
Abstract
Our previous work has shown that d-ribose (RIB)-induced depressive-like behaviors in mice. However, the relationship between variations in RIB levels and depression as well as potential RIB participation in depressive disorder is yet unknown. Here, a reanalysis of metabonomics data from depressed patients and depression model rats is performed to clarify whether the increased RIB level is positively correlated with the severity of depression. Moreover, we characterize intestinal epithelial barrier damage, gut microbial composition and function, and microbiota-gut-brain metabolic signatures in RIB-fed mice using colonic histomorphology, 16 S rRNA gene sequencing, and untargeted metabolomics analysis. The results show that RIB caused intestinal epithelial barrier impairment and microbiota-gut-brain axis dysbiosis. These microbial and metabolic modules are consistently enriched in peripheral (fecal, colon wall, and serum) and central (hippocampus) glycerophospholipid metabolism. In addition, three differential genera (Lachnospiraceae_UCG-006, Turicibacter, and Akkermansia) and two types of glycerophospholipids (phosphatidylcholine and phosphatidylethanolamine) have greater contributions to the overall correlations between differential genera and glycerophospholipids. These findings suggest that the disturbances of gut microbiota by RIB may contribute to the onset of depressive-like behaviors via regulating glycerophospholipid metabolism, and providing new insight for understanding the function of microbiota-gut-brain axis in depression.
Similar content being viewed by others


Introduction
D-ribose (RIB) is a naturally occurring monosaccharide that is found in riboflavin-containing foods such as wheat bran, eggs, and meat. Meanwhile, because RIB can bypass part of the pentose pathway to produce d-ribose-5-phosphate for the production of energy, it has been utilized as a daily nutritional or energy supplement1, notably for patients with chronic fatigue syndrome and coronary artery disease2,3. RIB is also a crucial component of several important biomolecules including adenosine and adenosine triphosphate, which are involved in a variety of metabolic activities4. However, as described by the European Food Safety Authority, the toxicological effects of RIB should not be ignored5. Several studies have reported that RIB can be involved in the onset of encephalopathy6,7.
Depression is one of the most prevalent serious mental disorders, characterized by a lack of interest, pessimism, appetite loss, and even suicidal behavior8,9. A new epidemiology study estimates that 7% of people experience depression annually, with a lifetime prevalence of over 15%10. Finding relevant risk variables is crucial for depression prevention and screening. Given that RIB has not been widely reported in depressive disorder and that a high-sugar diet may be an environmental risk factor for depression11,12, we recently gave normal mice prolonged RIB supplementation and found that these mice exhibited depressive-like behaviors and histological alterations, including obviously condensed and deeply-stained pyramidal cells in the hippocampus13. This finding implies that RIB has a significant impact on the development of depression. The key scientific concerns that we will research, however, are whether the variation in RIB level was associated with depression and the underlying biological mechanism of RIB implicated in depressive illness.
Of note, some other monosaccharides, such as fructose and glucose, have been linked to changes in the gut microbiota, leading to microbial metabolite disorder in rodents14,15. A high intake of sugar can cause enteric dysbacteriosis. The latter increases the permeability of the intestinal mucosa, and results in abnormalities in intestinal immunity and glucolipid metabolism15. Moreover, hyperglycemia would increase the permeability of the intestinal barrier, giving microbes a better chance to enter the body and causing the proliferation of pathogenic bacteria16. Nearly every aspect of host physiology may be influenced by gut microbiota, from controlling gut metabolism to influencing mood and behavior via the “microbiota-gut-brain” (MGB) axis17,18. Our groups previously found that depression was linked to altered gut microbiomes19, and germ-free mice exhibit depressive-like behaviors after receiving gut microbiota from depressed patients19,20. Furthermore, a recent study demonstrates that exogenous RIB can affect gut microbial architecture21. As a result, we proposed that changes in microbiota might account for the connection between RIB intake and depression.
At first, we reanalyzed the metabonomics data from our earlier studies to clarify the change of RIB in the urine of depressed patients and in the hippocampus of depression model rats. Then, in the current study, to further investigate the possible mechanism of RIB-induced depression, eight weeks of RIB-fed mice were constructed. The intestinal barrier impairment was evaluated using hematoxylin and eosin, immunohistochemistry, and electron microscopy. The distinct gut microbiota was initially identified by 16S rRNA gene sequencing analysis. Moreover, by systematic analysis of relevant biological samples, including peripheral (fecal, colon wall, and serum) and central (hippocampus) specimens from the RIB-fed mice and control mice, comparative untargeted metabolomics was used to capture the functions of the altered gut microbiome. Finally, by integrating these multi-omics data, we sought to understand how the gut microbiota contributed to the development of depressive-like behaviors and to pinpoint a putative way between the gut and the brain in RIB-fed mice.
Results
RIB was significantly increased in depressed patients and depression model rats
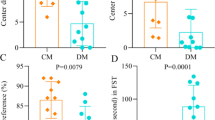
Compared to healthy controls (HC), the relative abundance of RIB was significantly increased in the urine of depressed patients (p = 0.014; Fig. 1a). Meanwhile, we found that the levels of RIB in males (p = 0.021) and females (p = 0.030) with depression were both significantly different from that in their respective HC. Depression model rats have greater RIB levels in their hippocampus than control (CON) rats (p = 0.008; Fig. 1b). These group-level comparisons were conducted using Student’s t-test. The results of Spearman’s correlation analysis showed that although there was no significant correlation between sucrose preference and RIB levels (r = −0.453, p = 0.068; Fig. 1c) in the depression model rats, the immobility time was significantly positively correlated with RIB levels (r = 0.682, p = 0.003; Fig. 1d). The results indicate that a tight connection may exist between elevated RIB and depressive illness.

