
Abstract
There have been many reports on the genetic mechanism in rhesus macaques (RMs) for environmental adaptation to high altitudes, but the synergistic involvement of gut microbiota in this adaptation remains unclear. Here we performed fecal metagenomic and metabolomic studies on samples from high- and low-altitude populations to assess the synergistic role of gut microbiota in the adaptation of RMs to high-altitude environments. Microbiota taxonomic annotation yielded 7471 microbiota species. There were 37 bacterial species whose abundance was significantly enriched in the high-altitude populations, 16 of which were previously reported to be related to the host’s dietary digestion and energy metabolism. Further functional gene enrichment found a stronger potential for gut microbiota to synthesize energy substrate acetyl-CoA using CO2 and energy substrate pyruvate using oxaloacetate, as well as a stronger potential to transform acetyl-CoA to energy substrate acetate in high-altitude populations. Interestingly, there were no apparent differences between low-altitude and high-altitude populations in terms of genes enriched in the main pathways by which the microbiota consumed the three energy substrates, and none of the three energy substrates were detected in the fecal metabolites. These results strongly suggest that gut microbiota plays an important energy compensatory role that helps RMs to adapt to high-altitude environments. Further functional enrichment after metabolite source analysis indicated the abundance of metabolites related to the degradation of exogenous toxins was also significantly higher in high-altitude populations, which suggested a contributory role of gut microbiota to the degradation of exogenous toxins in wild RMs adapted to high-altitude environments.
Similar content being viewed by others

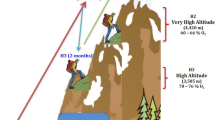

Introduction
The extreme cold, high ultraviolet radiation, low oxygen content, and poor food resources of high-altitude environments pose great challenges to animal survival1,2. The gut microbiota plays an important role in a variety of physiological activities in animals, such as cardiovascular activity, food digestion, energy metabolism, nutrient homeostasis, immune regulation, and maintenance of body temperature3,4,5,6,7,18,19. Among them, the production of SCFAs is related to diet, microbiota composition, and the host19. The macronutrient composition of the diet determines the amount and source of fermentable substrates for the gut microbiota and is a major driver of gut microbiota structure and function19,20. The SCFAs produced by gut microbiota when fermenting host foods are mainly acetate, propionate, and butyrate, which mediate energy metabolism and physiological regulatory processes in the host21,22. There are also a few branched-chain fatty acids (BCFAs), that although in low abundance, can also mediate metabolic processes in the host23,24,25. The gut microbiota of high-altitude ruminants helps the host adapt to the high-altitude environment by reducing the emission of methane and elevating the production of volatile fatty acids11.
Nonhuman primates (NHPs) are the closest biological relatives of humans, originated from tropical lowlands and are primarily found in warm tropical and subtropical environments, with a few species having radiated into temperate alpine forests26,27. The structure and composition of gut microbiota in NHPs are closely related to factors, such as dietary composition28,29, habitat occupancy30, social interactions30, eco-environmental factors31, and health status32,33. The mechanisms of high-altitude adaptation in NHPs, including physiological, behavioral, and genetic, have been widely studied34,35,36. However, less is known about the role of gut microbiota in high-altitude adaptation.
The gut microbiota of NHPs responds to changes in season and differences in habitat environment9,28. The plasticity of the gut microbiota in Ethiopian geladas (Theropithecus gelada) largely provides dietary and metabolic flexibility to the host and may be a key factor in allowing them to thrive in a changing environment37. There have been many reports on the interaction between gut microbiota composition and environmental adaptation in nonhuman primates based on the 16S rRNA genes of the microbiota; however, studies on the interaction between the functional gene composition of gut microbiota and metabolite composition in the environmental adaptation of NHPs are largely lacking.
Rhesus macaques (RMs) are the most widely distributed small omnivorous NHPs and are currently a widely used biological model in medical and biological research given their high physiological similarity to humans14. In China, wild RMs are widely distributed from sea level to the Qinghai Tibetan Plateau (altitude exceeding 3000 m). Given its strong adaptability, the RM is an ideal model for investigating the adaptation mechanisms involving primate gut microbiota in the host to high-altitude environments38. Previous studies have also found significant differences in the composition of the gut microbiota between RMs from different habitats, with the highest diversity and the largest number of specific microbiota in high-altitude populations9,14. Related studies are mainly based on 16S rRNA gene sequencing and the characterization of gut microbiota composition in RMs at the genus or family level. Less is known about the functional and metabolite composition of gut microbiota, or the interactive mechanisms between gut microbiota and host adaptation to high-altitude environments. Therefore, we collected fresh fecal samples in March 2021 from four wild RM populations on the southeast edge of the Qinghai Tibetan Plateau, from Pamlin (HA population) and ** the host improve the metabolic capacity of food19,20,21,22,23,24. The pyruvate, propionate, butyrate, starch, sucrose, and pentose phosphate pathways are important for the fermentation of unabsorbed carbohydrates into SCFAs25. Meanwhile, butyrate is preferentially used as an energy source for the intestinal mucosa, propionate contributes to gluconeogenesis in the liver, and acetate has the highest concentration in blood61. Acetate production pathways are widely distributed in anaerobic bacteria15, while the production pathways of propionate, butyrate, and lactic acid are more conservative and substrate-specific62. Among the SCFAs produced by gut microbiota, butyrate is the most preferred source of energy in this respect, but its synthesis depends mainly on carbohydrates in the intestine22,63. In human gut microbiota, a variety of fermentation strategies have been developed to further generate energy. Pyruvate