
Abstract
Complete mitochondrial genomes (mitogenomes) can provide useful information for phylogenetic relationships, gene rearrangement, and evolutionary traits. In this study, we determined the complete mitochondrial DNA sequence of the herbivorous crab Grapsus albolineatus. It is a typical metazoan mitochondrial genome. The total size is 15,583 bp, contains the entire set of 37 genes, and has an AT-rich region. Then, 23 of the 37 genes were encoded by the heavy (+) strand while 14 are encoded by the light (−) strand. Compared with the pan-crustacean ground pattern, two tRNA genes (tRNA-His and tRNA-Gln) were rearranged and the tandem duplication/random loss model was used to explain the observed gene rearrangements. The phylogenetic results showed that all Grapsidae crabs clustered together as a group. Furthermore, the monophyly of each family was well supported, with the exception of Menippidae. In general, the results obtained in this study will contribute to the better understanding of gene rearrangements in Grapsidae crab mitogenomes and provide new insights into the phylogeny of Brachyura.
Similar content being viewed by others


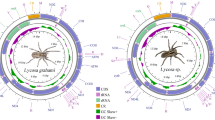
Introduction
Brachyura crab is the largest clade in the Decapod crustacean group, with more than 7250 known species, including 98 families of marine, freshwater, and terrestrial habitats, most of which are economically important1. However, the phylogenetic relationships among members of Brachyura and their evolutionary origin continue to be controversial due to the high morphological similarity and ecological diversity2,3,4. Initially, Brachyura was divided into Podotremata, Heterotremata, and Thoracotremata5. Subsequently, it was segmented into Dromiacea and Eubrachyura (including Thoracotremata, Raninoida, and Heterotremata)6. However, the latest classification scheme divides Brachyura into Cyclodorippoida, Eubrachyura, Dromicea, and Raninoida7,8. Although the phylogenetic relationship within Brachyura is still uncertain, the current classification system has been recognized by most scholars.
According to WoRMS (http://www.marinespecies.org/), the family Grapsidae has 8 genera and 49 species in total. However, only five species sequences of Grapsidae have been published4,9,10,11,12. The herbivorous crab (Grapsus albolineatus) is one of the marine crustaceans that live on rocky shores which belongs to the phylum Arthropod, subphylum Crustacea, order Decapoda, infraorder Brachyura, clade Thoracotremata, family Grapsidae, genus Grapsus. They are mainly distributed in Japan, Hawaii, Australia and China’s Guangdong, Hainan Island, ** the gene layout based on the complete mitochondrial sequences of 70 species. Through comparison and analysis with the ancestor of Decapoda (Fig. 3A), we found that G. albolineatus and another 5 species from Grapsidae have a trnH translocation4,9,10,11,12,13, which the trnH shifted into trnE and trnF instead of the usual location between nad5 and nad4 (Fig. 3C). It is widely believed that the tandem duplication/random loss model (TDRL) can explain the movement of trnH, occur from tandem duplication in the region between trnE and nad4, followed by deletions of redundant genes producing trnH-trnF-nad5. Additionally, 45 species from 14 families (Grapsidae, Mictyridae, Ocypodidae, Bythograeidae, Calappidae, Dotillidae, Matutidae, Menippidae, Oziidae, Xanthidae, Oregoniidae, Geryonidae, Portunidae, and Carpiliidae) had the same gene rearrangement, which are consistent with the ancestral of Brachyura (Fig. 3B). However, the gene order in 4 families (Sesarmidae, Varunidae, Macrophthalmidae, and Xenograpsidae)30,32 displayed 4 patterns of gene rearrangements. The family Sesarmidae observed trnQ and trnI invertred, which has been described in previous studies (Fig. 3D)3,19,20,33. The gene order of the Varunidae (Grapsoidea) and Macrophthalmidae (Ocypodoidea) have the same high level rearrangementa (Fig. 3E). It is worth noting that the two families come from two different superfamilies, but they form a sister clade in phylogenetic trees. The gene order of the Xenograpsidae have a more complex rearrangement and such within-genus rearrangements were infrequent34 (Fig. 3F,G), which seems to be related to their particular habitat. Xenograpsidae have been found thus far only in shallow-water, volcanically active, and sulphur-rich hydrothermal vents35.