a The relative abundance of ribose was significantly increased in patients with major depressive disorder (MDD; n = 126) compared to healthy controls (HC; n = 125); the relative abundance of RIB in male (n = 63) and female (n = 63) MDD patients was significantly different from of that in their respective HC (male, n = 77; female, n = 48). b The relative abundance of RIB was significantly increased in chronic social defeat stress (CSDS) model rats (n = 9) compared to control (CON) rats (n = 8). c There was a negative relationship between the percentage of sucrose preference (SP) and the relative abundance of RIB. d There was a significant positive correlation between immobility time (IT) and the relative abundance of RIB. Data are the means ± standard error of mean. The group-level comparisons were conducted using Student’s t-test to assess the changes of RIB in different groups, and Spearman’s correlation analysis was used to assess the relationships between RIB level and depressive-like behaviors.
RIB-fed mice exhibit depressive-like behaviors
After eight weeks of RIB treatment, there were no significant differences in the mouse body weight (p = 0.279; Fig. 2a) and fasting blood glucose (p = 0.741; Fig. 2b) between the CON and RIB groups. However, the result of sucrose preference was significantly lower in the RIB group than in the CON group (p = 0.001; Fig. 2c). In open field test, there was no difference in the total distance between the two groups (p = 0.203; Fig. 2d), whereas the central distance in the RIB group was significantly lower than that in the CON group (p = 0.022; Fig. 2e). Moreover, in tail suspension test, the RIB group had significantly higher immobility time than the CON group (p = 0.033; Fig. 2f). These group-level comparisons were conducted using Student’s t test. These results further confirm that RIB-fed induced depressive-like behaviors in mice.

a, b Both body weight (a) and fasting blood (b) were similar between the two groups. c Sucrose preference (%) was significantly lower in the RIB group. d Total distance was similar between the CON and RIB groups. e Center distance was significantly lower in the RIB group. f Compared to the CON group, the RIB group had a significantly higher immobility time. Data are the means ± standard error of mean, n = 13 per group. Student’s t test (data was normally distributed) or nonparametric Mann–Whitney U test (data was not normally distributed) was used to analyze the data.
RIB impairs the intestinal epithelial barrier
Homeostasis in the gut is important for brain function. We investigated whether RIB feeding perturbed colonic homeostasis, including colonic barrier and gut microbiota. There was no significant difference in the length of the colon between the two groups (Student’s t test, p = 0.696; Fig. 3a), whereas hematoxylin and eosin staining revealed the thickness of the muscularis mucosae significantly thinner, and loss of crypts and glands in the RIB-fed mice (Fig. 3b). Electron microscopy also showed severe mitochondrial swelling, injury of tight junction and gap junction domains, reduced numbers of the desmosome, and increased distance between adjacent epithelial cells in the colon of RIB-fed mice (Fig. 3c). Moreover, immunohistochemistry analysis indicated that the expression of Occludin (Fig. 3d) and mucin 2 (MUC2; Fig. 3e) were obviously decreased in RIB-fed mice.

a Representative images of colons and quantification of colon length were statistically analyzed (Data are the means ± standard error of mean, n = 13 per group), and no significant difference in the length of the colon between the two groups (Student’s t test, p = 0.696). b Representative hematoxylin–eosin (H&E) staining image, scale bar = 100 µm. c Representative transmission electron micrographs of colon epithelial cells (scale bar = 2 µm). The second row’s enlarged images were from the first row in the area indicated with a dotted line box (scale bar = 1 µm). M mitochondria, TJ tight junction, De desmosome. d, e Immunohistochemistry analysis for Occludin (D) and mucin 2 (MUC2; E) in colon tissue (scale bar = 50 µm).
Gut microbiome alterations in RIB-fed mice
Next, gut microbiota diversity and composition in response to RIB were analyzed using 16S rRNA gene sequencing. There were no significant differences in alpha diversity between the two groups (Supplementary Fig. S1). The results of principal coordinate analysis showed that there were significant differences in beta diversity between the two groups (p = 0.001; Fig. 4a). As shown in Fig. 4b, Firmicutes and Bacteroidetes were the two major bacterial phyla in both groups and the relative abundance of Verrucomicrobiota was significantly higher in RIB-fed mice than in CON (p = 0.005). The relative abundances of each phylum were described in Supplementary Data 1.

a Principal coordinate analysis model showed that there were significantly differential gut microbiota compositions between CON and RIB-fed mice. b Firmicutes, Bacteroidetes, and Verrucomicrobiota were the three major bacterial phyla in both groups. c In total, 22 differential genera responsible for the discrimination between CON and RIB-fed mice were identified using linear discriminant analysis effective size. d These differential genera were significantly involved in four amino acid metabolism pathways and six lipid metabolism pathways (Student’s t test was used). The relative abundance of glycerophospholipid metabolism was highest among these pathways. A1: Phenylalanine, tyrosine, and tryptophan biosynthesis; A2: Lysine degradation; A3: Valine, leucine, and isoleucine biosynthesis; A4: Histidine metabolism; L1: Secondary bile acid biosynthesis; L2: Glycerophospholipid metabolism; L3: Arachidonic acid metabolism; L4: Primary bile acid biosynthesis; L5: Fatty acid elongation; L6: Fatty acid biosynthesis. e Correlations between differential genera and depressive-like behaviors (Spearman’s correlation analysis was used). CD center distance, IT immobility time, SP sucrose preference.
Using linear discriminant analysis effective size, 22 differential genera responsible for the discrimination between CON and RIB-fed mice were identified (Fig. 4c and Supplementary Data 2). Most of the differential genera (n = 14, 63.63%) belonged to phylum Firmicutes. Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis (Fig. 4d) showed that these differential genera were significantly involved in four amino acid metabolism pathways (Phenylalanine, tyrosine, and tryptophan biosynthesis (up-regulate in RIB mice, p = 0.031); Lysine degradation (up-regulate in RIB mice, p = 0.030); Valine, leucine, and isoleucine biosynthesis (up-regulate in RIB mice, p = 0.037); Histidine metabolism (up-regulate in RIB mice, p = 0.029)) and six lipid metabolism pathways (Secondary bile acid biosynthesis (down-regulate in RIB mice, p = 0.031); Glycerophospholipid metabolism (down-regulate in RIB mice, p = 0.032); Arachidonic acid metabolism (up-regulate in RIB mice, p = 0.033); Primary bile acid biosynthesis (down-regulate in RIB mice, p = 0.031); Fatty acid elongation (up-regulate in RIB mice, p = 0.035); and Fatty acid biosynthesis (up-regulate in RIB mice, p = 0.030)).