can be decomposed and metabolized into succinate, lactic acid, or acetyl-CoA. These intermediates can be further metabolized by hosts to produce acetate, propionate, and butyrate at the same time23,64. In this study, we found that high-altitude populations, acting through the m00422, m00631, and m00003 modules, can produce the energy substrates acetyl-CoA and pyruvate, mediate the energy metabolism of the host and provide energy compensation to the host. Meanwhile, the gut microbiota of RMs had a significantly higher potential to synthesize acetate at high altitudes, and a significantly higher potential to synthesize butyrate at low altitudes. However, the gene enrichment of the microbiota itself for these SCFA breakdown-related modules did not differ significantly between high- and low-altitude populations. Further, metabolite analysis did not detect these energy substrates, and the detected SCFAs did not differ significantly between the high- and low-altitude populations, indicating that these SCFAs and energy substrates are more likely to be absorbed and utilized by the host. These results are compatible with food acquisition and environmental stress in populations of wild RMs from different altitudes. Low-altitude populations are more likely to obtain foods with a high carbohydrate content and have more residual carbohydrates in their intestines than high-altitude populations; therefore, the potential to catalyze the formation of butyrate is higher as a result of the richer carbohydrate in the food of low-altitude populations. The gut microbiota at high altitudes shows high potential for acetate production. Meanwhile, pyruvate and acetyl-CoA are produced through the gut microbiota own metabolic pathways, which provide energy substrates for the host’s tricarboxylic acid cycle and fatty acid metabolic pathways. This also corresponds to the fact that the abundance of the microbiota associated with acetate production was significantly higher in the high-altitude populations. A recent study in other mammals also found that elevated acetate levels induced by altered microbiota drive hosts to seek food and energy accumulation by activating the parasympathetic nervous system to promote the secretion of insulin and gastric hunger hormones65. Another study in obese mice also found that oral butyrate reduced body weight largely due to increased energy expenditure and lipid oxidation66. This also appears to account for the higher potential for gut microbiota in high-altitude populations to directly provide energy compensation to the host through acetate synthesis and by stimulating food intake, further promoting energy accumulation. Moreover, the gut microbiota of high-altitude populations had a significantly higher abundance of genes associated with the metabolism of methanol than that of low-altitude populations. Methanol, as a by-product of microbiota metabolism, can serve as a substrate for microbiota methanogenesis and acetate64,67. Our results shows that the potential of microbiota in high-altitude populations to use methanol decomposition to produce energy is also higher than that in low-altitude populations, and their utilization of intestinal metabolites is more refined than that in low-altitude populations.
Wildlife gut microbiota is commonly exposed to environmental contaminants and is involved in the process of degrading environmental contaminants68. The gastrointestinal tract is the main route for xenobiotics to enter the body, and the gut microbiota has a high metabolic potential for xenobiotics. Gut microbiota can directly metabolize xenobiotic compounds of exogenous origin or affect the absorption, distribution, metabolism, and elimination of xenobiotics in the host, thus changing the toxicity of xenobiotics to the host68,69,70. PAHs are a broad class of organic pollutants present in the environment and toxic to humans and animals71. Soils act as a reservoir of PAHs mainly derived from atmospheric deposition and plant to microbiota synthesis72,73. Toluene is a common volatile organic compound (VOC) that is toxic to animals. Dead plant litter produces toluene, which is then transported to the soil74,75,76. In this study, we performed functional enrichment analysis of differential metabolites derived from microbiota production and found that among 4 statistically significant metabolic pathways, metabolites associated with cyclic aromatic hydrocarbon degradation and toluene degradation were significantly enriched in high-altitude populations compared with low-altitude populations. Based on biological and statistical analysis, we found that multiple responses of various bacterial communities (such as Streptomyces vietnamensis, Tsukamurella paulometabola, Nocardia brasiliensis, Candidatus Formimonas warabiya, and Rhizobium leguminosarum) in these two pathways play an important role,, in promoting the degradation of these exogenous toxins through R01631, R01635, and R05375 reactions in high-altitude populations. Meanwhile, our metagenomic screening based on KO genes in microbiota also revealed significantly higher tphB (K18076) gene richness in the polycyclic aromatic hydrocarbon degradation pathway (terephthalate degradation M00624) module that prompted the R01633 reaction to proceed. These results indicate that the degradation of PAHs, occurs at a significantly higher rate in high-altitude populations than in low-altitude populations. This means that high-altitude RMs can eat many plant roots and stems in cold seasons when there is a lack of food. As we observed during the long-term monitoring of the HB population60 and sample collection, high-altitude populations will dig and eat many plant roots when food resources are scarce in winter, and PAHs and toluene are widely enriched in soil and plant roots. The degradation of these exogenous toxicants by gut microbiota in high-altitude populations is important. In addition, our gut microbe-based functional enrichment analysis also revealed that high-altitude populations were significantly enriched in genes involved in the conversion of L-serine to L-cysteine in the cysteine biosynthesis (M00021) module, while related studies found that L-cysteine, an amino acid detoxification drug, is involved in cellular reduction processes and phospholipid metabolism in the liver, protects hepatocytes from damage, and promotes liver function recovery pharmacological effects77, implying an important role for their gut microbiota in the detoxification of the host’s own products via metabolites. These results suggest that as part of RMs adaptation to high-altitude environments, the gut microbiota contributes to hosts degradation of exogenous toxicants.
