA, Moon SY, Kong Y, Cho SY, Rhyu MG. CsRn1, a novel active retrotransposon in a parasitic trematode, Clonorchis sinensis, discloses a new phylogenetic clade of Ty3/gypsy-like LTR retrotransposons. Mol Biol Evol. 2001;18(8):1474–83.
Goodwin TJ, Poulter RT. A group of deuterostome Ty3/ gypsy-like retrotransposons with Ty1/ copia-like pol-domain orders. Mol Gen Genomics. 2002;267(4):481–91.
Volff J-N, Körting C, Altschmied J, Duschl J, Sweeney K, Wichert K, et al. Jule from the Fish **phophorus Is the First Complete Vertebrate Ty3/Gypsy Retrotransposon from the Mag Family. Mol Biol Evol. 2001;18(2):101–11.
Tubío JM, Naveira H, Costas J. Structural and evolutionary analyses of the Ty3/gypsy group of LTR retrotransposons in the genome of Anopheles gambiae. Mol Biol Evol. 2005;22(1):29–39.
McCarthy EM, Liu J, Lizhi G, McDonald JF. Long terminal repeat retrotransposons of Oryza sativa. Genome Biol. 2002;3(10):research0053.1.
Kaminker JS, Bergman CM, Kronmiller B, Carlson J, Svirskas R, Patel S, et al. The transposable elements of the Drosophila melanogaster euchromatin: a genomics perspective. Genome Biol. 2002;3(12):research0084.1.
**-Shan X, Qing-You X, Jun L, Guo-Qing P, Ze-Yang Z. Survey of long terminal repeat retrotransposons of domesticated silkworm (Bombyx mori). Insect Biochem Mol Biol. 2005;35(8):921–9.
Bowen NJ, McDonald JF. Genomic analysis of Caenorhabditis elegans reveals ancient families of retroviral-like elements. Genome Res. 1999;9(10):924–35.
Michaille JJ, Mathavan S, Gaillard J, Garel A. The complete sequence of mag, a new retrotransposon in Bombyx mori. Nucleic Acids Res. 1990;18(3):674.
Springer MS, Davidson EH, Britten RJ. Retroviral-like element in a marine invertebrate. Proc Natl Acad Sci U S A. 1991;88(19):8401–4.
Butler M, Goodwin T, Poulter R. An unusual vertebrate LTR retrotransposon from the cod Gadus morhua. Mol Biol Evol. 2001;18(3):443–7.
Simmen MW, Bird A. Sequence analysis of transposable elements in the sea squirt, Ciona intestinalis. Mol Biol Evol. 2000;17(11):1685–94.
Abe H, Ohbayashi F, Shimada T, Sugasaki T, Kawai S, Mita K, et al. Molecular structure of a novel gypsy-Ty3-like retrotransposon (Kabuki) and nested retrotransposable elements on the W chromosome of the silkworm Bombyx mori. Mol Gen Genet. 2000;263(6):916–24.
Copeland CS, Brindley PJ, Heyers O, Michael SF, Johnston DA, Williams DL, et al. Boudicca, a retrovirus-like long terminal repeat retrotransposon from the genome of the human blood fluke Schistosoma mansoni. J Virol. 2003;77(11):6153–66.
Terrat Y, Bonnivard E, Higuet D. GalEa retrotransposons from galatheid squat lobsters (Decapoda, Anomura) define a new clade of Ty1/copia-like elements restricted to aquatic species. Mol Gen Genomics. 2008;279(1):63–73.
De La Chaux N, Wagner A. BEL/Pao retrotransposons in metazoan genomes. BMC Evol Biol. 2011;11:154.
Copeland CS, Mann VH, Morales ME, Kalinna BH, Brindley PJ. The Sinbad retrotransposon from the genome of the human blood fluke, Schistosoma mansoni, and the distribution of related Pao-like elements. BMC Evol Biol. 2005;5:20.
Wicker T, Keller B. Genome-wide comparative analysis of copia retrotransposons in Triticeae, rice, and Arabidopsis reveals conserved ancient evolutionary lineages and distinct dynamics of individual copia families. Genome Res. 2007;17(7):1072–81.
Du C, Swigonová Z, Messing J. Retrotranspositions in orthologous regions of closely related grass species. BMC Evol Biol. 2006;6:62.
Chénais B, Caruso A, Hiard S, Casse N. The impact of transposable elements on eukaryotic genomes: from genome size increase to genetic adaptation to stressful environments. Gene. 2012;509(1):7–15.
Halanych KM, Bacheller JD, Aguinaldo AM, Liva SM, Hillis DM, Lake JA. Evidence from 18S ribosomal DNA that the lophophorates are protostome animals. Science. 1995;267(5204):1641–3.