Linear representation of gene arrangements of an (A) ancestor of Decapoda, (B) ancestor of Brachyura, (C) gene arrangement of Grapsus albolineatus and 13 familes, (D) gene arrangement of Sesarmidae, (E) gene arrangement of Varunidae amd Macrophthalmidae, (F) gene arrangement of Xenograpsus testudinatus, and (G) gene arrangement of Xenograpsus testudinatus. Gene arrangement of all genes are transcribed from left to right. The green box indicates the duplicated gene. 16S rRNA and 12S rRNA are the large and small ribosomal RNA subunits, respectively. The rearranged gene blocks are underlined and compared with ancestral gene arrangement of Brachyura. The genes encoded on the light strand are highlighted in red.
Phylogenetic relationships
In the present study, the phylogenetic relationships were analyzed based on the sequences of the 13 PCGs to clarify the relationships in Brachyura. G. albolineatus and other 68 known brachyuran specie were analyzed, with P. nigrofascia and P. gracilipes as outgroups. The two phylogenetic trees (Maximum Likelihood (ML) tree and Bayesian Inference (BI) tree) resulted in identical topological structuring with different supporting value. Then, only one topology (ML) with both support values was presented displayed (Fig. 4). Both trees showed that all the species of Grapsidae clustered together as a solid monophyletic group and consist of three sister clades ((Grapsus + Pachygrapsus) + Metapograpsus). It is obvious that G. albolineatus had the closest relationship with G. tenuicrustatus, and that these two species form a sister clade with high support values (BI posterior probabilities PP = 1, ML bootstrap BP = 100), constituting a Grapsus group. However, recent molecular studies, including our dataset, have not reached an agreement about closest relatives in Grapsidae. Our phylogenetic tree showed that Grapsidae and Dotillidae form a sister clade, which was in concordance with Wang et al.10. While Wang et al. and Ng, N. K. et al. found that Grapsidae do not have any close relatives9,35, Li et al.36 found that Grapsidae and Ocypodidae form a sister clade.

The phylogenetic tree was inferred from the nucleotide sequences of 13 mitogenome PCGs using BI and ML methods. Numbers on branches indicate posterior probability (BI) and bootstrap support (ML). The node marked with a solid citcle indicates 100 ML bootstrap support (BS) and 100% BI posterior probability (PP).
Among the 21 families included in our phylogenetic tree, except Menippidae, each family in the tree forms a monophyletic clade with high nodal support values. At a higher level of classification, most Brachyura superfamilies were found to be monophyletic, except Ocypodoidea, Grapsoidea and Eriphioidea, which is in line with previous studies9,10,37. It showed that Grapsidea was divided into three clades (((Seasamidae + Gecarcinidae + Xengrapsidae) + Grapsidae) + Varunidae), Ocypodoidea was divided in three clades ((Ocypodidae + Dotillidae) + Macrophthalmidae + Mictyrisae) and Eriphioidea was divided into two clades (Oziidae + Menippidae). Within Thoracotremata, the superfamilies Ocypodoidea and Grapsoidea supported paraphletic and 9 families showed the following relayionship: ((((Seasamidae + Gecarcinidae) + Xengrapsidae) + Ocypodidae) + (Grapsidae + Dotillidae) + (Varunidae + Macrophthalmidae) + Mictyrisae) (Fig. 4).
The main phylogenetic structure of our tree is consistent with previous results, but some controversial findings were observed. Here, the families Macrophthalmidae and Varunidae were grouped into one clade, and Mictyridae as basal group which supports the previous findings revealed in Wang et al. and Zhang et al.9,33. However, previous researchers revealed that Macrophthalmidae and Varunidae were grouped into one clade, then into another clade with Varunidae ((Macrophthalmidae + Varunidae) + Mictyridae)38,39, which was conflict with our results. The classification of Grapsoidea and Ocypodoidea has long been controversial. Previous studies based on morphological characteristics considered them to be monophyletic branches. However, an increasing number of molecular studies, including ours, challenge the inconsistent views on the traditional classification system that are put forward. Although the polyphyly of Grapsoidea, Ocypodoidea, and Eriphioidea is well supported, the phylogenetic relationships of these superfamilies need to be further analyzed by integrating additional molecular data32,33,34,35,36. Previous studies on mitochondrial phylogeny have confirmed the importance of mitochondrial genomic data in elucidating the Grapsidae phylogeny13,19. On the contrary, many families contained only one representative, which may produce unstable phylogenetic relationships. Therefore, it is necessary to perform further mitogenome sequence studies to obtain a more comprehensive taxon sampling and understand the phylogeny and evolution of Grapsidae.