In conclusion, this study revealed the synergistic involvement of the gut microbiota in the adaptation of wild RMs to high-altitude environments (Fig. 7). Wild RMs adapted to high energy demands and high-quality food deprivation in a high-altitude environment had a diverse gut microbiota. And the abundance of multiple species of bacteria associated with acetate synthesis and those associated with fiber degradation was significantly higher than in low-altitude populations. We found a stronger potential for gut microbiota to synthesize the energy substrate acetyl-CoA using CO2 and the energy substrate pyruvate using oxaloacetate, as well as a stronger potential to transform acetyl-CoA to the energy substrate acetate in high-altitude populations. These energy substrates provide the host with enhanced energy compensation at high altitudes. Acetate production by gut microbiota promotes host insulin and gastric ghrelin secretion, driving food behaviors seeking and further energy accumulation. Meanwhile, in high-altitude populations, the gut microbiota can effectively help the host degrade PAHs consumed when feeding on complex foods such as plant rhizomes. In addition, we revealed biological and statistical links between multiple distinct gut microbiota and metabolites, providing fundamental data for understanding the ways gut microbiota affect the host.

cooF anaerobic carbon-monoxide dehydrogenase iron sulfur subunit, cdhD acetyl-CoA decarbonylase/synthase, CODH/ACS complex subunit delta, cdhE acetyl-CoA decarbonylase/synthase, eda 2-dehydro-3-deoxyphosphogluconate aldolase, kdgK 2-dehydro-3-deoxygluconokinase, E4.1.1.32 phosphoenolpyruvate carboxykinase, E4.1.1.49 phosphoenolpyruvate carboxykinase, ppdK pyruvate, orthophosphate dikinase, korA 2-oxoglutarate/2-oxoacid ferredoxin oxidoreductase subunit alpha, ACSS1_2 acetyl-CoA synthetase, EC:2.3.1.8 putative phosphotransacetylase. Red indicates that relative abundance was significantly higher in high-altitude populations. Green indicates that abundance was significantly higher in low-altitude populations. These macaque photos were taken by ourselves. These images and every element of these images were created by us.
Methods
Fecal sample collection
Fecal samples from RMs were collected from the wild natural habitat in March 2021. The samples were collected from Pamulin (HA; 101.182061°E, 30.101192°N) and **’elou Town (HB; 100.715649°E, 29.936961°N) in Yajiang County, Ganzi Prefecture, Sichuan Province at an altitude of more than 3000 m and from Baidicheng (LA; 109.577491°E, 31.040904°N) and Simianshan (LB; 106.405604°E, 28.644834°N) of the Chongqing municipality at an altitude of less than 1000 m (Fig. 8a). Yajiang County is located on the southeast edge of the Qinghai Tibet Plateau, with high-altitude climate characteristics of low temperature, low oxygen, and high ultraviolet radiation. The vegetation in the RMs habitat is mainly alpine coniferous forest, alpine hardwood forest, and alpine meadow, so it is difficult for RMs to obtain food. Simianshan and Baidicheng in Chongqing are located on the southeast edge of the Sichuan Basin, a subtropical humid monsoon climate area with a mild climate and high oxygen content. The vegetation in this habitat is mainly broad-leaved evergreens and broad-leaved deciduous trees, and it is less difficult for RMs to obtain food. Comparison of the monthly average temperature and precipitation data for the 4 sampled sites based on ArcGIS 10.0 and WorldClim’s (version 2.1)78 published monthly climate data for 1970–2000 (resolution 30 s) revealed that the climate of the habitats of the two high-altitude populations (Yajiang Pamulin Temple and **’elou Town) was similar, with the lowest average temperature in January (−6.1 °C/−4.9 °C) and the highest average in July (−10.7 °C/−11.7 °C), and precipitation mainly falling in 6–9 months, with the highest monthly precipitation of 150 mm/152 mm showing distinct high-altitude climatic features. Although separated by a wide geographic distance, the habitats of the two low-altitude populations (Baidicheng and Simianshan, Chongqing) also have similar climates, with the lowest average temperature in January (5.5 °C/3.8 °C), the highest average temperature in July/August (27.6 °C/21.2 °C), the highest monthly precipitation of 212 mm/174 mm, and significantly higher temperature and humidity than the two high-altitude population habitats (Fig. 8b). According to the observations made during sampling, the total number of RM groups sampled at the four sites ranged from 40 to 50, and all sampled individuals were considered adult individuals based on follow-up observations and comparison of stool size. After defecation by the wild RMs, fecal samples were immediately collected with sterile gloves then stored in liquid nitrogen and transported to the laboratory. The samples for DNA preparation and metabolome detection for metagenomic sequencing were taken from the center of the fecal samples under sterile conditions, while samples for DNA preparation for individual identification and gender identity were taken from the surface. After collection, the samples were transported in liquid nitrogen and stored at -80 °C until analysis. There was no direct contact with RM during fecal collection, and our study had no impact on the health or welfare of RMs.

a Distribution of sample collection sites. b Monthly average temperature and precipitation data for 1970–2000 at the sampling site obtained based on WorldClim (version 2.1). The boundary data comes from the data published by China National Basic Geographic Information Center. Triangles represent sample points.
