
Abstract
Background
Transposable elements (TEs) can represent one of the major sources of genomic variation across eukaryotes, providing novel raw materials for species diversification and innovation. While considerable effort has been made to study their evolutionary dynamics across multiple animal clades, molluscs represent a substantially understudied phylum. Here, we take advantage of the recent increase in mollusc genomic resources and adopt an automated TE annotation pipeline combined with a phylogenetic tree-based classification, as well as extensive manual curation efforts, to characterize TE repertories across 27 bivalve genomes with a particular emphasis on DDE/D class II elements, long interspersed nuclear elements (LINEs), and their evolutionary dynamics.
Results
We found class I elements as highly dominant in bivalve genomes, with LINE elements, despite less represented in terms of copy number per genome, being the most common retroposon group covering up to 10% of their genome. We mined 86,488 reverse transcriptases (RVT) containing LINE coming from 12 clades distributed across all known superfamilies and 14,275 class II DDE/D-containing transposons coming from 16 distinct superfamilies. We uncovered a previously underestimated rich and diverse bivalve ancestral transposon complement that could be traced back to their most recent common ancestor that lived ~ 500 Mya. Moreover, we identified multiple instances of lineage-specific emergence and loss of different LINEs and DDE/D lineages with the interesting cases of CR1- Zenon, Proto2, RTE-X, and Academ elements that underwent a bivalve-specific amplification likely associated with their diversification. Finally, we found that this LINE diversity is maintained in extant species by an equally diverse set of long-living and potentially active elements, as suggested by their evolutionary history and transcription profiles in both male and female gonads.
Conclusions
We found that bivalves host an exceptional diversity of transposons compared to other molluscs. Their LINE complement could mainly follow a “stealth drivers” model of evolution where multiple and diversified families are able to survive and co-exist for a long period of time in the host genome, potentially sha** both recent and early phases of bivalve genome evolution and diversification. Overall, we provide not only the first comparative study of TE evolutionary dynamics in a large but understudied phylum such as Mollusca, but also a reference library for ORF-containing class II DDE/D and LINE elements, which represents an important genomic resource for their identification and characterization in novel genomes.
Similar content being viewed by others

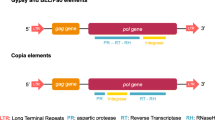
Background
Transposable elements (TEs) are selfish genetic elements that replicate independently from the replication of the host genome [1, 2]. They are widespread and ubiquitous across all branches of the eukaryotic tree of life and, although showing a remarkable sequence diversity across organisms, the conservation of common catalytic domains responsible for their replication suggests that their emergence could be traced back to the eukaryotic most recent common ancestor or even predate it [3].
TE classification is not straightforward, although many efforts have been undertaken to try to reconcile their diversity in a systematic framework. Two main classes are generally recognized: class I, which includes all TEs replicating via RNA intermediates, and class II, which embodies TEs moving via DNA intermediates [4]. This latest distinction still represents the only unambiguous classification of TEs. Conversely, the within-class diversity is much more complicated to analyze, since it can be performed both with mechanistic and homology-based criteria [5]. For example, considering the way TEs replicate and reintegrate, all class I elements use a “copy-and-paste” mechanism, while class II exhibits several models: the classical “cut-and-paste,” or the “peel-and-paste” (also known as rolling-circle replication) or even the “self-synthesizing” model (reviewed in [5]). The current classification scheme, which is also implemented in the main TE database, Repbase [6], is based on homology and structural similarities [7]. Class I elements mainly include long terminal repeat (LTR) elements and long interspersed nuclear elements (LINEs, also indicated as non-LTR elements) which encode for a reverse transcriptase (RT), an endonuclease (EN), and other domains used to reintegrate in the host genome. Class II elements, on the other hand, include terminal inverted repeat (TIR) elements, Helitrons, and Mavericks (also known as Polintons). In addition, both classes include non-autonomous elements (short interspersed nuclear elements, SINEs, and miniature inverted-repeats transposable elements, MITEs), TEs usually with a smaller size, which do not code for the enzymes necessary for replication/reintegration but parasitize those encoded by their autonomous counterparts [7]. Besides this commonly accepted scheme, further classification efforts are less clear. Generally speaking, when taking into consideration coding TEs, the clustering pattern after a phylogenetic analysis of their ORF(s) is taken as an indication of clades that should be considered possible families, groups of elements, or clades [5].
Although a common approach, the phylogenetic framework has limitations in this context both because of the sometimes unclear homology of TE ORFs and the genomic turnover of paralogous TE lineages blurring the phylogenetic signal [8].
The same replicative dynamics of TEs may impact their phylogenetic clustering: in fact, based on studies on mutation distribution on non-autonomous class I Alu sequences in the human genome, two distinct models have been formulated to explain how TEs replicate [9]. The first model, named “master gene model,” implies that one or few copies give origin to all other copies in the genome producing new, so-called families each time a master copy mutates. This way, new families are generated in different timeframes. On the contrary, in the other model, termed “transposons model,” each new copy can produce other copies with the outcome of getting several families produced nearly at the same time.
The rate at which TEs replicate can be a function of several different factors, including the ability of the host genome to limit their uncontrolled proliferation. In particular, the successful invasion of a genome by TEs can be dependent on a complex interplay among TE features, host genome biology, repression mechanisms interfering with TE functionality, and the extent of selective pressures on the outcome of TE insertions [10]. Despite this, some TE lineages managed to reach very high copy numbers in the host genomes, apparently esca** such controlling mechanisms. A suitable model to explain these dynamics has been formulated on the well-studied human SINE family Alu and on their autonomous counterparts L1 LINEs. These elements show several subfamilies that evolved following a master gene model in different hominid lineages during the last few million years. However, their origin seems to predate their species-specific expansions by far, with little or no transposition for tens of million years. Han et al. [1), a pattern consistent with an ongoing transposition of these elements [35].
We also added SINE families in the same RepeatMasker run, and we obtained their reliable genome occurrence in the 13 species selected for in-depth SINE mining (see the “Richness, diversity, and distribution of RT-containing LINEs” and “Construction of a manually curated library for LINE, SINEs, and DDE/D-related transposons” sections). SINE genome occurrence can greatly vary between and within species belonging to different bivalve orders (Table 2). The genomes of A. marissinica (6.02%), T. granosa (3.69%), S. broughtonii (4.37%), and B. platifrons (4.68%) host a relatively high number of SINEs while on the contrary, we observed a great reduction in the genome of C. gigas (0.08%) and S. glomerata (0.31%). Different SINE types successfully colonize different bivalve genomes: the Deu family was found to be dominant in A. marissinca (72% of the overall SINE complement), C. sinensis (94%), and S. broughtonii (55%), while the V family is dominant in the B. platifrons genome (67%) and the Meta in S. constricta (54%) and S. grandis (50%). Finally, in T. granosa, both Deu and V families occupy a considerable proportion of the overall SINE complement of respectively 30% and 46%. Finally, we did not find any evidence of a significant correlation between SINE and LINE genomic occurrence (Spearman’s rho = 0.31, p = 0.33).