Materials and methods
Sampling and DNA extraction
A specimen of G. albolineatus was collected from Yangjiang, Guangdong Province, China (21°28′45″ N, 111°16′35″ E). The specimen was immediately preserved in absolute ethanol after collection and then stored at −20 °C. This specimen was identified by morphology and fresh tissues were dissected from the operculum and preserved in absolute ethanol before DNA extraction. The total genomic DNA was extracted using the salt-extraction procedure with a slight modification40 and stored at −20 °C.
Genome sequencing, assembly, and annotation
The mitogenomes of G. albolineatus was sequenced by Origin gene Co. Ltd., Shanghai, China and was sequenced on the Illumina HiSeq X Ten platform. HiSeq X Ten libraries with an insert size of 300–500 bp were generated from the genomic DNA. About 10 Gb of raw data was generated for each library. Low-quality reads, adapters, and sequences with high “N” ratios and length less than 25 bp were removed. The clean reads were assembled using the software NOVOPlasty (https://github.com/ndierckx/NOVOPlasty)42, annotated, and manually corrected on the basis of the complete mitogenome sets assembled de novo by using MITOS tools (http://mitos2.bioinf.uni-leipzig.de/index.py)43. To confirm the correct sequences, we compared the assembled mitochondrial genes with those of other Grapsus species and identified the mitogenomic sequences by checking the cox1 barcode sequence with NCBI BLAST43. The abnormal start and stop codons were determined by comparing them with the start and stop codons of other marine gastropods. Then, the reads were reconstructed using the de novo assembly program. The complete mtDNA was annotated using the software Sequin version 16.0 (https://trace.ncbi.nlm.nih.gov/Traces/sra). The mitogenome map of the G. albolineatus was drawn using the online tool CGView Server (http://cgview.ca/)45. The secondary structures predicted of the tRNA genes were plotted by using MITOS Web Server. The relative synonymous codon usage (RSCU) values and substitution saturation for the 13 PCGs, calculated by DAMBE 545, were analyzed with MEGA 746. The GC-skews and AT-skews were used to determine the base compositional difference and strand asymmetry among the samples. According to the following formulas46, composition skew values were calculated as AT-skew = A − T/A + T and GC skew = G − C/G + C. Substitution saturation for the 13 PCGs was calculated by DAMBE 545.
Phylogenetic analysis
The phylogenetic relationships within Brachyura were reconstructed using the sequences of the 13 PCGs of a total of 57 complete mitogenome sequences downloaded from the GenBank database (https://www.ncbi.nlm.nih.gov/genbank/) and adding two species of Paguridae to serve as the outgroup (Table 1). The phylogenetic relationships were analyzed with Maximum Likelihood (ML) by using IQ-TREE 1.6.2 and Bayesian Inference (BI) methods in MrBayes 3.2 version program47,48,49. The ML analysis was inferred with 1000 ultrafast likelihood bootstrap replicates by using IQ-TREE 1.6.2. The best-fit model for each partition was GTR + F + R6, selected according to the Bayesian information criterion (BIC). BI was performed in MrBayes 3.2, and the best-fit evolutionary models were determined using MrMTgui50. MrMTgui was used to associate PAUP, ModelTest, and MrModelTest across platforms. MrBayes settings for the best-fit model (GTR + I + G) were selected by Akaike Information Criterion (AIC) in MrModelTest 2.351,52. The Bayesian phylogenetic analyses were performed using the parameter values estimated with the commands in MrModelTest or ModelTest (nst = 6, rates = invgamma)53. With three hot chains and one cold chain, they were run simultaneously twice by Markov Chain Monte Carlo (MCMC) sampling, and the posterior distribution was estimated. The MCMC chains were set for 2,000,000 generations and sampled every 1000 steps, with a relative burn-in of 25%. The convergence of the independent runs was evaluated by mean standard deviation of the split frequencies (< 0.01). The phylogenetic trees were visualized and edited using Figure Tree v1.4.3 software54.