Samples collected from the four locations were analyzed for microsatellite molecular markers for individual identification and for SRY/ZFX genes for sex determination. The partial SRY gene (364 bp) on the Y chromosome and the partial ZFX gene (183 bp) on the X chromosome were used for sex determination. The primers were designed by Primer Premier 5 based on sequences in the GenBank database (accession numbers AF284311 and XM_028842412). The six microsatellite loci used for individual identification were amplified using the primers designed in our previous study for nested PCR. The primer pairs (Supplementary Table 4) under their respective annealing temperatures were used with 1.0 μl of template DNA or the product of the first round of PCR in a 20 μl PCR mixture (95 °C for 10 min initial denaturation; 35 cycles of denaturation at 94 °C for 30 s, 45 s annealing, and 30 s extension at 72 °C; followed by final extension for 10 min at 72 °C). All PCR products were examined by electrophoresis in 1.5% agarose gels containing ethidium bromide. Capillary electrophoresis analysis was used for PCR products of microsatellite loci. Sex determination was performed with five independent experiments per sample. Ten individual samples were screened per population by a male to female ratio, and a total of 40 fresh fecal samples were subjected to subsequent metagenomic and metabolomic testing (Fig. 7, Supplementary Table 5).
Metagenomic sequencing and quality control of raw data
DNA samples with a total content >25 ng, dispersion concentration above 500 bp, and no serious agglomeration below 500 bp were used for library preparation. The qPCR concentration of the final library was greater than 1.5 nm. There was no splice sequence, heteropeaks, or small fragments, and the library had a fragment size of 350 bp. After we completed Dnbseq-T7 platform (MGI Tech Co., Ltd. China) sequencing (Novogene Co., Ltd. China), adapters and low-quality reads in raw data were removed by Trimmomatic79, and potential RM sequences were removed by Bowtie280 based on the RM reference genome (assembly Mmul_10; GCA_003339765.3).
Assembly, taxonomic annotation, functional prediction, and quantification of genes
After quality control of the data obtained and the removal of host genes, mate pair assembly was performed using MEGAHIT (1.2.9)81, and the quality of the assembly results was assessed using QUAST (5.0.2)82. Gene prediction was done using Prodigal (2.6.3)83 with the parameter ‘-p meta’. The gene model of all genes predicted by CD-hit (4.8.1)84 was used to construct a non-redundant gene catalog parameter of ‘- as 0.9 - c 0.95 - G 0 - g 0 - T 0 - M 0’. SALMON (1.3.0)85 was used to quantify the non-redundant genes. Functional assignments of the protein sequences was done based on DIAMOND alignment against the eggNOG 5.0 database using eggNOG Mapper (2.0.0)86 by taking the best hit with the criterion of an E value <1e−3. The functional description of the portal gene responding to the searched sequence was the final annotation result to calculate the difference in gene composition; the next difference analysis was performed using normalized counts (TPM values). STAMP (2.1.3)87 was used to analyze the differences in gene composition between the low-altitude and high-altitude populations. After false discovery rate (FDR) corrections, P < 0.05 was used as the threshold for significance, revealing the differential genes of the gut microbiota between high- and low-altitude populations of RMs. The 8 GB MiniKraken database, pre-built using the abundance estimates of Kraken2 (2.1.1)88, was used to test the classification analysis based on non-redundant genes. LEfSe analysis was completed using LEfSe (1.1.2). Taxonomic composition, alpha diversity, beta diversity analysis, and visualization of all results were done using R (4.1.2). In order to investigate pathway modules known in microorganisms, we collected KEGG modules listed under “Microbial metabolism in diverse environments” (map01120)89.
Metabolite profiling from stool samples
Faecal samples (100 mg) were individually ground with liquid nitrogen, and the homogenate was resuspended with prechilled 80% methanol and 0.1% formic acid using vortexing. The samples were incubated on ice for 5 min then centrifuged at 15,000 g and 4 °C for 20 min. Some of the supernatants were diluted with UPLC-MS/MS grade water to a final concentration of 53% methanol. The samples were subsequently transferred to a fresh Eppendorf tube then centrifuged at 15,000 g and 4 °C for 20 min. Finally, the supernatant was injected into the UPLC-MS/MS system90.
UHPLC-MS analyses were performed using a Vanquish UHPLC system (Thermo Fisher, Germany) coupled with an Orbitrap Q ExactiveTM HF mass spectrometer (Thermo Fisher, Germany) at Novogene Co., Ltd. (Bei**g, China). Samples were injected onto a Hypersil gold column (100 ×2.1 mm, 1.9 μm) using a 17-min linear gradient at a flow rate of 0.2 mL/min. The eluents for the positive polarity mode were eluent A (0.1% FA in water) and eluent B (methanol). The eluents for the negative polarity mode were eluent A (5 mM ammonium acetate, pH 9.0) and eluent B (methanol). The solvent gradient was set as follows: 2% B, 1.5 min; 2%–100% B, 12.0 min; 100% B, 14.0 min; 100%–2% B, 14.1 min; and 2% B, 17 min. The Q Exactive TM HF mass spectrometer was operated in positive/negative polarity mode with a spray voltage of 3.2 kV, a capillary temperature of 320 °C, a sheath gas flow rate of 40 arb, and an aux gas flow rate of 10 arbs.
The raw data files generated by UHPLC-MS/MS were processed using Compound Discoverer 3.1 (CD3.1, Thermo Fisher) to perform peak alignment, peak picking, and quantitation for each metabolite. The main parameters were set as follows: retention time tolerance, 0.2 min; actual mass tolerance, 5 ppm; signal intensity tolerance, 30%; signal/noise ratio, 3; and minimum intensity. After that, peak intensities were normalized to the total spectral intensity. The normalized data was used to predict the molecular formula based on additive ions, molecular ion peaks, and fragment ions, then peaks were matched with the mzCloud (https://www.mzcloud.org/), mzVault, and MassList databases to obtain accurate qualitative and relative quantitative results. Statistical analyses were performed using the statistical software R (R version 3.4.3), Python (Python version 2.7.6), and CentOS (CentOS release 6.6). When the data were not normally distributed, normal transformations were attempted using the area normalization method.