Discussion
A comprehensive TE annotation for bivalves
The phylum Mollusca shows a high level of organism diversity and includes species that are important for both their ecological and economic value. Although genomic studies are accumulating and comparative analyses are becoming more common for these organisms, a deep analysis of the mobilome is still limited to single genomes or to a few comparative studies with only a handful of species [24, 25]. As could be expected, this also resulted in a scarce representativity of molluscan TEs in the public databases which makes their automated annotation less reliable. As previously shown, high-quality, manually curated repeat libraries are considered necessary for a consistent, reliable repeat annotation and characterization in novel genomes [26, 27]. In the present analysis, we decided to focus our efforts on bivalves, which represent 27 out of the 39 analyzed genomes, due to the recent, increasing genome sequencing efforts for this class. The inclusion of five gastropod genomes, representative of their major lineages, together with two cephalopods, one polyplacophoran genome, and three annelids allowed us to identify the major shifts in TE composition that occurred during molluscan evolutionary history. To overcome the limitations of automatically generated TE sequence libraries, we set up a pipeline which included both automated, ORF-based extraction and classification and manual curation approaches and that has been used consistently across the analyzed genomes. In particular, the manual curation process allowed us to provide the first freely available and manually curated repeat library for bivalves, comprising DDE/D, LINEs, and a subset of SINE elements for a total of 1609 elements comprising all identified LINEs, with the exception of the low copy number R2 superfamily and 12 different DDE/D-related superfamilies. These new genomic resources could help future genome annotation projects and shed novel insight into TE evolutionary dynamics in bivalves. On the other hand, the ORF-based approach allows us to confidentially characterize both LINEs and DDE/D-related TE complements. As a comparison, concerning LINEs in the RepBase library v. 20181026, 1031 sequences are deposited for molluscs, with 796 of them belonging to well-characterized C. gigas. Fifty-nine of these are annotated as LINEs and, more specifically: one R2, two CR1, 12 CR1-Zenon, 14 L1-Tx1, and 27 RTE-X. In the present analysis, we also found multiple Proto2, RTE-BovB, and L2/L2-2 elements. Regarding DDE/D transposons, out of 422 total sequences coming from RepBase for C. gigas, 92 possess an ORF longer than 300 amino acids, and they belong to 13 different superfamilies. With our approach, we manage to identify ORF-derived signatures coming from all of them, with the expectation of Zator, Merlin, and Sola1 for which only two sequences are deposited for each superfamily in RepBase. Overall, these results suggest that our ORF-based approach successfully captures in a flexible way most of the diversity of coding TEs in non-model species.
We also paid particular attention to filter out possible misannotations from the automatically generated TE sequence libraries, such as the inclusion of repetitive genes, tandem repeats, degenerate, and low-copy number families, which are hard to correctly annotate and classify. This approach is probably quite conservative, indeed in some instances, it provided different estimates of the overall TE content compared to published genome papers. For example, in Mytilus edulis, our study estimated 47% of TE content vs the 56% provided in [36]; the same holds for S. glomerata (42% in the present study vs 45% in [37]) and for A. granulata (18% here vs 23% in [38]). In other instances, though, our analysis provided almost the same estimates as in the previous analyses, as in M. coruscus (49% here vs 47% in [39]), A. immaculata (41% here vs 40% in [40]), and M. mercenaria (51% here vs 49% in [41]).
TEs have been shown to be one of the major contributors to genome size evolution in metazoan lineages, such as insects [42] and vertebrates [43], and in angiosperms as well [44]. Our analyses provided further support for this hypothesis finding a positive correlation between TE content and assembly size also in molluscs. Across bivalves, the TE content varies greatly, ranging from ~ 20% in the Pectinida M. yessoensis up to ~ 60% in the Mytilida M. philippinarum. Different sequencing technologies and sequencing depths could potentially contribute to such differences; however, it must be noted that also for Illumina sequenced genome, we observed a high TE content, such as for the M. philippinarum and B. platifrons. It is interesting that the low TE load found across all analyzed Pectinida species. In fact, this order includes the most TE-poor bivalve species, with almost twofold less TE content compared to Mytilida and Ostreida. Similar occurrences of interspersed repeats were already observed for this lineage during whole genome sequencing projects [45,46,47,48], and transposable elements hosted by M. yessoensis were found to be generally less active in recent times compared to what was observed in the Pacific and pearl oysters [46]. This low TE activity was suggested to be the reason behind their conserved genome architecture that could resemble that of bilaterian ancestors [46]. However, as well described in birds, low TE content and apparent lack of activity could also originate from nonallelic homologous recombination which could physically remove TEs and other repetitive regions from the genome without implying a general genomic stability [49].
Concerning class I elements, LTR elements in general occupy a low proportion of host genomes as previously observed by [24], while we found LINE elements as the richest retroelements. They contribute from 1.61 to 10.84% respectively in C. virginica and M. coruscus genomes using automatically generated TE sequence libraries and between 6.18% for A. marissinica and 0.82% for P. fucata using manually curated libraries. A similar scenario occurs also for SINE elements, whose genome coverage can greatly vary between different bivalve species using both automatic and manually curated libraries. In both instances, we identify the genomes of A. marissinica, B. platifrons, and Arcida as richer in SINEs compared to other analyzed bivalves, but we did not find any evidence of a general increase of the SINE complement coupled with an increase of their autonomous counterparts LINEs.
Class II and RC elements generally outnumber other TEs, especially in bivalves where DNA elements were found significantly enriched compared to all retroposons. This is strikingly different from what is observed in mammals, where retroposons constitute the most successful TE group, but similar to what is observed in actinopterygian fishes where class II elements greatly dominate the overall TE content [43]. Moreover, we found that non-autonomous counterparts (MITEs) occupy a considerable proportion of host genomes suggesting the high proliferation of small, non-autonomous copies. Within the most rich superfamilies of DDE/D ORF-derived signatures in bivalve genomes, we identified TcMariner and hAT lineages. Interestingly, the same superfamilies were also found to be the richest of ORF signatures in all other analyzed molluscs and to be ubiquitous even when using the automatically generated TE sequence libraries. Both TcMar and hAT superfamilies were found anciently expanded across cephalopods in a recent study from [25], possibly suggesting their high representativeness as a plesiomorphic state of molluscs. On the other hand, we could identify notable examples of bivalve-specific expansion, such as for Academ and RC elements. The former seems to be poorly represented in non-bivalve genomes, with only few ORF identified in the ringworms C. teleta and H. robusta and few insertions annotated in non-bivalve molluscs when using automatically generated libraries. RC elements can occupy up to 12% of the analyzed Crassostrea species. As a comparison, RC have a more patchy distribution in arthropod genomes, generally contributing to a smaller extent of the genome size with only few lineage-restricted expansions (e.g., Drosophila and Musca domestica [42, 50]). Also in plants, where they were first discovered, they are usually less represented, covering a maximum of 6% of the maize genome [ In the present study, we performed the first comparative analysis of transposable element evolutionary dynamics across molluscs with a particular emphasis on bivalves, an ecologically and economically important group. Despite genomic resources still being limited to few representative species compared to other clades, such as insects, the relatively low taxon sampling allowed us to deeply characterize for the first time their LINE and class II DDE/D-related complement. Moreover, because a high-quality repeat library is essential for the analyses of new genomes, our reference set of classified LINEs and DDE/D elements can be used to improve genome annotations and/or to easily classify novel elements across other lophotrochozoans. We also want to emphasize the necessity to extend similar analyses to other classes of transposons, empowering the scientific community with novel and high-quality genomic resources. While TEs have been hypothesized to be involved in the evolution of multiple bivalve genomic oddities, such as high levels of gene presence-absence variation [79] and of hemizygosity [80], the ability to identify their possible role deeply and consistently in sha** bivalve genome evolution will be limited as long as the great majority of elements are unclassified, fragmented, or not freely accessible for the scientific community. With our approach, we discovered a diverse set of LINEs and DDE/D that were likely already greatly diversified in the most recent common ancestor of bivalves. The restricted emergence of the bivalve-rich Proto2, RTE-X, CR1-Zenon, and Academ elements could have contributed to bivalve fast radiation providing novel raw genomic material for their diversification. Moreover, we found that this LINE diversity seems to be maintained across extant species by an equally diverse set of potentially contemporary active families that could follow a stealth driver model of evolution. Indeed, multiple families seem to be able to survive and co-exist for a long period of time in the host genome without triggering the evolution of sequence-specific repression mechanisms, resembling what was previously observed in multiple non-mammalian vertebrates such as lizards and fishes. Finally, despite their relatively low genome occurrence, several LINE superfamilies/clades/types emerged, and others contracted in a lineage-specific manner during the diversification of bivalves. Therefore, this highly diverse LINE complement, despite being less represented than class II elements, is a rather dynamic portion of bivalve genomes and can play important roles in local adaptations and lineage-specific evolutionary dynamics. Thirty-six molluscs and three annelid genomes were downloaded from publicly available resources (NCBI, GigaDB, Dryad, MolluscDB, dbSROG, and Phaidra, see Additional file 1: Table S1), giving preference to bivalve assemblies representative of their major clades. Concerning molluscs, we selected 27 genomes belonging to bivalves, five to gastropods, two to cephalopods, and one to the polyplacophoran A. granulata. The species tree was manually reconstructed following the phylogenetic relationships found in recent phylogenomic studies [81,82,83,84] as well as the reference phylogeny presented in MolluscDB [85]. For each analyzed genome, we compiled species-specific repeat libraries using a combination of structural and homology-based methods. RepeatModeler v. 2.0.1 [86] with the LTR pipeline extension which includes the structural-based LTRharvest [87] and LTR_retrivier packages [88], MITE Tracker [89], and HelitronScanner v. 1.1 [2: Fig. S1. To have a more precise picture of the representation of different superfamilies and clades of both LINEs and DDE/D class II elements, we applied an ORF-based extraction and classification pipeline. Firstly, insertion sites resulting from RepeatCraft analyses were extracted with the bedtools suite [98] together with 1000 bp at both ends to correct for possible partial/fragmented annotations due to the likely incomplete status of automated generated consensus sequences [26]. ORFinder was then used to identify and extract non-overlap** open reading frames (-n) with a required methionine as the start codon and a minimum ORF length of at least 300 amino acids (i.e., 900 nucleotides; -ml 900). To further characterize both class II DDE/D-related transposons and LINE elements, we used an HMM-based approach. For the former, we started from the amino acid sequences corresponding to DDE/D domains found in the 17 superfamilies described in [31]. All sequences coming from each superfamily (namely hAT, Tc1/Mariner, PIF/Harbinger, CMC, Merlin, MULE, P, Kolobok, Novosib, Sola1, Sola2, Sola3, PiggyBac, Transib, Academ, Ginger, Zator) were downloaded and separately aligned with MAFFT v. 7.475 [99] (E-INS-i strategy), and from each alignment, we build up a superfamily-specific HMM profile using the hmmbuild function from the HMMER3 package [100]. The collection of all 17 profiles was then used as a target database for hmmscan homology searches (E-value < 1E − 5) against all extracted ORFs provisionally annotating each element based on the corresponding best hit. To avoid misclassification of Ginger elements due to their high homology to Gypsy-encoded integrases [101] and to confirm the classification of all ORFs, we additionally blasted all significant hits against the full RepeatPep library (Blastp; E-value 1E − 05), imitating a reciprocal best-hit approach. Sequences with a best hit against a different superfamily compared to our previous HMM-based classification were considered as miss-classified and discarded. For LINE elements, we started with an RPSblast search on the same set of extracted and translated ORFs against the complete CDD database (E-value < 1E − 05). Sequences with a significant hit against RT-related profiles were considered as putative retrotransposons (see Additional file 28: Table S8 for a list of CDD entries). To distinguish between LTR- and LINE-derived RT-containing ORFs, all LINE and LTR elements from the Repeatpeps library were extracted and separately aligned with MAFFT v. 7.475 (l-INS-i strategy) together with the seed sequences of the RVT_1 Pfam HMM profile (PF00078) to manually identify boundaries of the RT domain. We extracted LINE and LTR RTs from the resulting alignments, and we built two class-specific HMM profiles with the hmmbuild function from the HMMER3 package. The two profiles were then used as a target database for hmmscan (E-value < 1E − 5) homology searches of our previously identified RT-containing ORFs. Sequences with the best hit against the LTR-specific RT profile were considered as putative LTR and therefore discarded from subsequent analyses. LINE elements were considered autonomous when both RT and EN domains (see Additional file 28: Table S8 for a list of CDD entries) were present on the same ORF (i.e., non-intervening stop codons). Sequences missing the EN domain were classified as RT-only LINEs. To test the interplay between assembly quality and the ability to identify RT-containing and autonomous LINEs as well as DDE/D-related transposons, we checked for a correlation between a number of identified elements and contig/scaffold N50 with Spearman’s rank correlation tests. All confirmed LINEs (regardless of being autonomous or RT-only) and DDE/D-containing transposons were clustered at the nucleotide level using CD-HIT and following the 80–80 rule (same parameter set used for repeat library construction). Therefore, hereafter, we will refer to clusters as groups of TEs related by high nucleotide homology along their coding sequence to distinguish them from the canonical transposon families which ideally should take into consideration the elements along their entire length [7]. For LINE elements only, we additionally called “low-copy number clusters” clusters with less than 5 members and as “singleton cluster” sequences that did not fall in any cluster. For class II elements, we avoid such classification because non-autonomous members of a family can replicate through the genome parasitizing their autonomous counterparts. Moreover, while the presence of a complete ORF can give some first insight on which superfamilies/clades could have been more active in recent/mid times, on the other hand, it must be noted that this approach is not able to identify non-autonomous elements thus greatly underestimating the number of short Class II transposons. ORF-containing LINE elements were classified using a phylogenetic approach. We adopted the superfamily classification scheme proposed by [7] and the clade classification proposed by [29], as in [102], while we use the “type” term to refer to the RepeatMasker or Dfam classification schemes [103]. Starting from previously identified clusters (> 5 members), we extracted the amino acid sequence of the RT domain based on the coordinates of the RPSblast hits. RT segments were aligned with MAFFT v. 7.475 (g-INS-i strategy) and cleaned from columns with gaps in more than 50% of the sequences using TrimAl [104]. Cons from the EMBOSS package [105] was then used to build up a consensus sequence from the resulting alignment setting the parameter plurality to 3. RT consensus sequences were then aligned together with reference LINE sequences from [29] and a subset of LTR and LINE elements from the Repeatpeps library, using MAFFT and a g-INS-i strategy. Poorly aligned sequences were removed from the alignment using TrimaAl (-resoverlap 0.75 -seqoverlap 80). Because of the short RT domain, the deep divergence time of LINE superfamilies, and the consequently difficulties in identifying stable LINE phylogenies (e.g., [29, 30, 106]), we used a combination of neighbor-joining, unconstrained maximum likelihood (ML), and constrained ML tree inferences. Each topology was then statistically tested in a ML framework to produce a confident phylogeny useful for LINE classification. We performed (a) a neighbor-joining (NJ) clustering with Clearcut v. 1.0.9 [107], reshuffling the distance matrix and using a traditional neighbor-joining algorithm (–shuffle and –neighbor options, respectively); (b) 5 unconstrained maximum likelihood (ML) tree searches with IQtree v. 2.1.3 [108] and the corresponding best-fit evolutionary model identified by ModelFinder2 [109]; (c) 6 constrained ML tree searches forcing the full NJ topology (FullNJ constraint, one run); and (d) only the monophyly of LINEs superfamilies, as inferred by the NJ tree, with the exception of Jockey and I superfamilies which were constrained in a single, comprehensive monophyletic clade (SupFAM constraint, 5 runs). For the unconstrained and the SupFAM-constrained ML tree inferences (analyses b and d, respectively), nodal support was estimated with 1000 UltraFastBootstrap replicates [110]. All ML topologies were tested using the Kishino-Hasegawa test [111], Shimodaira-Hasegawa test [112], expected likelihood weights [113], and approximately unbiased (AU) test [114]. As an additional confirmation of our classification and to avoid the inclusion of Penelope-like elements, we (a) blasted each consensus RT (blastp; E-value < 1E − 5) against all protein sequences from the RepeatPeps library extracting the best-hit for each query sequence and (b) used the online implementation of RTClass1 [29] on a random subset of 111 RT sequences covering all identified clades. Low-copy numbers, singletons, and clusters removed by TrimAl were classified based on Blastp best-hit (E-value < 0.05) against tree-based classified clusters and the whole RepeatPeps library for competing purposes. For the low-copy clusters, one representative (i.e., the longest) sequence was used. For bivalve species, and excluding the poorly represented R2 superfamily, the correlation between the number of RT-containing LINEs and the number of clusters in each identified LINE clade was tested for each superfamily separately with Spearman’s rank correlation tests. To have a first insight into the SINE composition of bivalves, we selected 13 species (namely, A. marissinca, C. sinensis, C. gigas, S. glomerata, T. granosa, S. broughtonii, M. coruscus, B. platifrons, S. constricta, S. grandis, P. maximus, M. yessoensis, M. nervosa) representative of Venerida, Ostreida, Arcida, Mytilida, Adepedonta, Pectinida, and Unionida, to mine additional SINE candidates using SINE_Scan v1.1.1 [115]. This software collects and validates SINE candidates based on copy number across the genome, presence of target site duplications (TSDs), and trRNA-related heads. All representative elements were merged with consensus sequences classified as SINEs by RepeatModeler in the corresponding species-specific repeat library (see the “Class-level mollusc mobilome characterization using automatically generated TE sequence libraries” section) and subjected to manual validation and curation as described in the following section. After this process, curated consensus sequences were annotated using the RepeatClassifier utility from the RepeatModeler package. We selected a set of the previously found LINEs RT, SINEs, and DDE/D-containing clusters for manual refinement, following [27] guidelines. For LINEs, we selected all clusters with at least one autonomous element (i.e., encoding for an ORF with both RT and EN domains without interrupting stop codons) and five other sequences (both autonomous and/or RT-only) while for DDE/D elements, we required only the presence of at least five elements in the corresponding cluster. These criteria were chosen in order to prioritize the manual curation of sequences that likely possess one or more autonomous copies across the genome and thus could potentially be recently mobilized or mobilize their non-autonomous counterparts. Members of LINEs and DDE/D-related clusters were aligned at the nucleotide level using MAFFT (–auto strategy). CIAlign [116] was then used to remove insertions found in less than 50% of the sequences and to construct a nucleotide consensus sequence (–remove-insertions and –make-consensus option). At this set of LINEs and DDE/D preliminary consensus, we also added all the aforementioned SINEs, and all sequences were subjected to a “blast-extend-extract” process with a minimum required query coverage and identity of 70, extending each hit by 3 kb and extracting the top 25 hits for each query sequence and building up a preliminary consensus sequence using CIAlign. Resulting alignments were manually inspected to (i) identify structural features (e.g., microsatellites for LINEs and SINEs at the 3′ end 5′ truncations for LINEs, terminal inverted repeats, and superfamily-specific motifs for DDE/D elements), (ii) identify boundaries of the elements searching for TSDs whenever possible, (iii) identify domain signatures using the CDD web server, and (iv) correct and extend as long as possible the consensus sequence. Additionally, for SINE only, we also required (a) the presence of a detectable tRNA-related region at the 5′ ends and predicted with tRNAScan-SE (sequence source: mixed; score cutoff 0.01 [117]) and (b) the presence of a central domain and/or a tail region after the tRNA-related head. It must be noted that the presence of TSDs to confirm the boundaries of the element was only required for SINEs and class II superfamilies that exhibit them (thus excluding for example the SPY group from the PIF-Harbinger superfamily; see [120] and bowtie2 [121] to align reads on extracted insertions. Raw counts were then normalized by the length of the corresponding family consensus sequences, and TPM values were calculated. Log2-transformed normalized counts were tested for a correlation with the number of previously identified 3′ anchored insertions with a minimum length of 100 bp for the corresponding family for each species, tissue, and biological replicate separately.Conclusions
Methods
Genomic resources and phylogeny construction
Mining and annotation of interspersed repeats
ORF-based annotation of RT containing LINEs and class II DDE/D elements
Tree-based classification of ORF-containing LINE elements
Additional prediction of SINEs in a subset of selected species
Manual curation of LINEs, SINEs, and DDE/D-related transposons
Availability of data and materials
All data generated or analyzed during this study are included in this published article, its supplementary information files, and publicly available repositories. Phylogenetic trees can be found in Additional file 29: Data S1 together with the multiple sequence alignment used to generate them in Additional file 30: Data S2. Manually curated families can be found in Additional files 31, 32, and 33 with a RepeatMasker formatted style as well as in the GitHub repository (https://github.com/CompBio-BO/Bivalvia_TEs) and in DFAM under Creative Commons CC0 1.0 public domain license. All supplementary data have been also deposited in a figshare database under the https://doi.org/10.6084/m9.figshare.22188280.v1 [122]. Scripts used to automatically generate the species-specific repeat libraries and to extract LINEs and DDE/D-related ORFs can be found in GitHub (https://github.com/jacopoM28/EvoTEs_BiV) and in Zenodo under the https://doi.org/10.5281/zenodo.7944844 [123].
Abbreviations
- TE:
-
Transposable elements
- LINEs:
-
Long interspersed nuclear elements
- RT:
-
Reverse transcriptase
- EN:
-
Endonuclease
- SINEs:
-
Short interspersed nuclear elements
- LTR:
-
Long terminal repeat
- TIR:
-
Terminal inverted repeats
- MITEs:
-
Miniature inverted-repeats transposable elements
- RC:
-
Rolling circle
- MULE:
-
Mutator-like elements
References
Werren JH, Nur U, Wu CI. Selfish genetic elements. Trends Ecol Evol. 1988;3(11):297–302.
Bourque G, Burns KH, Gehring M, Gorbunova V, Seluanov A, Hammell M, et al. Ten things you should know about transposable elements. Genome Biol. 2018;19(1):199.
Wells JN, Feschotte C. A Field guide to eukaryotic transposable elements. Annu Rev Genet. 2020;54:539–61.
Finnegan DJ. Eukaryotic transposable elements and genome evolution. Trends Genet. 1989;5:103–7.
Arkhipova IR. Using bioinformatic and phylogenetic approaches to classify transposable elements and understand their complex evolutionary histories. Mob DNA. 2017;8(1):19.
Bao W, Kojima KK, Kohany O. Repbase update, a database of repetitive elements in eukaryotic genomes. Mob DNA. 2015;6(1):11.
Wicker T, Sabot F, Hua-Van A, Bennetzen JL, Capy P, Chalhoub B, et al. A unified classification system for eukaryotic transposable elements. Nat Rev Genet. 2007;8(12):973–82.
Luchetti A, Mantovani B. Non-LTR R2 element evolutionary patterns: phylogenetic incongruences, rapid radiation and the maintenance of multiple lineages. PLoS ONE. 2013;8(2):e57076.
Deininger PL, Batzer MA, Hutchison CA, Edgell MH. Master genes in mammalian repetitive DNA amplification. Trends Genet. 1992;8(9):307–11.
Kelleher ES, Barbash DA, Blumenstiel JP. Taming the turmoil within: new insights on the containment of transposable elements. Trends Genet. 2020;36(7):474–89.
Han K, **ng J, Wang H, Hedges DJ, Garber RK, Cordaux R, et al. Under the genomic radar: the stealth model of Alu amplification. Genome Res. 2005;15(5):655–64.
Šatović E, Plohl M. Tandem repeat-containing MITEs in the clam Donax trunculus. Genome Biol Evol. 2013;5(12):2549–59.
Vera M, Bello X, Álvarez-Dios JA, Pardo BG, Sánchez L, Carlsson J, et al. Screening of repetitive motifs inside the genome of the flat oyster (Ostrea edulis): transposable elements and short tandem repeats. Mar Genomics. 2015;24:335–41.
Luchetti A, Šatović E, Mantovani B, Plohl M. RUDI, a short interspersed element of the V-SINE superfamily widespread in molluscan genomes. Mol Genet Genomics. 2016;291(3):1419–29.
Nishihara H, Plazzi F, Passamonti M, Okada N. MetaSINEs: broad distribution of a novel SINE superfamily in animals. Genome Biol Evol. 2016;8(3):528–39.
Šatović E, Plohl M. Two new miniature inverted-repeat transposable elements in the genome of the clam Donax trunculus. Genetica. 2017;145(4–5):379–85.
Biscotti MA, Barucca M, Canapa A. New insights into the genome repetitive fraction of the Antarctic bivalve Adamussium colbecki. PLoS ONE. 2018;13(3):e0194502.