To find out the differential genera significantly correlated with depressive-like behaviors, Spearman’s correlation analysis was conducted here. The results (Fig. 4e) showed that center distance was significantly correlated with Parasutterella (positive; r = 0.563, p = 0.032) and Christensenellaceae_R-7_group (negative; r = −0.513, p = 0.042), immobility time was significantly correlated with Akkermansia (negative; r = −0.519, p = 0.039), and sucrose preference was significantly correlated with nine differential genera (Candidatus_Stoquefichus (positive; r = 0.669, p = 0.005), Turicibacter (negative; r = −0.659, p = 0.006), Gordonibacter (positive; r = 0.627, p = 0.009), Eubacterium_siraeum_group (positive; r = 0.571, p = 0.021), Ileibacterium (negative; r = −0.551, p = 0.027), Clostridium_sensu_stricto_1 (negative; r = −0.541, p = 0.031), Alloprevotella (positive; r = 0.521, p = 0.039), Oscillibacter (positive; r = 0.518, p = 0.040), and Akkermansia (negative; r = −0.513, p = 0.042)).
Differential microbial metabolites in RIB-fed mice
In total, there were 1331 metabolites successfully annotated (Supplementary Data 3). The built orthogonal partial least-squares discriminant analysis (OPLS-DA) model using microbial metabolites in feces showed that the RIB-fed mice were separate from CON with no overlap, suggesting the divergent microbial metabolic phenotypes between the two groups (Fig. 5a). The results of 399-permutation testing demonstrated that this model was valid and not over-fitting (Supplementary Fig. S2). By analyzing the variable importance plot (VIP) from corresponding OPLS-DA loading plot and p-value from Student’s t test, we identified 246 differential microbial metabolites responsible for the discrimination between CON and RIB-fed mice (VIP > 1.0 and p < 0.05). Detailed information on these microbial metabolites was described in Supplementary Data 4. The heat map consisting of these differential microbial metabolites showed a consistent clustering pattern within the individual groups (Fig. 5b).

a OPLS-DA model showed that the two groups had significantly different fecal metabolic phenotypes. b There were 246 differential fecal metabolites (VIP > 1.0 from corresponding OPLS-DA loading plots and p < 0.05 from Student’s t test) responsible for the discrimination between CON and RIB groups. c KEGG pathway classification showed that these metabolites were mainly annotated into the metabolism category. d Using online software MetaboAnalyst, four significantly dysregulated metabolic pathways in KEGG metabolism category classifications at level 3 were identified via hypergeometric test (each dot represents a KEGG path, the dot size represents the impact value, and the dot color represents the p-value, the more important the differential metabolites were in this pathway, the larger the dot).
In addition, KEGG pathway classification showed that these differential microbial metabolites were mainly annotated into the metabolism category (Fig. 5c). Using online software MetaboAnalyst, four significantly affected metabolic pathways in KEGG metabolism category classifications at level 3 were identified via hypergeometric test (Fig. 5d): Tryptophan metabolism (p = 0.006), Isoflavonoid biosynthesis (p = 0.004), Purine metabolism (p = 0.016), and Glycerophospholipid metabolism (p = 0.041).
Differential metabolites in the colon, blood, and hippocampus
In total, 1063 metabolites were successfully annotated in the colon (Supplementary Data 5). The built OPLS-DA model showed divergent metabolic phenotypes in the colon between the two groups (Supplementary Fig. S3a). There were 189 differential metabolites responsible for separating RIB-fed mice from CON were identified (VIP > 1.0 and p < 0.05; Supplementary Data 6). KEGG pathway classification showed that these differential microbial metabolites were mainly annotated into the metabolism category; and using the online software MetaboAnalyst, seven significantly affected metabolic pathways in KEGG metabolism category classifications at level 3 were identified via hypergeometric test (Fig. 6a): Galactose metabolism (p = 0.031), Glycerophospholipid metabolism (p = 0.003), Sphingolipid metabolism (p = 0.007), Primary bile acid biosynthesis (p = 0.031), Thiamine metabolism (p = 0.013), Taurine and hypotaurine metabolism (p = 0.007), and Purine metabolism (p = 0.001).

a Seven significantly dysregulated metabolic pathways were found using hypergeometric tests in colon tissue. b Eight significantly dysregulated metabolic pathways were identified using hypergeometric tests in blood. c Three significantly dysregulated metabolic pathways were identified using hypergeometric tests in hippocampus. The online software MetaboAnalyst was used to conduct pathway analysis. Each dot represents a KEGG path, the dot size represents the impact value, and the dot color represents the p-value. The more important the differential metabolites were in this pathway, the larger the dot. d A heat map representation comprising all the differential metabolites from the colon, blood, and hippocampus showed a consistent clustering pattern within the individual groups.
Similarly, in blood, 665 metabolites were successfully annotated (Supplementary Data 7), and 104 differential metabolites responsible for separating RIB-fed mice from CON were identified (VIP > 1.0 and p < 0.05; Supplementary Fig. S3b and Supplementary Data 8). Using online software MetaboAnalyst, eight significantly affected metabolic pathways in KEGG metabolism category classifications at level 3 were identified via hypergeometric test (Fig. 6b): Phenylalanine metabolism (p = 0.014), Glycerophospholipid metabolism (p = 0.001), Primary bile acid biosynthesis (p = 0.014), alpha-Linolenic acid metabolism (p = 0.006), Taurine and hypotaurine metabolism (p = 0.003), d-Glutamine and d-glutamate metabolism (p = 0.041), Pyrimidine metabolism (p = 0.024), and Aminoacyl-tRNA biosynthesis (p = 0.001).