Metabolite statistical analysis
Data on the fecal metabolome were processed using the Novogene platform. The metabolites were annotated using the KEGG (https://www.genome.jp/kegg/pathway.html) and HMDB (https://hmdb.ca/metabolites) databases. Principal components analysis (PCA) and PLS-DA were performed using metaX91 (a flexible and comprehensive software for processing metabolomics data). We applied univariate analysis (t-test) to calculate the statistical significance (P-value). The metabolites with VIP > 1, a P-value <0.05, and a fold change (FC) > 1.5 or <0.7 were considered differential metabolites. Volcano plots were used to filter metabolites of interest, and were based on the log2 (FC) and -log10(P-value) of metabolites by ‘ggplot2’ in R. For clustering heat maps, the data were normalized using z-scores of the intensity areas of differential metabolites and were plotted using the ‘pheatmap’ package in R. The correlation between differential metabolites was calculated using the function ‘cor’ in R (method = Pearson). The statistical significance of correlations between differential metabolites was calculated using ‘cor.mtest’ in R. A P-value greater than 0.05 was considered as statistically significant, and correlation plots were plotted using the ‘corrplot’ package in R. The functions of these metabolites and metabolic pathways were studied using the KEGG database. Metabolic pathway enrichment was analyzed: when the ratio was x/n > y/N, the metabolic pathway was considered as enriched, and when the P-value of the metabolic pathway was <0.05, the metabolic pathway was considered as significantly enriched.
Metabolite traceability and association analysis with microbiota
The bioinformatics analysis process on the MetOrigin platform92 was used to trace the source of metabolites by searching and integrating the databases of seven metabolites (KEGG, HMDB, BIGG, ChEBI, FoodDB, drug database, and toxin and toxin target database [T3DB]). Further, functional enrichment analysis was performed according to the different sources of metabolites92. The reference metabolic pathway of the host was from the KEGG database, and the reference metabolic pathway of the microbiota community was from more than 6,800 microbiotas in the integrated database of the MetOrigin platform. The reference metabolic pathway shared by the host and the microorganism was obtained by integrating these two pathways. Combined with the differential microbiota data, the statistical links between the differential fecal metabolites and differential microbiota were first revealed by Spearman analysis, then the KEGG database was utilized to search for microbiotas that might be involved in the associated metabolic responses and to perform associations with metabolites. Sankey network analysis was further utilized to visually demonstrate the biological and statistical links between gut microbiota and fecal metabolites92. Finally, network summary analysis was performed to further reveal the statistical and biological significance of correlations between microbiota and metabolites in specific functional pathways92. We mapped the KO genes and metabolites with significant differences in the feces of RMs at high and low altitudes together in the “microbial metabolism in diverse environments” pathway using the KEGG Mapper–Color tool (https://www.genome.jp/kegg/mapper/color.html) to reveal the residues of metabolites in feces produced by the main modules of the functioning of RMs gut microbiota.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Code availability
Code and detailed information are available on github (https://github.com/junsongzhao-junsongzhao/Metagenome-metabolome-gut-microbiota-in-high-altitude-rhesus-macaque-Macaca-mulatta).
References
Ma, Y. et al. Gut microbiota adaptation to high altitude in indigenous animals. Biochem. Biophys. Res. Commun. 516, 120–126 (2019).
Guo, N. et al. Seasonal dynamics of diet-gut microbiota interaction in adaptation of yaks to life at high altitude. NPJ Biofilms Microbiomes 7, 38 (2021).
Simonson, T. S. et al. Genetic evidence for high-altitude adaptation in Tibet. Science 329, 72–75 (2010).
Zhu, H., Zhong, L., Li, J., Wang, S. & Qu, J. Differential expression of metabolism-related genes in Plateau Pika (Ochotona curzoniae) at different altitudes on the Qinghai-Tibet Plateau. Front. Genet. 12, 784811 (2021).
Yu, L. et al. Genomic analysis of snub-nosed monkeys (Rhinopithecus) identifies genes and processes related to high-altitude adaptation. Nat. Genet. 48, 947–952 (2016).
Bäckhed, F., Ley, R. E., Sonnenburg, J. L., Peterson, D. A. & Gordon, J. I. Host-bacterial mutualism in the human intestine. Science 307, 1915–1920 (2005).
Ley, R. E., Peterson, D. A. & Gordon, J. I. Ecological and evolutionary forces sha** microbial diversity in the human intestine. Cell 124, 837–848 (2006).
Yan, M. A., **n, X., Jiakai, F. A. N. & Benyin, Z. Effect of altitude on the diversity of gut microbiota of yaks grazing on the Qinghai-Tibet Plateau. Microbiol. China 49, 620–634 (2022).
Zhao, J. et al. Characterization of the gut microbiota in six geographical populations of Chinese Rhesus Macaques (Macaca mulatta), implying an adaptation to high-altitude environment. Microb. Ecol. 76, 565–577 (2018).
Li, L. & Zhao, X. Comparative analyses of fecal microbiota in Tibetan and Chinese Han living at low or high altitude by barcoded 454 pyrosequencing. Sci. Rep. 5, 14682 (2015).
Zhang, Z. et al. Convergent evolution of rumen microbiomes in high-altitude mammals. Curr. Biol. CB 26, 1873–1879 (2016).
Liu, Y.-X., Chen, T., Li, D., Fu, J. & Liu, S.-J. Integrated meta-omics for biology and environments. iMeta 1, e15 (2022).
Li, H., Zhou, R., Zhu, J., Huang, X. & Qu, J. Environmental filtering increases with elevation for the assembly of gut microbiota in wild pikas. Microb. Biotechnol. 12, 976–992 (2019).