Lee SI, Gim JA, Lim MJ, Kim HS, Nam BH, Kim NS. Ty3/Gypsy retrotransposons in the Pacific abalone Haliotis discus hannai: characterization and use for species identification in the genus Haliotis. Genes Genomics. 2018;40(2):177–87.
Puzakov MV, Puzakova LV, Cheresiz SV. An analysis of IS630/Tc1/mariner transposons in the genome of a Pacific oyster. Crassostrea gigas J Mol Evol. 2018;86(8):566–80.
Šatović E, Luchetti A, Pasantes JJ, García-Souto D, Cedilak A, Mantovani B, et al. Terminal-repeat retrotransposons in miniature (TRIMs) in bivalves. Sci Rep. 2019;9(1):19962.
Puzakov MV, Puzakova LV, Cheresiz SV. The Tc1-like elements with the spliceosomal introns in mollusk genomes. Mol Genet Genomics. 2020;295(3):621–33.
Vojvoda Zeljko T, Pavlek M, Meštrović N, Plohl M. Satellite DNA-like repeats are dispersed throughout the genome of the Pacific oyster Crassostrea gigas carried by Helentron non-autonomous mobile elements. Sci Rep. 2020;10(1):15107.
Metzger MJ, Paynter AN, Siddall ME, Goff SP. Horizontal transfer of retrotransposons between bivalves and other aquatic species of multiple phyla. Proc Natl Acad Sci U S A. 2018;115(18):E4227–35.
Thomas-Bulle C, Piednoël M, Donnart T, Filée J, Jollivet D, Bonnivard É. Mollusc genomes reveal variability in patterns of LTR-retrotransposons dynamics. BMC Genomics. 2018;19(1):821.
Marino A, Kizenko A, Wong WY, Ghiselli F, Simakov O. Repeat age decomposition informs an ancient set of repeats associated with coleoid cephalopod divergence. Front Genet. 2022;13:793734.
Platt RN II, Blanco-Berdugo L, Ray DA. Accurate transposable element annotation is vital when analyzing new genome assemblies. Genome Biol Evol. 2016;8(2):403–10.
Goubert C, Craig RJ, Bilat AF, Peona V, Vogan AA, Protasio AV. A beginner’s guide to manual curation of transposable elements. Mob DNA. 2022;13(1):7.
Gundappa MK, Peñaloza C, Regan T, Boutet I, Tanguy A, Houston RD, et al. Chromosome-level reference genome for European flat oyster (Ostrea edulis L.). Evol Appl. 2022;15(11):1713–29.
Kapitonov VV, Tempel S, Jurka J. Simple and fast classification of non-LTR retrotransposons based on phylogeny of their RT domain protein sequences. Gene. 2009;448(2):207–13.
Kojima KK. Structural and sequence diversity of eukaryotic transposable elements. Genes Genet Syst. 2020;94(6):233–52.
Yuan YW, Wessler SR. The catalytic domain of all eukaryotic cut-and-paste transposase superfamilies. Proc Natl Acad Sci U S A. 2011;108(19):7884–9.
Jurka J, Kapitonov VV, Pavlicek A, Klonowski P, Kohany O, Walichiewicz J. Repbase update, a database of eukaryotic repetitive elements. CGR. 2005;110(1–4):462–7.
Ivancevic AM, Kortschak RD, Bertozzi T, Adelson DL. LINEs between species: evolutionary dynamics of LINE-1 retrotransposons across the eukaryotic tree of life. Genome Biol Evol. 2016;8(11):3301–22.
Feschotte C, Pritham EJ. DNA transposons and the evolution of eukaryotic genomes. Annu Rev Genet. 2007;41:331–68.
Meyer A, Schloissnig S, Franchini P, Du K, Woltering JM, Irisarri I, et al. Giant lungfish genome elucidates the conquest of land by vertebrates. Nature. 2021;590(7845):284–9.
Corrochano-Fraile A, Davie A, Carboni S, Bekaert M. Evidence of multiple genome duplication events in Mytilus evolution. BMC Genomics. 2022;23(1):340.
Powell D, Subramanian S, Suwansa-ard S, Zhao M, O’Connor W, Raftos D, et al. The genome of the oyster Saccostrea offers insight into the environmental resilience of bivalves. DNA Res. 2018;25(6):655–65.
Varney RM, Speiser DI, McDougall C, Degnan BM, Kocot KM. The iron-responsive genome of the chiton Acanthopleura granulata. Genome Biol Evol. 2021;13(1):evaa263.
Yang JL, Feng DD, Liu J, Xu JK, Chen K, Li YF, et al. Chromosome-level genome assembly of the hard-shelled mussel Mytilus coruscus, a widely distributed species from the temperate areas of East Asia. GigaScience. 2021;10(4):giab024.
Liu C, Ren Y, Li Z, Hu Q, Yin L, Wang H, et al. Giant African snail genomes provide insights into molluscan whole-genome duplication and aquatic–terrestrial transition. Mol Ecol Resour. 2021;21(2):478–94.
Song H, Guo X, Sun L, Wang Q, Han F, Wang H, et al. The hard clam genome reveals massive expansion and diversification of inhibitors of apoptosis in Bivalvia. BMC Biol. 2021;19(1):15.
Petersen M, Armisén D, Gibbs RA, Hering L, Khila A, Mayer G, et al. Diversity and evolution of the transposable element repertoire in arthropods with particular reference to insects. BMC Ecology and Evolution. 2019;19(1):11.
Chalopin D, Naville M, Plard F, Galiana D, Volff JN. Comparative analysis of transposable elements highlights mobilome diversity and evolution in vertebrates. Genome Biol Evol. 2015;7(2):567–80.
Wang D, Zheng Z, Li Y, Hu H, Wang Z, Du X, et al. Which factors contribute most to genome size variation within angiosperms? Ecol Evol. 2021;11(6):2660–8.
Li Y, Sun X, Hu X, Xun X, Zhang J, Guo X, et al. Scallop genome reveals molecular adaptations to semi-sessile life and neurotoxins. Nat Commun. 2017;8(1):1721.
Wang S, Zhang J, Jiao W, Li J, Xun X, Sun Y, et al. Scallop genome provides insights into evolution of bilaterian karyotype and development. Nat Ecol Evol. 2017;1(5):1–12.
Li C, Liu X, Liu B, Ma B, Liu F, Liu G, et al. Draft genome of the Peruvian scallop Argopecten purpuratus. GigaScience. 2018;7(4):giy031.
Kenny NJ, McCarthy SA, Dudchenko O, James K, Betteridge E, Corton C, et al. The gene-rich genome of the scallop Pecten maximus. GigaScience. 2020;9(5):giaa037.
Kapusta A, Suh A, Feschotte C. Dynamics of genome size evolution in birds and mammals. Proc Natl Acad Sci U S A. 2017;114(8):E1460–9.
Thomas J, Vadnagara K, Pritham EJ. DINE-1, the highest copy number repeats in Drosophila melanogaster are non-autonomous endonuclease-encoding rolling-circle transposable elements (Helentrons). Mob DNA. 2014;5(1):18.
**ong W, He L, Lai J, Dooner HK, Du C. HelitronScanner uncovers a large overlooked cache of Helitron transposons in many plant genomes. Proc Natl Acad Sci U S A. 2014;111(28):10263–8.
Eickbush TH, Furano AV. Fruit flies and humans respond differently to retrotransposons. Curr Opin Genet Dev. 2002;12(6):669–74.
Volff JN, Bouneau L, Ozouf-Costaz C, Fischer C. Diversity of retrotransposable elements in compact pufferfish genomes. Trends Genet. 2003;19(12):674–8.
Howe K, Clark MD, Torroja CF, Torrance J, Berthelot C, Muffato M, et al. The zebrafish reference genome sequence and its relationship to the human genome. Nature. 2013;496(7446):498–503.
Boissinot S, Sookdeo A. The evolution of LINE-1 in vertebrates. Genome Biol Evol. 2016;8(12):3485–507.