In the hippocampus, 762 metabolites were successfully annotated (Supplementary Data 9), and 80 differential metabolites responsible for separating RIB-fed mice from CON were identified (VIP > 1.0 and p < 0.05; Supplementary Fig. S3c and Supplementary Data 10). Using online software MetaboAnalyst, three significantly affected metabolic pathways in KEGG metabolism category classifications at level 3 were identified via hypergeometric test (Fig. 6c): Glycerophospholipid metabolism (p = 0.001), Pyrimidine metabolism (p = 0.019), and Purine metabolism (p = 0.002). A heat map representation comprising all the differential metabolites from colon, blood, and hippocampus showed a consistent clustering pattern within the individual groups (Fig. 6d).
Metabolomic correlations with behavioral phenotypes
As the samples of the MGB axis were diverse, only a limited number of metabolites (n = 17) were shared by these detected samples (feces, colon, blood, and hippocampus). This phenomenon was also observed in our previous nonhuman primate model of depression22. As such, the different components of a given metabolic pathway might synergistically modulate the function of the MGB axis in different tissues. Thus, weighted correlation network analysis was used here to cluster the identified differential metabolites into the metabolic modules of the MGB axis. The results showed that there were seven different modules, in which four modules (blue, red, black, and turquoise) were significantly correlated with at least one type of depressive-like behavior (Fig. 7a).

a Spearman’s correlations between behavioral phenotypes and metabolomic modules. SP was significantly correlated with three metabolic modules (blue, red, and black), and IT was significantly correlated with the turquoise metabolic module. Red and green squares indicated positive and negative correlations, respectively. b The differential metabolites in the turquoise metabolic module were significantly correlated with IT mainly belonged to lipid metabolism, especially PE and PC. c The differential metabolites in three metabolic modules (blue, red, and black) were significantly correlated with SP mainly belonging to PC, fatty acyls, organic compounds, and carboxylic acids and derivatives. Differential metabolites belonging to lipid metabolism were marked using different colors except for the gray, and other differential metabolites were marked using gray circles. Circle size indicated the abundance of the metabolites belonging to this node. BW body weight, CD center distance, CL cardiolipin, FB fasting blood, IT immobility time, PA phosphatidic acid, PC phosphatidylcholine, PE phosphatidylethanolamine, PI phosphatidylinositol, PS phosphatidylserine, SP sucrose preference, TD total distance.
Module-trait analysis showed that the differential metabolites in the turquoise metabolic module significantly correlated with immobility time were mainly involved in peripheral and central glycerophospholipid metabolism within the MGB axis (Fig. 7b); and the differential metabolites in the other three metabolic modules (blue, red, and black) significantly correlated with sucrose preference were mainly involved in peripheral and central glycerophospholipid and fatty acyls metabolism within the MGB axis (Fig. 7c). Details regarding the module and chemical class of each compound were shown in Supplementary Data 11.
Correlations between differential genera and glycerophospholipids
The abovementioned findings indicated that glycerophospholipid metabolism might play an important role in the crosstalk of gut microbiota and the brain. Thus, we further analyzed the potential correlations between differential genera and glycerophospholipids using Spearman’s correlation analysis. The results (Fig. 8) showed that there were significant correlations between differential genera and glycerophospholipids, especially metabolites belonging to phosphatidylcholine (PC) and phosphatidylethanolamine (PE).

The results of Spearman’s correlation analysis showed that the differential bacteria taxa mainly correlated with differential metabolites belonging to PE and PC. The numbers associated with the metabolite names were just codes, for example, we used PC39 to represent 1-heptadecanoyl-glycero-3-phosphate (detailed information about the codes representing metabolites is shown in Supplementary Data 12). PC phosphatidylcholine, PE phosphatidylethanolamine.
Further analysis found that correlations between three differential genera (Lachnospiraceae_UCG-006, Turicibacter, and Akkermansia) and two types of glycerophospholipids (PC and PE) had greater contributions to the overall correlation between differential genera and glycerophospholipids (Fig. 9). These results indicated that glycerophospholipid metabolism, especially PC and PE, might be the important bridge of gut microbiota in affecting brain functions.

Correlations between three differential genera (Lachnospiraceae_UCG-006, Turicibacter, and Akkermansia) and two types of glycerophospholipids (PC and PE) had greater contributions to the overall correlation between these differential genera and differential metabolites. The numbers associated with the metabolite names were just codes, for example, we used PC39 to represent 1-heptadecanoyl-glycero-3-phosphate (detailed information about the codes representing metabolites is shown in Supplementary Data 12). PC phosphatidylcholine, PE phosphatidylethanolamine. Spearman’s correlation analysis was used here.
Discussion
Dietary sugars, like fructose and glucose, are associated with psychosis-related higher brain dysfunctions12,15. Our previous study provided evidence that another simple sugar, RIB, could lead to depressive-like behaviors, and we demonstrated in mice that this was connected with altered hippocampus metabolic and transcriptome profiles13, but how the brain is affected by RIB remains poorly understood. In this study, we clarified that the RIB level was significantly increased in the depressed patients and depression model rats, and there was an obvious correlation between the change of RIB and the severity of depression disorder. However, these studies only involved untargeted metabolomics analysis, which provides relative metabolite abundance rather than absolute quantification23. The results further suggested that high levels of RIB were correlated with depression. We observed the RIB-fed mice were characterized by intestinal epithelial barrier impairment, alterations of microbial composition, function, and metabolic pathways of the MGB axis. Meanwhile, the altered microbial and metabolic modules linked the gut microbiome with dysregulation of peripheral and hippocampus glycerophospholipid metabolism in RIB-fed mice. To our knowledge, this is the first report of RIB influencing gut microbiota, and gut dysbiosis may be responsible for mediating the depressive-like behaviors seen in RIB-fed mice by regulating the MGB metabolism.