Wu, Y. et al. Characterisation of the gut microbial community of rhesus macaques in high-altitude environments. BMC Microbiol. 20, 68 (2020).
Thursby, E. & Juge, N. Introduction to the human gut microbiota. Biochemical J. 474, 1823–1836 (2017).
Louis, P., Hold, G. L. & Flint, H. J. The gut microbiota, bacterial metabolites and colorectal cancer. Nat. Rev. Microbiol. 12, 661–672 (2014).
Arnoriaga-Rodríguez, M. et al. Gut bacterial ClpB-like gene function is associated with decreased body weight and a characteristic microbiota profile. Microbiome 8, 59 (2020).
Heilbronner, S., Krismer, B., Brötz-Oesterhelt, H. & Peschel, A. The microbiome-sha** roles of bacteriocins. Nat. Rev. Microbiol. 19, 726–739 (2021).
Krautkramer, K. A., Fan, J. & Bäckhed, F. Gut microbial metabolites as multi-kingdom intermediates. Nat. Rev. Microbiol. 19, 77–94 (2021).
Boets, E. et al. Systemic availability and metabolism of colonic-derived short-chain fatty acids in healthy subjects: a stable isotope study. J. Physiol. 595, 541–555 (2017).
Cummings, J. H., Pomare, E. W., Branch, W. J., Naylor, C. P. & Macfarlane, G. T. Short chain fatty acids in human large intestine, portal, hepatic and venous blood. Gut 28, 1221–1227 (1987).
den Besten, G. et al. The role of short-chain fatty acids in the interplay between diet, gut microbiota, and host energy metabolism. J. lipid Res. 54, 2325–2340 (2013).
Koh, A., De Vadder, F., Kovatcheva-Datchary, P. & Bäckhed, F. From dietary fiber to host physiology: short-chain fatty acids as key bacterial metabolites. Cell 165, 1332–1345 (2016).
Smith, E. A. & Macfarlane, G. T. Dissimilatory amino Acid metabolism in human colonic bacteria. Anaerobe 3, 327–337 (1997).
Macfarlane, G. T., Gibson, G. R., Beatty, E. & Cummings, J. H. Estimation of short-chain fatty acid production from protein by human intestinal bacteria based on branched-chain fatty acid measurements. FEMS Microbiol. Lett. 101, 81–88 (1992).
Estrada, A. et al. Impending extinction crisis of the world’s primates: Why primates matter. Sci. Adv. 3, e1600946 (2017).
Hanya, G. et al. Dietary adaptations of temperate primates: comparisons of Japanese and Barbary macaques. Primates J. Primatol. 52, 187–198 (2011).
Gomez, A. et al. Temporal variation selects for diet-microbe co-metabolic traits in the gut of Gorilla spp. ISME J. 10, 514–526 (2016).
Bueno de Mesquita, C. P. et al. Structure of Chimpanzee Gut Microbiomes across Tropical Africa. mSystems 6, e0126920 (2021).
Umanets, A. et al. Occupancy strongly influences faecal microbial composition of wild lemurs. FEMS Microbiol. Ecol. 94, 1–13 (2018).
Sharma, A. K. et al. The primate gut mycobiome-bacteriome interface is impacted by environmental and subsistence factors. NPJ Biofilms Microbiomes 8, 12 (2022).
Yang, S. et al. The gut microbiome and antibiotic resistome of chronic diarrhea rhesus macaques (Macaca mulatta) and its similarity to the human gut microbiome. Microbiome 10, 29 (2022).
Zheng, P. et al. The gut microbiome modulates gut-brain axis glycerophospholipid metabolism in a region-specific manner in a nonhuman primate model of depression. Mol. Psychiatry 26, 2380–2392 (2021).
Yamada, A. & Muroyama, Y. Effects of vegetation type on habitat use by crop-raiding Japanese macaques during a food-scarce season. Primates J. Primatol. 51, 159–166 (2010).
Ley, R. E. et al. Evolution of mammals and their gut microbes. Science 320, 1647–1651 (2008).
Mortola, J. P. & Wilfong, D. Hematocrit of mammals (Artiodactyla, Carnivora, Primates) at 1500m and 2100m altitudes. Zoology 125, 10–23 (2017).
Baniel, A. et al. Seasonal shifts in the gut microbiome indicate plastic responses to diet in wild geladas. Microbiome 9, 26 (2021).
Jiang, X. L. & Wang, Y. X. Ma SLJZR. Taxonomic revision and distribution of subspecies of rhesus monkey (Macaca mulatta) in China. Zool. Res. 12, 241–247 (1991).
Lian, X., Zhang, T., Cao, Y., Su, J. & Thirgood, S. Group size effects on foraging and vigilance in migratory Tibetan antelope. Behav Process. 76, 192–197 (2007).
Liu, X. et al. EPAS1 gain-of-function mutation contributes to high-altitude adaptation in Tibetan horses. Mol. Biol. Evol. 36, 2591–2603 (2019).
Ma, Y. F. et al. Population genomics analysis revealed origin and high-altitude adaptation of Tibetan pigs. Sci. Rep. 9, 11463 (2019).
Moon, J. Y., Choi, M. H. & Kim, J. Metabolic profiling of cholesterol and sex steroid hormones to monitor urological diseases. Endocr. Relat. Cancer 23, R455–R467 (2016).
Weigel, N. L. & Moore, N. L. Kinases and protein phosphorylation as regulators of steroid hormone action. Nucl. Receptor Signal. 5, e005 (2007).