Plazzi F, Puccio G, Passamonti M. Burrowers from the past: mitochondrial signatures of Ordovician bivalve infaunalization. Genome Biol Evol. 2017;9(4):956–67.
Kocot KM, Poustka AJ, Stöger I, Halanych KM, Schrödl M. New data from Monoplacophora and a carefully-curated dataset resolve molluscan relationships. Sci Rep. 2020;10(1):101.
Zhang HH, Peccoud J, Xu MRX, Zhang XG, Gilbert C. Horizontal transfer and evolution of transposable elements in vertebrates. Nat Commun. 2020;11(1):1362.
Arriagada G, Metzger MJ, Muttray AF, Sherry J, Reinisch C, Street C, et al. Activation of transcription and retrotransposition of a novel retroelement, Steamer, in neoplastic hemocytes of the mollusk Mya arenaria. Proc Natl Acad Sci U S A. 2014;111(39):14175–80.
Peccoud J, Loiseau V, Cordaux R, Gilbert C. Massive horizontal transfer of transposable elements in insects. Proc Natl Acad Sci U S A. 2017;114(18):4721–6.
Reiss D, Mialdea G, Miele V, de Vienne DM, Peccoud J, Gilbert C, et al. Global survey of mobile DNA horizontal transfer in arthropods reveals Lepidoptera as a prime hotspot. PLoS Genet. 2019;15(2): e1007965.
Wei W, Gilbert N, Ooi SL, Lawler JF, Ostertag EM, Kazazian HH, et al. Human L1 retrotransposition: cisPreference versus trans complementation. Mol Cell Biol. 2001;21(4):1429–39.
Ivancevic AM, Kortschak RD, Bertozzi T, Adelson DL. Horizontal transfer of BovB and L1 retrotransposons in eukaryotes. Genome Biol. 2018;19(1):85.
Galbraith JD, Ludington AJ, Sanders KL, Suh A, Adelson DL. Horizontal transfer and subsequent explosive expansion of a DNA transposon in sea kraits (Laticauda). Biol Lett. 2021;17(9):20210342.
Lydeard C, Cummings KS. Unionidae Rafinesque, 1820, and the general Unionida. In: Freshwater mollusks of the world: a distribution atlas. Baltimore: Johns Hopkins University Press; 2019. p. 202–9.
Barnhart MC, Haag WR, Roston WN. Adaptations to host infection and larval parasitism in Unionoida. J North Am Benthol Soc. 2008;27(2):370–94.
Pritham EJ. Transposable elements and factors influencing their success in eukaryotes. J Hered. 2009;100(5):648–55.
Szitenberg A, Cha S, Opperman CH, Bird DM, Blaxter ML, Lunt DH. Genetic drift, not life history or RNAi, determine long-term evolution of transposable elements. Genome Biol Evol. 2016;8(9):2964–78.
Galbraith JD, Kortschak RD, Suh A, Adelson DL. Genome stability is in the eye of the beholder: CR1 retrotransposon activity varies significantly across avian diversity. Genome Biol Evol. 2021;13(12):evab259.
Ip JCH, Xu T, Sun J, Li R, Chen C, Lan Y, et al. Host–endosymbiont genome integration in a deep-sea chemosymbiotic clam. Mol Biol Evol. 2021;38(2):502–18.
Furano AV. The biological properties and evolutionary dynamics of mammalian LINE-1 retrotransposons. Prog Nucleic Acid Res Mol Biol. 2000;64:255–94.
Cordaux R, Batzer MA. The impact of retrotransposons on human genome evolution. Nat Rev Genet. 2009;10(10):691–703.
Le Rouzic A, Capy P. The first steps of transposable elements invasion: parasitic strategy vs. genetic drift. Genetics. 2005;169(2):1033–43.
Le Rouzic A, Boutin TS, Capy P. Long-term evolution of transposable elements. Proc Natl Acad Sci U S A. 2007;104(49):19375–80.
Kijima TE, Innan H. Population genetics and molecular evolution of DNA sequences in transposable elements. A simulation framework. Genetics. 2013;195(3):957–67.
Fallet M, Luquet E, David P, Cosseau C. Epigenetic inheritance and intergenerational effects in mollusks. Gene. 2020;729:144166.
Männer L, Schell T, Provataris P, Haase M, Greve C. Inference of DNA methylation patterns in molluscs. Philos Trans R Soc Lond B Biol Sci. 1825;2021(376):20200166.
Wei KHC, Mai D, Chatla K, Bachtrog D. Dynamics and impacts of transposable element proliferation in the Drosophila nasuta species group radiation. Mol Biol Evol. 2022;39(5):msac080.
Gerdol M, Moreira R, Cruz F, Gómez-Garrido J, Vlasova A, Rosani U, et al. Massive gene presence-absence variation shapes an open pan-genome in the Mediterranean mussel. Genome Biol. 2020;21(1):275.
Calcino AD, Kenny NJ, Gerdol M. Single individual structural variant detection uncovers widespread hemizygosity in molluscs. Philos Trans R Soc Lond B Biol Sci. 1825;2021(376):20200153.
González VL, Andrade SCS, Bieler R, Collins TM, Dunn CW, Mikkelsen PM, et al. A phylogenetic backbone for Bivalvia: an RNA-seq approach. Proc Biol Sci. 1801;2015(282):20142332.
Combosch DJ, Collins TM, Glover EA, Graf DL, Harper EM, Healy JM, et al. A family-level tree of life for bivalves based on a Sanger-sequencing approach. Mol Phylogenet Evol. 2017;107:191–208.
Lemer S, Bieler R, Giribet G. Resolving the relationships of clams and cockles: dense transcriptome sampling drastically improves the bivalve tree of life. Proc Biol Sci. 1896;2019(286):20182684.
Regan T, Stevens L, Peñaloza C, Houston RD, Robledo D, Bean TP. Ancestral physical stress and later immune gene family expansions shaped bivalve mollusc evolution. Genome Biol Evol. 2021;13(8):evab177.
Liu F, Li Y, Yu H, Zhang L, Hu J, Bao Z, et al. MolluscDB: an integrated functional and evolutionary genomics database for the hyper-diverse animal phylum Mollusca. Nucleic Acids Res. 2021;49(D1):D1556.
Flynn JM, Hubley R, Goubert C, Rosen J, Clark AG, Feschotte C, et al. RepeatModeler2 for automated genomic discovery of transposable element families. Proc Natl Acad Sci U S A. 2020;117(17):9451–7.
Ellinghaus D, Kurtz S, Willhoeft U. LTRharvest, an efficient and flexible software for de novo detection of LTR retrotransposons. BMC Bioinformatics. 2008;9(1):18.
Ou S, Jiang N. LTR_retriever: a highly accurate and sensitive program for identification of long terminal repeat retrotransposons. Plant Physiol. 2018;176(2):1410–22.
Crescente JM, Zavallo D, Helguera M, Vanzetti LS. MITE Tracker: an accurate approach to identify miniature inverted-repeat transposable elements in large genomes. BMC Bioinformatics. 2018;19(1):348.
Peng J, Li Q, Xu L, Wei P, He P, Zhang X, et al. Chromosome-level analysis of the Crassostrea hongkongensis genome reveals extensive duplication of immune-related genes in bivalves. Mol Ecol Resour. 2020;20(4):980–94.
Qi H, Li L, Zhang G. Construction of a chromosome-level genome and variation map for the Pacific oyster Crassostrea gigas. Mol Ecol Resour. 2021;21(5):1670–85.
Farhat S, Bonnivard E, Pales Espinosa E, Tanguy A, Boutet I, Guiglielmoni N, et al. Comparative analysis of the Mercenaria mercenaria genome provides insights into the diversity of transposable elements and immune molecules in bivalve mollusks. BMC Genomics. 2022;23(1):192.
Ou S, Su W, Liao Y, Chougule K, Agda JRA, Hellinga AJ, et al. Benchmarking transposable element annotation methods for creation of a streamlined, comprehensive pipeline. Genome Biol. 2019;20(1):275.