Conclusions
In this study, the mitogenome of G. albolineatus was sequenced by next-generation sequencing, thereby generating new mitochondrial data for Grapsidae and confirming its ancestral gene order. The G. albolineatus mitogenome is a typical closed-circular molecule including 13 PCGs, 22 tRNA genes, two rRNA genes, and a CR. The AT-skew and GC-skew are both negative in the mitogenome of G. albolineatus, showing an obvious bias towards the use of T’s and C’s, consistent with published findings in most Brachyura crabs. G. albolineatus exhibits a novel gene rearrangement, which is similar to G. tenuicrustatus, P. crassipes, P. marmoratu, M. frontalis, and M. quadridentatus. Compared with the pan-crustacean ground pattern, the trnH of G. albolineatus shifted into trnE and trnF instead of the usual location between nad5 and nad4. By adding 62 Brachyura mitochondrial genomes, rearrangement and the phylogeny of Brachyura was reanalyzed. The phylogenetic analyses indicated that G. albolineatus has close relationships with G. tenuicrustatus, P. crassipesand, P. marmoratu, M. frontalis, and M. quadridentatus, belonging to Grapsoidea, part of the Grapsidae family.
Data availability
The complete mitogenome of Grapsus albolineatus has been submitted to GenBank under the accession number of MZ262276. The data that support the finding of this study are openly available in Microsoft OneDrive at https://1drv.ms/u/s!Apz_mHDHDJqiUHXhxzoLR0_NEHf?e=u7Ne8W.
References
Basso, A. et al. Thehighly rearranged mitochondrial genomes of the crabs Maja crispata and Maja squinado (Majidae) and gene order evolution in Brachyura. Sci. Rep. 7(1), 4096 (2017).
Liu, H. et al. Novel insights into mitochondrial gene rearrangement in thrips (Insecta: Thysanoptera) from the grass thrips, Anaphothrips obscurus. Sci. Rep. 7(1), 4284 (2017).
Li, Q., Xu, C., Wan, C. & Liu, G. The complete mitochondrial genome of red-clawed crab Chiromantes haematochir (Sesarmidae: Grapsidae). Mitochond. DNA B Resour. 4(1), 53–54 (2019).
Guan, M. et al. The whole mitochondrial genome of the mangrove crab, Metopograpsus frontalis (Miers, 1880) (Decapoda, Grapsidae) and its phylogenetic relationship. Mitochond. DNA B Resour. 3(1), 368–369 (2018).
Gregersen, H. M., Oram, P. & Spears, J. Priorities for forestry and agroforestry policy research. Class. Rev. 54(1), 138–139 (1992).
Martin, J. W. & Davis, G. W. An updated classification of the recent Crustacea. Natural history museum of Los Angeles county. Science 39, 1–124 (2001).
Ming, T. L. et al. Evolutionary history of true crabs (Crustacea: Decapoda: Brachyura) and the origin of freshwater crabs. Mol. Biol. Evol. 31(5), 1173–1187 (2014).
Ahyong, S. T. et al. Phylogenetics of the brachyuran crabs (Crustacea: Decapoda): the status of Podotremata based on small subunit nuclear ribosomal RNA. Mol. Phylog. Evol. 45(2), 576–586 (2007).
Wang, Z. et al. Characterization of the complete mitochondrial genome of Uca lacteus and comparison with other Brachyuran crabs. Genomics 112(1), 10–19 (2020).
Wang, Q. et al. Comparative mitochondrial genomic analysis of Macrophthalmus pacificus and insights into the phylogeny of the Ocypodoidea and Grapsoidea. Genomics 112(1), 82–91 (2020).
Sung, J. M., Lee, J. H. & Kim, S. K. The complete mitochondrial genome of Grapsus tenuicrustatus (Herbst, 1783) (Decapoda, Grapsidae). Mitochond. DNA B Resour. 1(1), 441–442 (2016).
Yu, Y. Q., Ma, W. M., Yang, W. J. & Yang, J. S. The complete mitogenome of the lined shore crab Pachygrapsus crassipes Randall 1840 (Crustacea: Decapoda: Grapsidae). Mitochond. DNA B Resour. 25(4), 263–274 (2014).
Kennish, R. & Williams, G. A. Feeding preferences of the herbivorous crab Grapsus albolineatus: the differential influence of algal nutrient content and morphology. Mar. Ecol. Prog. Ser. 147(1/3), 87–95 (1997).
Li, M. H. Fluctuating asymmetry and intersexuality in the shore crab Grapsus albolineatus near a coastal landfill site in northern Taiwan. Br. Mar. Sci. 70(1), 75–88 (2002).
Schubart, C. D., Cannicci, S., Vannini, M. & Fratini, S. Molecular phylogeny of grapsoid crabs (Decapoda, Brachyura) and allies based on two mitochondrial genes and a proposal for refraining from current superfamily classification. J. Zool. Syst. Evol. Res. 44(3), 193–199 (2006).