Metzger MJ, Paynter AN, Siddall ME, Goff SP. Horizontal transfer of retrotransposons between bivalves and other aquatic species of multiple phyla. Proc Natl Acad Sci U S A. 2018;115(18):E4227–35.
LLUY_1.0 - Genome - Assembly - NCBI. Available from: https://www.ncbi.nlm.nih.gov/assembly/GCA_009193005.1. Cited 2020 Apr 14
Ellinghaus D, Kurtz S, Willhoeft U. LTRharvest, an efficient and flexible software for de novo detection of LTR retrotransposons. BMC Bioinformatics. 2008;9(1):18.
Xu Z, Wang H. LTR_FINDER: an efficient tool for the prediction of full-length LTR retrotransposons. Nucleic Acids Res. 2007;35(Web Server issue):W265–8.
Lerat E. Identifying repeats and transposable elements in sequenced genomes: How to find your way through the dense forest of programs. Heredity. 2010;104:520–33 Nature Publishing Group.
Ou S, Jiang N. LTR_retriever: A Highly Accurate and Sensitive Program for Identification of Long Terminal Repeat Retrotransposons. Plant Physiol. 2018;176(2):1410–22.
zhangrengang/TEsorter: TEsorter: lineage-level classification of transposable elements using conserved protein domains. Available from: https://github.com/zhangrengang/TEsorter. Cited 2020 Apr 14
Llorens C, Futami R, Covelli L, Domínguez-Escribá L, Viu JM, Tamarit D, et al. The Gypsy Database (GyDB) of mobile genetic elements: release 2.0. Nucleic Acids Res. 2011;39(Database issue):D70–4.
Vershinin AV, Ellis TH. Heterogeneity of the internal structure of PDR1, a family of Ty1/copia-like retrotransposons in pea. Mol Gen Genet. 1999;262(4-5):703–13.
Neogi U, Engelbrecht S, Claassen M, Jacobs GB, van Zyl G, Preiser W, et al. Mutational Heterogeneity in p6 Gag Late Assembly (L) Domains in HIV-1 Subtype C Viruses from South Africa. AIDS Res Hum Retrovir. 2016;32(1):80–4.
Cao L, Yin G, Cao Z, Bing X, Ding W. Identification and characterization of a LTR retrotransposon from the genome of Cyprinus carpio var. Jian. Genetica. 2016;144(3):325–33.
Katoh K, Standley DM. Article Fast Track MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol Biol Evol. 2013;30(4):772–80.
Nguyen L-T, Schmidt HA, Von Haeseler A, Minh BQ. IQ-TREE: A Fast and Effective Stochastic Algorithm for Estimating Maximum-Likelihood Phylogenies. Mol Biol Evol. 2015;32(1):268–74.
FigTree. Available from: http://tree.bio.ed.ac.uk/software/figtree/. Cited 2020 May 19.
Jukes TH, Cantor CR. Evolution of protein molecules. Mammalian protein metabolism III. New York: Academic Press; 1969. p. 21–132.
Ma J, Bennetzen JL. Rapid recent growth and divergence of rice nuclear genomes. Proc Natl Acad Sci U S A. 2004;101(34):12404–10.
Acknowledgements
I would like to thank the members of the Molette Lab for their valuable information and guidance. Also Dr. Joanna Solinska and Dr. Jason Upton provided helpful insights in this project. Lastly, I would like to thank Dr. Shujun Ou for his continued help throughout the course of this project. This is a Mollette Biology Laboratory contribution 107 and Auburn University Marine Biology Program contribution 205.
Funding
Project funded by Schneller Endowed Chair fund to KMH and NSF IOS-0843473.
Author information
Authors and Affiliations
Contributions
Both authors were involved in study design, interpretation and writing. Aroh performed bioinformatic analyses. The authors read and approved this manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Table S1.
Details (size, domain present e.t.c.) of elements excluded from further analysis. Table S2. Details of each LTR-retrotransposons found in Lamellibrachia luymesi genome, including element sizes, LTR sizes and pair identity, TSD sizes and sequences,motif sequences.
Additional file 2.
Integrase sequence alignment file.
Additional file 3.
Ribonuclease H sequence alignment file.
Additional file 4.
Reverse transcriptase sequence alignment file.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Aroh, O., Halanych, K.M. Genome-wide characterization of LTR retrotransposons in the non-model deep-sea annelid Lamellibrachia luymesi. BMC Genomics 22, 466 (2021). https://doi.org/10.1186/s12864-021-07749-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-021-07749-1