Tarailo-Graovac M, Chen N. Using RepeatMasker to identify repetitive elements in genomic sequences. Curr Protoc Bioinformatics. 2009;Chapter 4:4.10.1-4.10.14.
Campbell MS, Law M, Holt C, Stein JC, Moghe GD, Hufnagel DE, et al. MAKER-P: A tool kit for the rapid creation, management, and quality control of plant genome annotations. Plant Physiol. 2014;164(2):513–24.
Fu L, Niu B, Zhu Z, Wu S, Li W. CD-HIT: accelerated for clustering the next-generation sequencing data. Bioinformatics. 2012;28(23):3150–2.
Wong WY, Simakov O. RepeatCraft: a meta-pipeline for repetitive element de-fragmentation and annotation. Bioinformatics. 2019;35(6):1051–2.
Quinlan AR, Hall IM. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26(6):841–2.
Katoh K, Standley DM. MAFFT Multiple Sequence Alignment Software Version 7: improvements in performance and usability. Mol Biol Evol. 2013;30(4):772–80.
Eddy SR. Accelerated profile HMM searches. PLoS Comput Biol. 2011;7(10):e1002195.
Bao W, Kapitonov VV, Jurka J. Ginger DNA transposons in eukaryotes and their evolutionary relationships with long terminal repeat retrotransposons. Mob DNA. 2010;1:3.
Metcalfe CJ, Casane D. Modular organization and reticulate evolution of the ORF1 of Jockey superfamily transposable elements. Mob DNA. 2014;5(1):19.
Wheeler TJ, Clements J, Eddy SR, Hubley R, Jones TA, Jurka J, et al. Dfam: a database of repetitive DNA based on profile hidden Markov models. Nucleic Acids Res. 2013;41(D1):D70-82.
Capella-Gutierrez S, Silla-Martinez JM, Gabaldon T. trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics. 2009;25(15):1972–3.
Rice P, Longden I, Bleasby A. EMBOSS: the European Molecular Biology Open Software Suite. Trends Genet. 2000;16(6):276–7.
Moura Gama J, Ludwig A, Gazolla CB, Guizelini D, Recco-Pimentel SM, Bruschi DP. A genomic survey of LINE elements in Pipidae aquatic frogs shed light on Rex-elements evolution in these genomes. Mol Phylogenet Evol. 2022;168:107393.
Sheneman L, Evans J, Foster JA. Clearcut: a fast implementation of relaxed neighbor joining. Bioinformatics. 2006;22(22):2823–4.
Minh BQ, Schmidt HA, Chernomor O, Schrempf D, Woodhams MD, von Haeseler A, et al. IQ-TREE 2: new models and efficient methods for phylogenetic inference in the genomic era. Mol Biol Evol. 2020;37(5):1530–4.
Kalyaanamoorthy S, Minh BQ, Wong TKF, von Haeseler A, Jermiin LS. ModelFinder: fast model selection for accurate phylogenetic estimates. Nat Methods. 2017;14(6):587–9.
Hoang DT, Chernomor O, von Haeseler A, Minh BQ, Vinh LS. UFBoot2: improving the ultrafast bootstrap approximation. Mol Biol Evol. 2018;35(2):518–22.
Kishino H, Hasegawa M. Evaluation of the maximum likelihood estimate of the evolutionary tree topologies from DNA sequence data, and the branching order in hominoidea. J Mol Evol. 1989;29(2):170–9.
Shimodaira H, Hasegawa M. Multiple comparisons of log-likelihoods with applications to phylogenetic inference. Mol Biol Evol. 1999;16(8):1114.
Strimmer K, Rambaut A. Inferring confidence sets of possibly misspecified gene trees. Proc Biol Sci. 2002;269(1487):137–42.
Shimodaira H. An approximately unbiased test of phylogenetic tree selection. Syst Biol. 2002;51(3):492–508.
Mao H, Wang H. SINE_scan: an efficient tool to discover short interspersed nuclear elements (SINEs) in large-scale genomic datasets. Bioinformatics. 2017;33(5):743–5.
Tumescheit C, Firth AE, Brown K. CIAlign: A highly customisable command line tool to clean, interpret and visualise multiple sequence alignments. PeerJ. 2022;10:e12983.
Chan PP, Lowe TM. tRNAscan-SE: searching for tRNA genes in genomic sequences. Methods Mol Biol. 2019;1962:1–14.
Han MJ, **ong CL, Zhang HB, Zhang MQ, Zhang HH, Zhang Z. The diversification of PHIS transposon superfamily in eukaryotes. Mob DNA. 2015;6:12.
Bushnell B. BBMap: a fast, accurate, splice-aware aligner. Berkeley: Lawrence Berkeley National Lab. (LBNL); 2014. Report No.: LBNL-7065E.
Lerat E, Fablet M, Modolo L, Lopez-Maestre H, Vieira C. TEtools facilitates big data expression analysis of transposable elements and reveals an antagonism between their activity and that of piRNA genes. Nucleic Acids Res. 2017;45(4):e17.
Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9(4):357–9.
Martelossi J. Transposable element characterization in molluscs. Figshare. 2023.
Martelossi J. EvoTEs_BiV. Zenodo. 2023. https://doi.org/10.5281/zenodo.7944844.
Acknowledgements
We thank the EVO·COM lab members Mariangela Iannello, Giobbe Forni, and Giovanni Piccinini for the useful comments and discussions about the analyses. We would also like to thank Prof. David Adelson, another anonymous reviewer, and the BMC Biology AE Dr. Ruslan Kalendar for providing insightful comments on the first version of the manuscript which helped us to greatly improve the overall quality of the manuscript.
Funding
This work was supported by the Canziani bequest funded to F.G. and A.L. and the ‘Ricerca Fondamentale Orientata’ (RFO) funding from the University of Bologna to F.G. and A.L.
Author information
Authors and Affiliations
Contributions
JM, AL, and FG designed the study. JM collected the data and performed the bioinformatic analyses. JM, FN, SS, and DP curated the data. JM wrote the first version of the manuscript and additional supplementary files. JM, FG, AL, and FN revised the manuscript. All authors read and approved the final version of the manuscript.
Authors’ information
@JacopoM4 (Jacopo Martelossi); @Filo_nicoo (Filippo Nicolini); @f_ghiselli (Fabrizio Ghiselli); @AndreaLuchett10 (Andrea Luchetti).
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Table S1.
Source and information of the 39 analyzed assemblies.
Additional file 2: Fig. S1.
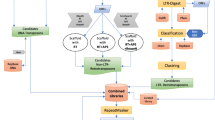
Schematic representation of the workflow used to create automatically generated repeat libraries and mined LINEs and DDE/D related transposons with ORF evidence (see “Mining and annotation of interspersed repeats” and “ORF-based annotation of RT containing LINEs and Class II DDE/D elements” sections).
Additional file 3: Table S2.
Details about the number of de-novo sequences mined from software and used for automatic construction of species-specific repeats libraries (see “Mining and annotation of interspersed repeats” section).
Additional file 4: Table S3.
Transposable element genomic content of each transposon class using species-specific automatically generated TE sequence libraries (see “Mining and annotation of interspersed repeats” section).
Additional file 5: Fig. S2.
(A) Copy number and (B) consensus length distribution of RepeatModeler “Unknown” consensus sequences (see “Mining and annotation of interspersed repeats” section).
Additional file 6: Fig. S3.
Genome occupancy of each TE class in the 39 analyzed genomes using automatically generated TE libraries (see “Mining and annotation of interspersed repeats” section). Significant comparison are highlighted by asterisks (Pairwise Wilcoxon rank test with Bonferroni correction; * = p < 0.05, ** = ps < 0.01).
Additional file 7: Fig. S4.
K-mean clustering obtained using 3 centers and based on the number of insertions for each transposon type as annotated by RepeatMasker using automatically generated TE libraries (see “Mining and annotation of interspersed repeats” section).
Additional file 8: Table S4.