Boore, J. L. Animal mitochondrial genomes. Nucl. Acids Res. 27(8), 1767–1780 (1999).
Sato, M. & Sato, K. Maternal inheritance of mitochondrial DNA by diverse mechanisms to eliminate paternal mitochondrial DNA. BBA Biomemb. 1833(8), 1979–1984 (2013).
Mun, H. T. et al. Comparative mitogenomics of the Decapoda reveals evolutionary heterogeneity in architecture and composition. Sci. Res. 9(2), 221–229 (2019).
Zhang, Y. et al. Gene rearrangements in the mitochondrial genome of Chiromantes eulimene (Brachyura: Sesarmidae) and phylogenetic implications for Brachyura. Int. J. Biol. Macromol. 162, 704–714 (2020).
Wang, Z. et al. Complete mitochondrial genome of Parasesarma affine (Brachyura: Sesarmidae): Gene rearrangements in Sesarmidae and phylogenetic analysis of the Brachyura. Int. J. Biol. Macromol. 118, 31–40 (2018).
Maynard, S. J. & Smith, N. H. Recombination in animal mitochondrial DNA. Mol. Biol. Evol. 12, 23–33 (2002).
Moritz, C., Dowling, T. E. & Brown, W. M. Evolution of animal mitochondrial DNA: relevance for population biology and systematics. Annu. Rev. Ecol. S. 18(1), 269–292 (1987).
Arndt, A. & Smith, M. J. Mitochondrial gene rearrangement in the sea cucumber genus Cucumaria. Mol. Biol. Evol. 15(8), 1009–1016 (1998).
Postaire, B., Bruggemann, J. H., Magalon, H. & Faure, B. Evolutionary dynamics in the southwest indian ocean marine biodiversity hotspot: a perspective from the rocky shore gastropod genus Nerita. PLoS ONE 9(4), e95040 (2014).
Grantham, R., Gautier, C. & Gouy, M. Codon catalog usage and the genome hypothesis. Nucl. Acids Res. 8(1), 49–62 (1980).
Tan, M. H., Gan, H. M., Lee, Y. P., Linton, S. & Austin, C. M. ORDER within the chaos: insights into phylogenetic relationships within the Anomura (Crustacea: Decapoda) from mitochondrial sequences and gene order rearrangements. Mol. Phylogenet. Evol. 127, 320 (2018).
Zhang, K. Z. et al. Novel gene rearrangement in the mitochondrial genome of Muraenesox cinereus and the phylogenetic relationship of Anguilliformes. Sci. Rep. 11(1), 2411 (2021).
Gong, L. et al. Novel gene rearrangement in the mitochondrial genome of Coenobita brevimanus (Anomura: Coenobitidae) and phylogenetic implications for Anomura. Genomics 112(2), 1804–1812 (2020).
Gong, L., Lü, Z. M., Guo, B. Y., Ye, Y. Y. & Liu, L. Q. Characterization of the complete mitochondrial genome of the tidewater goby, Eucyclogobius newberryi (Gobiiformes, Gobiidae, Gobionellinae) and its phylogenetic implications. Conserv. Genet. Resour. 10(1), 93–97 (2018).
Ng, N. K., Suzuki, H., Shih, H. T. & Dewa, S. I. The hydrothermal crab, xenograpsus testudinatus ng, huang & ho, 2000 (crustacea: decapoda: brachyura: Grapsidae) in southern japan. Proc. Biol. Soc. Wash. 127(2), 391–399 (2014).
Tan, M. H., Gan, H. M., Lee, Y. P. & Austin, C. M. The complete mitogenome of the ghost crab ocypode ceratophthalmus (pallas, 1772) (crustacea: decapoda: ocypodidae). Mitochond. DNA. 2123 (2016).
Kim, S. J., Kim, H. S. & Ju, S. J. Mitochondrial genome of the hydrothermal vent crab austinograea alayseae (crustacea: bythograeidae): genetic differences between individuals from tofua arc and manus basin. Mitochond. DNA. 25(4), 251–252 (2014).
Zhang, Y., Gong, L., Lu, X., Jiang, L. & Zhang, X. Gene rearrangements in the mitochondrial genome of Chiromantes eulimene (Brachyura: Sesarmidae) and phylogenetic implications for Brachyura. Int. J. Biol. Macromol. 162, 704–714 (2021).