We found that the RIB-fed mice had considerably impaired intestinal barrier as compared to the CON. The gut barrier function was regulated by the gut microbiota24,25. Gut homeostasis also has a significant role in maintaining the host’s health. Previous clinical studies have found that depressed individuals’ gut microbiomes have changed significantly26,27. As a result, we deduced that the RIB might disrupt the gut microbiota, and that the gut dysbiosis would subsequently lead to depression via the MGB axis. Using 16 S rRNA gene sequencing, we discovered that the RIB-fed mice were characterized by 22 differential bacteria taxa on the Genera level, especially Akkermansia, Turicibacter, and Lachnospiraceae_UCG-006. Akkermansia belonged to the Verrucomicrobiota phylum. Khan S et al.28 observed that feeding dietary simple sugars like glucose and fructose would enhance the abundance of Akkermansia, which is consistent with our current findings. The mucus-degrading bacterium Akkermansia would regulate intestinal homeostasis and preserve the integrity of the gut barrier29. The increased Akkermansia might be the cause of intestinal barrier impairment in RIB-fed mice. Interestingly, recent research has shown that Akkermansia has beneficial roles in human health30; nevertheless, there is also strong evidence that Akkermansia promotes the etiology of colitis31. Similarly, Akkermansia was detected in much higher abundance in individuals with severe depressive symptoms, according to Zhang et al.32, while Ding et al.33 suggested that Akkermansia might ameliorate chronic stress-induced depressive-like behaviors. These contradictory findings might be the consequence of differences in participants, sequencing, and analytical approaches. Turicibacter and Lachnospiraceae_UCG-006 belonged to phylum Firmicutes. Furthermore, we found that 63.63% of the differential genera belonged to the phylum Firmicutes. This result is in line with our earlier research that 44.44% of differential genera in depressed individuals also belonged to the phylum Firmicutes34. At the phylum level, disturbances of Firmicutes have been identified as a possible hallmark of depression19,35. Accordingly, these results suggested that RIB would induce the gut microbiota disordered, and that the bacterial phylum Firmicutes disturbances might be a significant contributing factor to RIB-caused depressive-like behaviors.
According to previous research36, RIB levels in human urine were positively correlated with serum triglyceride levels, and Sprague-Dawley rats given RIB injection had considerably higher hepatic triglyceride levels, suggesting that RIB might regulate lipid metabolism. Besides, lipids are crucial for brain neuronal activitySucrose preference test Mice were adapted to a 1% sucrose solution for 3 days before being presented with two bottles that contained water or 1% sucrose solution on testing day. For sucrose preference, mice were given water and 1% sucrose solution, and 12 h consumption was quantified. The consumption of sucrose preference was calculated as follows: sucrose intake/total fluid consumption52. Mice were placed in an open field apparatus (50 × 50 × 40 cm) for 5 min 30 s, with the first 30 s used for adaptation. The locomotor activity was analyzed using the Noldus automated tracking system. The test was performed, with the murine caudal tip adhered on a suture and suspended 30 cm above the ground. Each test lasted for 5 min 30 s, with the first 30 s used for adaption. The immobility duration of each mouse was counted by the Noldus automated tracking system. Mice were dissected at the end of the experiments, and the entire colon was removed to measure its length from the colon-cecal junction to the anal verge. Excised colon tissue was fixed in 4% paraformaldehyde and embedded in paraffin, similar to the earlier description13, the blocks were serially cut into 4-μm-thick sections and stained with hematoxylin and eosin. Histological images were captured using an E100 microscope (Nikon, Tokyo, Japan) to explore the examine colon tunica mucosa, tunica submucosa, and tunica muscularis thickness. Excised colon tissues were dissected into small pieces (1 × 1 × 1 mm), and fixed in 1% osmium tetroxide for 2 h. Following the dehydration steps, the tissues were embedded in Epon 812 resin (Electron Microscopy Sciences; Hartfield, PA, US), sliced, and stained with 2% uranyl acetate and lead citrate. Finally, the slices were observed with an electron microscope (HT7700, HITACHI, Tokyo, Japan), and the images were captured on a Morada camera (Münster, Germany). Briefly, 4% paraformaldehyde-fixed and paraffin-embedded colon tissues were sectioned into 4μm sections. The intestinal epithelial barrier markers Occludin (1:200, Abcam, ab216327) and colonic MUC2 (1:2000, Abcam, ab272692) were then detected by immunohistochemistry53. All images were captured using a Nikon DS-U3 microscope (Nikon, Tokyo, Japan). The EZNA-soil DNA kit (Omega Bio‐Tek, USA) was used to extract microbial DNA from frozen fecal samples in accordance with the manufacturer’s protocols and our previous studies19,54. DNA concentration and purity were assessed using the NanoDrop 2000 UV–vis spectrophotometer, and the quality of the DNA was verified using 1% agarose gel electrophoresis. The 16S rRNA gene of bacteria was amplified by Polymerase Chain Reaction using primers 338 F (5′‐ACTCCTACGGGAGGCAGCAG‐3′) and 806 R (5′‐GGACTACHVGGGTWTCTAAT‐3′) to target the V3–V4 hypervariable regions. AxyPrep DNA Gel Extraction Kit (Axygen Biosciences, Union City, CA, USA) was used to extract and purify amplicons from 2% agarose gels. Purified amplicons were quantified using QuantiFluor‐ST (Promega, USA) and paired‐end sequenced (2 × 250) on an Illumina MiSeq platform using standard protocols in Shanghai Majorbio Bio‐pharm Technology Co., Ltd. Trimmomatic was used to quality-filter raw fastq files, and short reads were quickly joined by adjusting their length. Using UPARSE (https://drive5.com/uparse/), the remaining high‐quality sequences were grouped into OTUs at 97% similarity. The ribosomal database project classifier algorithm (https://rdp.cme.msu.edu/) was