Results of ORF-based LINE annotation. N.Clusters=Number of cluster; N.RT=Number of RT-containing LINEs; RT.Clusters.Min5=Number of cluster with at least five members; RT.Seq.Clusters.Min5=Number of sequences contained in cluster with a size greater than 5; N.AE=number of putative autonomous LINEs. See “ORF-based annotation of RT containing LINEs and Class II DDE/D elements” section.
Additional file 9: Fig. S5.
(A) Positive linear relationship between number of identified Reverse Transcriptase (RT) -containing LINE loci and number of autonomous elements (i.e., possessing both an RT and an Endonuclease domain) (Spearman’s rho=0.89, p < 0.01); (B) Positive linear relationship between scaffold N50 and number of identified Reverse Transcriptase containing LINE loci (Spearman’s rho=0.35, p < 0.05); (C) Positive linear relationship between scaffold N50 and number of identified LINE autonomous elements (Spearman’s rho=0.34, p < 0.05). See “ORF-based annotation of RT containing LINEs and Class II DDE/D elements” section.
Additional file 10: Table S5.
Sequences included in phylogenetic analyses and used as reference to annotate molluscs LINEs (see “Tree-based classification of ORF-containing LINE elements” section). Reference = sequence obtained from Kapitov et al (2009) or from the RepeatPep library. RTClass = Subset of Mollusc LINEs annotated with RTClass1.
Additional file 11: Fig. S6.
Superfamilies relationships obtained with Neighbor-Joining (A) and unconstrained Maximum Likelihood analyses (B). For the latter, only the run with the highest log-likelihood is shown (See Additional File 12: Table S6). All trees with nodal support values can be found in Additional File 29: Data S1. See “Tree-based classification of ORF-containing LINE elements” section.
Additional file 12: Table S6.
Results of Maximum likelihood (ML) topology test between constrained and unconstrained tree searches (see “Tree-based classification of ORF-containing LINE elements” section). Tree with the highest log likelihood is highlighted in bold. FullNJ=ML tree obtained with a full constrain on the topology recovered by Neighbour-Joining; SupFAM=ML tree constrained only on the superfamilies relationships obtained by Neighbour-Joining. Plus signs denote accepted topologies by the respective topology test.
Additional file 13: Fig. S7.
L1 superfamily subtree extracted from the SupFAM tree #2. Numbers on nodes represent UltraFast Bootstrap values. The outer-left annotation refers to the classification scheme proposed by RepBase and based on [29] while the outer-right based on the RepeatMasker “type” and obtained through blastp against the RepeatPep library. See “Tree-based classification of ORF-containing LINE elements” section.
Additional file 14: Fig. S8.
RTE superfamily subtree extracted from the SupFAM tree #2. Numbers on nodes represent UltraFast Bootstrap values. The outer-left annotation refers to the classification scheme proposed by RepBase and based on [29] while the outer-right based on the RepeatMasker “type” and obtained through blastp against the RepeatPep library. See “Tree-based classification of ORF-containing LINE elements” section.
Additional file 15: Fig. S9.
Jockey superfamily subtree extracted from the SupFAM tree #2. Numbers on nodes represent ultrafast bootstrap values. The outer-left annotation refers to the classification scheme proposed by RepBase and based on [29] while the outer-right based on the RepeatMasker “type” and obtained through blastp against the RepeatPep library. See “Tree-based classification of ORF-containing LINE elements” section.
Additional file 16: Fig. S10.
I superfamily subtree extracted from the SupFAM tree #2. Numbers on nodes represent UltraFast Bootstrap values. The outer-left annotation refers to the classification scheme proposed by RepBase and based on [29] while the outer-right based on the RepeatMasker “type” and obtained through blastp against the RepeatPep library. See “Tree-based classification of ORF containing LINE elements” section.
Additional file 17: Fig. S11.
R2 superfamily subtree extracted from the SupFAM tree #2. Numbers on nodes represent UltraFast Bootstrap values. The outer-left annotation refers to the classification scheme proposed by RepBase and based on [29] while the outer-right based on the RepeatMasker “type” and obtained through blastp against the RepeatPep library. See “Tree-based classification of ORF-containing LINE elements” section.
Additional file 18: Fig. S12.
Number of RT-containing LINE clusters annotated in each analyzed genome and subdivided by clade following [29] and by RepeatMasker “type” classification in parenthesis. Clades are grouped by superfamily following [103] and the aforementioned SupFam tree #2. Note that the L2-2 clade includes Crack, Daphne, L2A and L2B elements. See “Tree-based classification of ORF-containing LINE elements” section.
Additional file 19: Fig. S13.
Scatterplot of number of RT-containing LINEs and clusters for L1, Jockey, RTE and I superfamilies. The R2 superfamily was not included because of the low number of data points. Each point represents a clade/type. See “Tree-based classification of ORF-containing LINE elements” section.
Additional file 20: Fig. S14.
Number of autonomous elements annotated in each analyzed genome and subdivided by clade following [29] and by RepeatMasker “type” classification in parenthesis. Clades are grouped by superfamily following [103] and the aforementioned SupFam tree #2. Note that the L2-2 clade includes Crack, Daphne, L2A and L2B elements. See “ORF-based annotation of RT containing LINEs and Class II DDE/D elements” and “Tree-based classification of ORF-containing LINE elements” sections.
Additional file 21: Fig. S15.
Length distribution of manually curated LINE families. Each bar represents an element and colors denote different LINE superfamilies/types. See “Manual curation of LINEs, SINEs, and DDE/D-related transposons” section.
Additional file 22: Fig. S16.
Length distribution of manually curated DDE/D transposon families. Each bar represents an element and colors denote different superfamilies. See “Manual curation of LINEs, SINEs, and DDE/D-related transposons” section.
Additional file 23: Fig. S17.
Phylogenetic tree of curated bivalves LINEs RTE families. Numbers on nodes represent UltraFast Bootstrap values. See “Genome annotation of LINEs and SINEs using manually curated libraries and phylogenetic inference of curated LINE families” section.
Additional file 24: Fig. S18.
Phylogenetic tree of curated bivalves LINEs I families. Numbers on nodes represent UltraFast Bootstrap values. See “Genome annotation of LINEs and SINEs using manually curated libraries and phylogenetic inference of curated LINE families” section.
Additional file 25: Fig. S19.
Phylogenetic tree of curated bivalves LINEs L1 families. Numbers on nodes represent UltraFast Bootstrap values. See “Genome annotation of LINEs and SINEs using manually curated libraries and phylogenetic inference of curated LINE families” section.
Additional file 26: Fig. S20.
Phylogenetic tree of curated bivalves LINEs Jockey families. Numbers on nodes represent UltraFast Bootstrap values. See “Genome annotation of LINEs and SINEs using manually curated libraries and phylogenetic inference of curated LINE families” section.
Additional file 27: Table S7.
Annotation of RT-containing LINEs included in phylogenetic analyses. Superfamily and clade classification are based on reference sequences extracted from RepBase and follow [7] and [29], RM Type=Classification based on best-hit blastp results against the full RepeatPep library extracted from the RepeatMasker package; RT members=Number of RT-containing LINEs included in each cluster.
Additional file 28: Table S8.
Conserved Domain Database identifier used to search for Reverse Transcriptase and Endonuclease signatures in extracted open reading frames.
Additional file 29: Data S1.
LINE phylogenetic trees.
Additional file 30: Data S2.
Multi Sequence Alignment used to generate all phylogenetic trees.
Additional file 31: Data S3.
Library of curated LINE families. All sequences have been classified following a RepeatMasker formatting style.
Additional file 32: Data S4.
Library of curated SINE families. All sequences have been classified following a RepeatMasker formatting style.
Additional file 33: Data S5.
Library of curated Class II DDE/D-related families. These families correspond to full length elements. All sequences have been classified following a RepeatMasker formatting style.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Martelossi, J., Nicolini, F., Subacchi, S. et al. Multiple and diversified transposon lineages contribute to early and recent bivalve genome evolution. BMC Biol 21, 145 (2023). https://doi.org/10.1186/s12915-023-01632-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12915-023-01632-z