Zhang, Q. et al. Genome resequencing identifies unique adaptations of Tibetan chickens to hypoxia and high-dose ultraviolet radiation in high-altitude environments. Genome Biol. Evol. 8, 765–776 (2016).
Domankevich, V., Eddini, H., Odeh, A. & Shams, I. Resistance to DNA damage and enhanced DNA repair capacity in the hypoxia-tolerant blind mole rat Spalax carmeli. J. Exp. Biol. 221, jeb174540 (2018).
Tap, J. et al. Gut microbiota richness promotes its stability upon increased dietary fibre intake in healthy adults. Environ. Microbiol. 17, 4954–4964 (2015).
Belzer, C. & de Vos, W. M. Microbes inside–from diversity to function: the case of Akkermansia. ISME J. 6, 1449–1458 (2012).
Holland, S. I., Ertan, H., Montgomery, K., Manefield, M. J. & Lee, M. Novel dichloromethane-fermenting bacteria in the Peptococcaceae family. ISME J. 15, 1709–1721 (2021).
Li, Z. et al. The complete genome sequence of Ethanoligenens harbinense reveals the metabolic pathway of acetate-ethanol fermentation: a novel understanding of the principles of anaerobic biotechnology. Environ. Int. 131, 105053 (2019).
Shkoporov, A. N. et al. Ruthenibacterium lactatiformans gen. nov., sp. nov., an anaerobic, lactate-producing member of the family Ruminococcaceae isolated from human faeces. Int. J. Syst. Evolut. Microbiol. 66, 3041–3049 (2016).
Tao, Y. et al. Complete genome sequence of Ruminococcaceae bacterium CPB6: a newly isolated culture for efficient n-caproic acid production from lactate. J. Biotechnol. 259, 91–94 (2017).
Endo, A., Tanno, H., Kadowaki, R., Fujii, T. & Tochio, T. Extracellular fructooligosaccharide degradation in Anaerostipes hadrus for co-metabolism with non-fructooligosaccharide utilizers. Biochem Biophys Res. Commun. 613, 81–86 (2022).
Yoshikawa, S. et al. Valerate production by Megasphaera elsdenii isolated from pig feces. J. Biosci. Bioeng. 125, 519–524 (2018).
Yang, M. et al. Synergistic cellulose hydrolysis dominated by a multi-modular processive endoglucanase from Clostridium cellulosi. Front. Microbiol. 7, 932 (2016).
Li, L. L., Taghavi, S., Izquierdo, J. A. & van der Lelie, D. Complete genome sequence of Clostridium sp. strain BNL1100, a cellulolytic mesophile isolated from corn stover. J. Bacteriol. 194, 6982–6983 (2012).
Wegmann, U. et al. Complete genome of a new Firmicutes species belonging to the dominant human colonic microbiota (‘Ruminococcus bicirculans’) reveals two chromosomes and a selective capacity to utilize plant glucans. Environ. Microbiol. 16, 2879–2890 (2014).
Ben David, Y. et al. Ruminococcal cellulosome systems from rumen to human. Environ. Microbiol. 17, 3407–3426 (2015).
Wang, N., Yan, Z., Liu, N., Zhang, X. & Xu, C. Synergy of cellulase systems between Acetivibrio thermocellus and Thermoclostridium stercorarium in Consolidated-Bioprocessing for Cellulosic Ethanol. Microorganisms 10, 502 (2022).
Tomazetto, G. et al. Complete genome analysis of Clostridium bornimense strain M2/40(T): A new acidogenic Clostridium species isolated from a mesophilic two-phase laboratory-scale biogas reactor. J. Biotechnol. 232, 38–49 (2016).
Zhang, K. et al. Diet and feeding behavior of a group of high-altitude rhesus macaques: high adaptation to food shortages and seasonal fluctuations. Curr. Zool. zoac047, 1–11 (2022).
Morrison, D. J. & Preston, T. Formation of short chain fatty acids by the gut microbiota and their impact on human metabolism. Gut Microbes 7, 189–200 (2016).
Louis, P. & Flint, H. J. Formation of propionate and butyrate by the human colonic microbiota. Environ. Microbiol. 19, 29–41 (2017).
Ríos-Covián, D. et al. Intestinal short chain fatty acids and their link with diet and human health. Front. Microbiol. 7, 185 (2016).
Oliphant, K. & Allen-Vercoe, E. Macronutrient metabolism by the human gut microbiome: major fermentation by-products and their impact on host health. Microbiome 7, 91 (2019).
Perry, R. J. et al. Acetate mediates a microbiome-brain-β-cell axis to promote metabolic syndrome. Nature 534, 213–217 (2016).
Gao, Z. et al. Butyrate improves insulin sensitivity and increases energy expenditure in mice. Diabetes 58, 1509–1517 (2009).
Dorokhov, Y. L., Shindyapina, A. V., Sheshukova, E. V. & Komarova, T. V. Metabolic methanol: molecular pathways and physiological roles. Physiol Rev. 95, 603–644 (2015).
Wang, D., Ren, J., Tan, Z. & You, J. Gut microbial profiles in nereis succinea and their contribution to the degradation of organic pollutants. Environ. Sci. Technol. 54, 6235–6243 (2020).
**, Y., Wu, S., Zeng, Z. & Fu, Z. Effects of environmental pollutants on gut microbiota. Environ. Pollut. 222, 1–9 (2017).
Claus, S. P., Guillou, H. & Ellero-Simatos, S. The gut microbiota: a major player in the toxicity of environmental pollutants? NPJ Biofilms Microbiomes 2, 16003 (2016).