used to examine the taxonomy of each 16S rRNA gene sequence. Alpha diversity was assessed to estimate the microbial communities’ diversity, including four parameters (Shannon, Simpson, Shao, and Phylogenetic diversity). Beta diversity analysis was performed to evaluate the difference in bacterial communities between CON and RIB using principal coordinate analysis plots. The linear discriminant analysis effective size is conducted to identify the key bacterial taxa responsible for discrimination between two groups. Similar to our previous studies55,56, peripheral and hippocampus tissue samples were prepared by homogenization, dissociation, and centrifugation. Serum samples were collected and centrifuged twice. L-2-chlorophenylalanine dissolved in methanol (0.02 mg/mL) was served as an internal standard. The untargeted liquid chromatography–mass spectrometry metabolomics analysis was conducted on a Thermo UHPLC-Q Exactive HF-X system equipped with an ACQUITY UPLC T3 column (100 mm × 2.1 mm i.d., 1.8 μm; Waters, USA). For the electrospray ionization positive mode, the mobile phases consisted of 0.1% formic acid in water (solvent A) and 0.1% formic acid in acetonitrile (solvent B). For the electrospray ionization negative mode, the mobile phases were water with 6.5 mM ammonium bicarbonate (solvent C) and 6.5 mM ammonium bicarbonate 95% methanol solution (solvent D). The flow rate was 0.40 mL/min and the column temperature was 40 °C. The injection volume was 2 μL. The mass spectrometric data was collected using the Thermo UHPLC-Q Exactive HF-X Mass Spectrometer equipped with an electrospray ionization source operating in positive mode and negative mode. The optimal conditions were set as follows: source temperature, 425 °C; sheath gas flow rate, 50 arb; Aux gas flow rate, 13 arb; ion-spray voltage floating, -3500 V (-) and 3500 V (+); Normalized collision energy, 20-40-60 V rolling for mass spectrometry/mass spectrometry. The raw data generated by liquid chromatography–mass spectrometry were processed by baseline filtering, peak identification, integration, retention time correction, peak alignment, and normalization using Progenesis QI (Waters Corporation). The quality control samples were used to validate the stability of the metabolomic analysis. Metabolites were identified by Human Metabolome Database (http://www.hmdb.ca), Metlin (https://metlin.scripps.edu), and self-built databases. The peak intensity was deemed as the expression level of metabolite57. Unsupervised principal component analysis and OPLS-DA were used to show overall metabolic differences and variation between groups. Metabolite sets enrichment analysis was further performed based on the differential expressed metabolites using MetaboAnalyst 5.0 (http://www.metaboanalyst.ca)58. The differential metabolites were also inspected into the KEGG database (http://www.genome.jp/kegg/), and then KEGG was used to annotate the functions in which these differential metabolites were closely involved. Statistical analyses were performed using SPSS 20 (Chicago, IL, US) and R studio (version 3.6.0, 2021). Data are presented as mean ± Standard Error of mean. p < 0.05 was considered statistically significant. The Student’s t-test, nonparametric Mann-Whitney U test, Chi-Square test, or Spearman’s correlation analysis was used when appropriate. Linear discriminant analysis effective size analysis was performed to identify the differential genera (linear discrimination analysis >2.0 and p < 0.05) responsible for the discrimination between groups. Phylogenetic Investigation of Communities by Reconstruction of Unobserved States was further used to predict the functions of these differential genera. Metabolites with VIP > 1.0 and p < 0.05 were viewed as differential metabolites by analyzing the OPLS-DA loading plot. To find out the key depressive-like behaviors-related metabolic modules of the MGB axis, the weighted correlation network analysis was used59,60. All data collection and analyses were performed blind to the conditions of the experiments. Sample sizes are defined in the corresponding figure legend. Especially, for the 16 S rRNA gene sequencing and metabolomics analysis, 16 mice were used (n = 8 in CON group, n = 8 in RIB group). For the detection of the hematoxylin and eosin, immunohistochemistry, and electron microscopy analysis, three biological independent animals were used.Open field test
Tail suspension test
Colonic histopathology
Transmission electron microscopy analysis
Histology
16S rRNA gene sequence analysis
Untargeted metabolomics analysis
Statistical and reproducibility
Data availability
Raw data for 16S rRNA sequencing have been deposited in NCBI, under SRA accession number PRJNA1052466. Raw metabolite data are available in the Metabolights Database (MTBLS9185). Source data for Figs. 1, 2, and 3a are provided in Supplementary Data 13. All other data are available from the corresponding author upon reasonable request.
References
Hellsten, Y., Skadhauge, L. & Bangsbo, J. Effect of ribose supplementation on resynthesis of adenine nucleotides after intense intermittent training in humans. Am. J. Physiol. Regul. Integr. Comp. Physiol. 286, R182–188, (2004).
Teitelbaum, J. E., Johnson, C. & St Cyr, J. The use of D-ribose in chronic fatigue syndrome and fibromyalgia: a pilot study. J. Altern. Complement. Med. 12, 857–862 (2006).
Perlmutter, N. S. et al. Ribose facilitates thallium-201 redistribution in patients with coronary artery disease. J. Nucl. Med. 32, 193–200 (1991).
Mauser, M., Hoffmeister, H. M., Nienaber, C. & Schaper, W. Influence of ribose, adenosine, and “AICAR” on the rate of myocardial adenosine triphosphate synthesis during reperfusion after coronary artery occlusion in the dog. Circ. Res. 56, 220–230 (1985).
Turck, D. et al. Safety of d-ribose as a novel food pursuant to Regulation (EU) 2015/2283. EFSA J. 16, e05265 (2018).
Yu, L. et al. D-ribose is elevated in T1DM patients and can be involved in the onset of encephalopathy. Aging 11, 4943–4969 (2019).
Wei, Y. et al. Ribosylation triggering Alzheimer’s disease-like Tau hyperphosphorylation via activation of CaMKII. Aging Cell 14, 754–763 (2015).