Wang, Q. et al. Insights into the evolution of Brachyura (Crustacea: Decapoda) from mitochondrial sequences and gene order rearrangements. Int. J. Biol. Macromol. 170, 2 (2021).
Ng, N. K. et al. Xenograpsidae, a new family of grapsoid crabs (Crustacea: Brachyura) associated with shallow water hydrothermal vents. Raffles Bull. Zool. 16, 233–256 (2007).
Li, Y., et al. Comparative mitochondrial genome analyses of sesarmid and other brachyuran crabs reveal gene rearrangements and phylogeny. J. Front. Genet. (2020)
**nting, L. et al. The complete mitochondrial genome of Calappa bilineata: the first representative from the family Calappidae and its phylogenetic position within Brachyura. J. Genom. 112, 3 (2020).
Xu, X. et al. The entire mitochondrial genome of Macrophthalmus Abbreviatus reveals insights into the phylogeny and gene rearrangements of Brachyura. Biochem. Genet. 59(3), 211–219 (2021).
Tan, M. H., Gan, H. M., Schultz, M. B. & Austin, C. M. MitoPhAST, a new automated mtogenomic phylogeny tool in the post-genomic era with a case study of 89 decapod mitogenomes including eight new freshwater crayfish mitogenomes. Mol. Phylogenet. Evol. 85, 180–188 (2015).
Aljanabi, S. M. & Martinez, I. Universal and rapid salt-extraction of high quality genomic DNA for PCR-based techniques. Nucleic Acids Res. 22, 4692–4693 (1997).
Dierckxsens, N., Mardulyn, P. & Smits, G. NOVOPlasty: de novo assembly of organelle genomes from whole genome data. Nucl. Acids Res. 45(4), e18 (2017).
Bernt, M. et al. MITOS: improved de novo metazoan mitochondrial genome annotation. Mol. Phylogenet. Evol. 69(2), 313–319 (2013).
Altschul, S. F. et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucl. Acids Res. 25(17), 3389–3402 (1997).
Grant, J. R. & Stothard, P. The CGView server: a comparative genomics tool for circular genomes. Nucl. Acids Res. 36, 181–184 (2008).
**a, X. DAMBE5: comprehensive software package for data analysis in molecular biology and evolution. Mol. Biol. Evol. 2013(30), 1720–1728 (2013).
Kumar, S., Stecher, G. & Tamura, K. MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 33(7), 1870–1874 (2016).
Perna, N. T. & Kocher, T. D. Patterns of nucleotide composition at fourfold degenerate sites of animal mitochondrial genomes. J. Mol. Evol. 41(3), 353–358 (1995).
Nguyen, L. T., Schmidt, H. A., Haeseler, A. & Minh, B. Q. IQTREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 32(1), 268–274 (2015).
Ronquist, F. et al. MrBayes 3.2: efcient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 61(3), 539–542 (2012).
Ma, X. M. Study on complete mitochondrial genome of Cypridopsis vidua and molecular phylogeny of ostracoda. PhD thesis, Shanghai, China: East China Normal University. (2016).
Huelsenbeck, J. P. & Ronquist, F. MRBAYES: Bayesian inference of phylogenetic trees. Bioinformatics 17(8), 754–755 (2001).
Nylander, J. A., Ronquist, F., Huelsenbeck, J. P. & Nieves, J. L. Bayesian phylogenetic analysis of combined data. Syst. Biol. 53(1), 47–67 (2004).
Posada, D. & Crandall, K. A. Modeltest: testing the model of DNA substitution. Bioinformatics 14(9), 817–818 (1998).
Rambaut, A. Fig Tree, version 1.4.3, http://tree.bio.ed.ac.uk/softw are/fgtree/accessed 1 July. (2016).
Funding
This work was financially supported by the Fundamental Research Funds for Zhejiang Provincial Universities and Research Institutes (No. 2021J005), the Foundation of Guangdong Provincial Key Laboratory of Marine Biotechnology (No. GPKLMB202103) and the Project of Bureau of Science and Technology of Zhoushan (No. 2020C21026 and No. 2021C21017).
Author information
Contributions
Conceptualization, J.L. and Y.Y., methodology, J.L. and L.X., software, J.L. and L.X., formal analysis, Y.M. and X.L., writing—original draft preparation, J.L. and L.X., writing—review and editing, J.L. and Y.Y., supervision, B.G., funding acquisition, J.L. and Y.Y. All authors have read and agreed to the published version of the manuscript.