Van de Wiele, T. et al. Human colon microbiota transform polycyclic aromatic hydrocarbons to estrogenic metabolites. Environ. Health Perspect. 113, 6–10 (2005).
Suess, M. J. The environmental load and cycle of polycyclic aromatic hydrocarbons. Sci. Total Environ. 6, 239–250 (1976).
Edwards, N. T. Polycyclic Aromatic Hydrocarbons (PAH’s) in the terrestrial environment—a review. J. Environ. Qual. 12, 427–441 (1983).
Wu, T., Zhao, X., Liu, M., Zhao, J. & Wang, X. Wheat straw return can lead to biogenic toluene emissions. J. Environ. Sci. 124, 281–290 (2021).
Isidorov, V. A., Vinogorova, V. T. & Rafałowski, K. HS-SPME analysis of volatile organic compounds of coniferous needle litter. Atmos. Environ. 37, 4645–4650 (2003).
Fedele, R., Galbally, I. E., Porter, N. & Weeks, I. A. Biogenic VOC emissions from fresh leaf mulch and wood chips of Grevillea robusta (Australian Silky Oak). Atmos. Environ. 41, 8736–8746 (2007).
Yin, J. et al. L-Cysteine metabolism and its nutritional implications. Mol. Nutr. Food Res. 60, 134–146 (2016).
Fick, S. E. & Hijmans, R. J. WorldClim 2: new 1-km spatial resolution climate surfaces for global land areas. Int. J. Climatol. 37, 4302–4315 (2017).
Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120 (2014).
Langmead, B. & Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 (2012).
Li, D. et al. MEGAHIT v1.0: a fast and scalable metagenome assembler driven by advanced methodologies and community practices. Methods. 102, 3–11 (2016).
Gurevich, A., Saveliev, V., Vyahhi, N. & Tesler, G. QUAST: quality assessment tool for genome assemblies. Bioinformatics 29, 1072–1075 (2013).
Hyatt, D., LoCascio, P. F., Hauser, L. J. & Uberbacher, E. C. Gene and translation initiation site prediction in metagenomic sequences. Bioinformatics 28, 2223–2230 (2012).
Fu, L., Niu, B., Zhu, Z., Wu, S. & Li, W. CD-HIT: accelerated for clustering the next-generation sequencing data. Bioinformatics 28, 3150–3152 (2012).
Patro, R., Duggal, G., Love, M. I., Irizarry, R. A. & Kingsford, C. Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 14, 417–419 (2017).
Huerta-Cepas, J. et al. eggNOG 5.0: a hierarchical, functionally and phylogenetically annotated orthology resource based on 5090 organisms and 2502 viruses. Nucleic Acids Res. 47, D309–d314 (2019).
Parks, D. H., Tyson, G. W., Hugenholtz, P. & Beiko, R. G. STAMP: statistical analysis of taxonomic and functional profiles. Bioinformatics. 30, 3123–3124 (2014).
Wood, D. E., Lu, J. & Langmead, B. Improved metagenomic analysis with Kraken 2. Genome Biol. 20, 257 (2019).
Yachida, S. et al. Metagenomic and metabolomic analyses reveal distinct stage-specific phenotypes of the gut microbiota in colorectal cancer. Nat. Med. 25, 968–976 (2019).
Want, E. J. et al. Global metabolic profiling of animal and human tissues via UPLC-MS. Nat. Protoc. 8, 17–32 (2013).
Wen, B., Mei, Z., Zeng, C. & Liu, S. metaX: a flexible and comprehensive software for processing metabolomics data. BMC Bioinforma. 18, 183 (2017).
Yu, G., Xu, C., Zhang, D., Ju, F. & Ni, Y. MetOrigin: discriminating the origins of microbial metabolites for integrative analysis of the gut microbiome and metabolome. iMeta 1, e10 (2022).
Guo, X. et al. CNSA: a data repository for archiving omics data. Database 2020, baaa055 (2020).
Chen, F. Z. et al. CNGBdb: China National GeneBank DataBase. Yi Chuan 42, 799–809 (2020).
Acknowledgements
This work was supported by the National Natural Science Foundation of China under Grant (31870355). We would like to extend our sincere gratitude to Mengshi Yu, Kechu Zhang, Fei Zhou, Lin Zhang, **nyu Chen and Huangkai Si for their assistance in sample collection. We would like to acknowledge that the elevation data used in Fig. 8 is from WorldClim’s shared data78, the boundary data comes from the data published by China National Basic Geographic Information Center. Map production is completed through ArcGIS 10.0 software.
Author information
Authors and Affiliations
Contributions
H.Xu, J.Z. and Y.Y. designed the experiment and wrote the first draft. J.Z., Y.Y., D.L. and W.Z. performed data analysis. J.Z., Y.Y., H.**ao and M.X. performed the experimental manipulations. Y.X., J.W., Q.N., M.Z. collected the fecal samples and performed preliminary preparation. All authors have helped in revision and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval
Before sample collection, all the animal work was approved by the Institutional Animal Care and Use Committee of the Sichuan Agricultural University (permit number SKY -2020116003). All field work was granted permission by the Administration for Wild Animal and Plant Protection and Nature Reserves and the Department of Forestry for the Sichuan provinces and Chongqing municipality.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhao, J., Yao, Y., Li, D. et al. Metagenome and metabolome insights into the energy compensation and exogenous toxin degradation of gut microbiota in high-altitude rhesus macaques (Macaca mulatta). npj Biofilms Microbiomes 9, 20 (2023). https://doi.org/10.1038/s41522-023-00387-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41522-023-00387-3
- Springer Nature Limited