Almulla, A., Abbas Abo Algon, A., Tunvirachaisakul, C., Al-Hakeim, H. & Maes, M. T helper-1 activation via interleukin-16 is a key phenomenon in the acute phase of severe, first-episode major depressive disorder and suicidal behaviors. J. Adv. Res. https://doi.org/10.1016/j.jare.2023.11.012 (2023).
Swainson, J. et al. Diet and depression: a systematic review of whole dietary interventions as treatment in patients with depression. J. Affect. Disord. 327, 270–278 (2023).
DelMastro, K. et al. Incidence of major depressive episode correlates with elevation of substate region of residence. J. Affect. Disord. 129, 376–379 (2011).
Harrell, C. S., Burgado, J., Kelly, S. D., Johnson, Z. P. & Neigh, G. N. High-fructose diet during periadolescent development increases depressive-like behavior and remodels the hypothalamic transcriptome in male rats. Psychoneuroendocrinology 62, 252–264 (2015).
Hirai, S. et al. High-sucrose diets contribute to brain angiopathy with impaired glucose uptake and psychosis-related higher brain dysfunctions in mice. Sci. Adv. 7, eabl6077 (2021).
Xu, K. et al. Chronic D-ribose and D-mannose overload induce depressive/anxiety-like behavior and spatial memory impairment in mice. Transl. Psychiatry 11, 90 (2021).
Do, M. H., Lee, E., Oh, M. J., Kim, Y. & Park, H. Y. High-glucose or -fructose diet cause changes of the gut microbiota and metabolic disorders in mice without body weight change. Nutrients 10, 761 (2018).
Li, J. M. et al. Dietary fructose-induced gut dysbiosis promotes mouse hippocampal neuroinflammation: a benefit of short-chain fatty acids. Microbiome 7, 98 (2019).
Thaiss, C. A. et al. Hyperglycemia drives intestinal barrier dysfunction and risk for enteric infection. Science 359, 1376–1383 (2018).
Mitra, S., Dash, R., Nishan, A., Habiba, S. & Moon, I. Brain modulation by the gut microbiota: from disease to therapy. J. Adv. Res. 53, 153–173 (2023).
Sanada, K. et al. Gut microbiota and major depressive disorder: a systematic review and meta-analysis. J. Affect. Disord. 266, 1–13 (2020).
Zheng, P. et al. Gut microbiome remodeling induces depressive-like behaviors through a pathway mediated by the host’s metabolism. Mol. Psychiatry 21, 786–796 (2016).
Li, B. et al. Metabolite identification in fecal microbiota transplantation mouse livers and combined proteomics with chronic unpredictive mild stress mouse livers. Transl. Psychiatry 8, 34 (2018).
Glowacki, R. W. P. et al. A ribose-scavenging system confers colonization fitness on the human gut symbiont bacteroides thetaiotaomicron in a diet-specific manner. Cell Host Microbe 27, 79–92 e79 (2020).
Zheng, P. et al. The gut microbiome modulates gut-brain axis glycerophospholipid metabolism in a region-specific manner in a nonhuman primate model of depression. Mol. Psychiatry 26, 2380–2392 (2021).
Stockard, B., Gauldin, C., Truog, W. & Lewis, T. Pharmacometabolomics profiling of preterm infants validates patterns of metabolism associated with response to dexamethasone treatment for bronchopulmonary dysplasia. Front. Pediatr. 10, 898806 (2022).
Ghosh, S., Whitley, C. S., Haribabu, B. & Jala, V. R. Regulation of intestinal barrier function by microbial metabolites. Cell. Mol. Gastroenterol. Hepatol. 11, 1463–1482 (2021).
Chen, G. et al. Fluoride induced leaky gut and bloom of Erysipelatoclostridium ramosum mediate the exacerbation of obesity in high-fat-diet fed mice. J. Adv. Res. 50, 35–54 (2023).
Zhao, H. et al. A pilot exploration of multi-omics research of gut microbiome in major depressive disorders. Transl. Psychiatry 12, 8 (2022).
Caso, J. et al. Gut microbiota, innate immune pathways, and inflammatory control mechanisms in patients with major depressive disorder. Transl. Psychiatry 11, 645 (2021).
Khan, S. et al. Dietary simple sugars alter microbial ecology in the gut and promote colitis in mice. Sci. Transl. Med. 12, eaay6218 (2020).
Aggarwal, V., Sunder, S. & Verma, S. R. Disease-associated dysbiosis and potential therapeutic role of Akkermansia muciniphila, a mucus degrading bacteria of gut microbiome. Folia Microbiol. (Praha) 67, 811–824 (2022).
Derrien, M., Belzer, C. & de Vos, W. M. Akkermansia muciniphila and its role in regulating host functions. Microb. Pathog. 106, 171–181 (2017).
Desai, M. S. et al. A dietary fiber-deprived gut microbiota degrades the colonic mucus barrier and enhances pathogen susceptibility. Cell 167, 1339–1353.e1321 (2016).
Zhang, X. et al. Taxonomic and metabolic signatures of gut microbiota for assessing the severity of depression and anxiety in major depressive disorder patients. Neuroscience 496, 179–189 (2022).
Ding, Y. et al. A next-generation probiotic: akkermansia muciniphila ameliorates chronic stress-induced depressive-like behavior in mice by regulating gut microbiota and metabolites. Appl. Microbiol. Biotechnol. 105, 8411–8426 (2021).
Zhong, Q. et al. Differential gut microbiota compositions related with the severity of major depressive disorder. Front. Cell. Infect. Microbiol. 12, 907239 (2022).
Naseribafrouei, A. et al. Correlation between the human fecal microbiota and depression. Neurogastroenterol. Motil. 26, 1155–1162 (2014).
Chen, Y. et al. D-ribose increases triglyceride via upregulation of DGAT in the liver. Sci. China Life Sci. 62, 858–861 (2019).
**ong, F. et al. Optimized integration of metabolomics and lipidomics reveals brain region-specific changes of oxidative stress and neuroinflammation in type 1 diabetic mice with cognitive decline. J. Adv. Res. 43, 233–245 (2023).
Muller, C. P. et al. Brain membrane lipids in major depression and anxiety disorders. Biochim. Biophys. Acta 1851, 1052–1065 (2015).
Bai, S. et al. Potential biomarkers for diagnosing major depressive disorder patients with suicidal ideation. J. Inflamm. Res. 14, 495–503 (2021).
Xu, K. et al. Altered MANF and RYR2 concentrations associated with hypolipidemia in the serum of patients with schizophrenia. J. Psychiatr. Res. 163, 142–149 (2023).
Chen, J. et al. Absence of gut microbiota affects lipid metabolism in the prefrontal cortex of mice. Neurol. Res. 41, 1104–1112 (2019).
Chen, J. J. et al. Effects of gut microbiota on the microRNA and mRNA expression in the hippocampus of mice. Behav. Brain Res. 322, 34–41 (2017).
Liu, X. et al. Plasma lipidomics reveals potential lipid markers of major depressive disorder. Anal. Bioanal. Chem. 408, 6497–6507 (2016).
Davis, M. T., Holmes, S. E., Pietrzak, R. H. & Esterlis, I. Neurobiology of chronic stress-related psychiatric disorders: evidence from molecular imaging studies. Chronic Stress (Thousand Oaks) 1, 2470547017710916 (2017).
Harper, D. G. et al. Tissue-specific differences in brain phosphodiesters in late-life major depression. Am. J. Geriatr. Psychiatry 22, 499–509 (2014).
Oliveira, T. G. et al. The impact of chronic stress on the rat brain lipidome. Mol. Psychiatry 21, 80–88 (2016).
Chen, J. et al. Differential urinary metabolites related with the severity of major depressive disorder. Behav. Brain Res. 332, 280–287 (2017).
Liu, L. et al. Hippocampal metabolic differences implicate distinctions between physical and psychological stress in four rat models of depression. Transl. Psychiatry 8, 4 (2018).
Liu, Y. et al. Social defeat stress causes depression-like behavior with metabolite changes in the prefrontal cortex of rats. PLoS ONE 12, e0176725 (2017).
Wu, B. et al. Gavage of D-Ribose induces Aβ-like deposits, Tau hyperphosphorylation as well as memory loss and anxiety-like behavior in mice. Oncotarget 6, 34128–34142 (2015).
Xu, K. et al. Validation of the targeted metabolomic pathway in the hippocampus and comparative analysis with the prefrontal cortex of social defeat model mice. J. Neurochem. 149, 799–810 (2019).
Willner, P., Towell, A., Sampson, D., Sophokleous, S. & Muscat, R. Reduction of sucrose preference by chronic unpredictable mild stress, and its restoration by a tricyclic antidepressant. Psychopharmacology 93, 358–364 (1987).
Xu, K. et al. HMGB1/STAT3/p65 axis drives microglial activation and autophagy exert a crucial role in chronic Stress-Induced major depressive disorder. J. Adv. Res. https://doi.org/10.1016/j.jare.2023.06.003 (2023).
Tian, T. et al. Multi-omics data reveals the disturbance of glycerophospholipid metabolism caused by disordered gut microbiota in depressed mice. J. Adv. Res. 39, 135–145 (2022).
Wu, Z. et al. Non-targeted metabolomics profiling of plasma samples from patients with major depressive disorder. Front. Psychiatry 12, 810302 (2021).
Fan, L. et al. Proteomic and metabolomic characterization of amygdala in chronic social defeat stress rats. Behav. Brain Res. 412, 113407 (2021).
Liang, L. et al. Metabolic dynamics and prediction of gestational age and time to delivery in pregnant women. Cell 181, 1680–1692.e1615 (2020).
Pang, Z. et al. MetaboAnalyst 5.0: narrowing the gap between raw spectra and functional insights. Nucleic Acids Res. 49, W388–W396 (2021).
Duan, W. et al. Chilling-induced peach flavor loss is associated with expression and DNA methylation of functional genes. J. Adv. Res. 53, 17–31 (2023).
Zillich, L. et al. Multi-omics signatures of alcohol use disorder in the dorsal and ventral striatum. Transl. Psychiatry 12, 190 (2022).
Acknowledgements
This study was supported by the National Key R&D Program of China (2017YFA0505700), the Non-profit Central Research Institute Fund of Chinese Academy of Medical Sciences (2019PT320002), the National Natural Science Foundation of China (81820108015, 82301720), the China Postdoctoral Science Foundation (2023MD734128), the Chongqing Postdoctoral Science Foundation (CSTB2023NSCQ-BHX0214), and the First Affiliated Hospital of Chongqing Medical University Postdoctoral Science Foundation (CYYY-BSHPYXM-202310).
Author information
Authors and Affiliations
Contributions
P.X., J.C., and K.X. conceived, designed, and supervised the overall project. K.X., Y.R., and S.Z. performed the whole experiments and acquired the data. J.C. conducted statistical analysis. J.F. helped with the behavior testing and related data analyses. Q.W. assisted with histology experiments. X.G. assisted with transmission electron microscopy experiments. K.X. and J.C. wrote the first version of the manuscript. All authors read, edited, and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Communications Biology thanks Wenzhi Hao and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Primary Handling Editor: Tobias Goris.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Xu, K., Ren, Y., Zhao, S. et al. Oral d-ribose causes depressive-like behavior by altering glycerophospholipid metabolism via the gut-brain axis. Commun Biol 7, 69 (2024). https://doi.org/10.1038/s42003-023-05759-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s42003-023-05759-1
- Springer Nature Limited
This article is cited by
-
Elevated SCN11A concentrations associated with lower serum lipid levels in patients with major depressive disorder
Translational Psychiatry (2024)