

Abstract
D6 PROTEIN KINASE (D6PK) is a polarly localized plasma-membrane-associated kinase from Arabidopsis thaliana that activates polarly distributed PIN-FORMED auxin transporters. D6PK moves rapidly to and from the plasma membrane, independent of its PIN-FORMED targets. The middle D6PK domain, an insertion between kinase subdomains VII and VIII, is required and sufficient for association and polarity of the D6PK plasma membrane. How D6PK polarity is established and maintained remains to be shown. Here we show that cysteines from repeated middle domain CXX(X)P motifs are S-acylated and required for D6PK membrane association. While D6PK S-acylation is not detectably regulated during intracellular transport, phosphorylation of adjacent serine residues, in part in dependence on the upstream 3-PHOSPHOINOSITIDE-DEPENDENT PROTEIN KINASE, promotes D6PK transport, controls D6PK residence time at the plasma membrane and prevents its lateral diffusion. We thus identify new mechanisms for the regulation of D6PK plasma membrane interaction and polarity.
Similar content being viewed by others

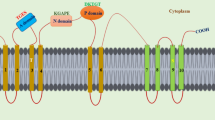

Main
The phytohormone auxin regulates essentially every aspect of plant growth and development. Auxin is transported, within the plant, from cell to cell by different auxin transporters1. The polar distribution of so-called ‘canonical’ PIN-FORMED (PIN) auxin efflux carriers can give directionality to auxin transport, and PIN polarity or activity regulation can explain auxin-controlled responses during development1,2,3,4.
‘Canonical’ PINs are activated by phosphorylation through serine/threonine kinases of the AGC1 and AGC3 families, kinases related to animal cyclic adenosine monophosphate- (cAMP-), cyclic guanosine monophosphate- (cGMP-) and Ca2+-dependent kinases5,6,42,43.
Analysis of S-acylation
The acyl biotin exchange assay was performed as previously described using 1 mg of total protein obtained from transgenic lines overexpressing YFP–D6PK44. Protein extracts were treated with NEM to modify free cysteines, which was followed by hydroxylamine treatment leading to the hydrolysation of S-acylations. Subsequently, the extract was labelled with sulfhydryl-reactive biotin–HPDP ((3aS,4S,6aR)-hexahydro-2-oxo-N-[6-[[1-oxo-3-(2-pyridinyldithio)propyl]amino]hexyl]-1H-thieno[3,4-d]imidazole-4-pentanamide), which reacts with the thiol groups of unmodified cysteines; the modified proteins were purified with streptavidin beads and subjected to anti-GFP immunoblot analysis. For the negative control, the hydroxylamine treatment was omitted; tubulin A (TUA), a known palmitoylation substrate detected with aTUA (anti-tubulin alpha chain, AS10680, Agrisera), served as a positive control27. To assess dynamic S-acylation of YFP–D6PK, an acyl resin-assisted capture assay was performed using soluble and membrane fractions obtained after ultracentrifugation of 1 mg total at 100,000 g protein as previously described45. 2-Bromo palmitate (2-BP) treatments were performed by treating 6-day-old light-grown seedlings for 2 h with 2-BP, as specified and diluted from a 100 mM stock solution prepared in ethanol (Sigma), before examining the seedlings with an Olympus FV1000 confocal laser scanning microscope (Olympus). Protein fractionation, immunoblot and quantification of 2-BP-treated samples were performed as described above.
Immunoblot analysis and immunostaining
For immunoblot analysis of plant protein extracts, YFP–D6PK or its variants were examined by SDS–PAGE using 40 µg total protein and, where specified, after subcellular fractionation through 1 h ultracentrifugation at 100,000 g. SDS–PAGE were blotted and probed with anti-GFP (aGFP; 1:3,000, laboratory stock), aTUA (AS10 680; 1:2,000, Agrisera) or anti-uridine diphosphate (UDP) glucose pyrophosphorylase (aUGP, AS05 086; 1:1500, Agrisera) primary antibodies and an anti-rabbit horse radish peroxidase-conjugated secondary antibody (1:100,000; A9169; Sigma). Chemiluminescence was detected with a Fujifilm LAS 4000 mini (Fuji) and quantified using the measure and profile functions of the Fiji (ImageJ) software38.
To assess D6PK phosphorylation at S310/S311, an anti-aS310p/S311p antibody was raised in rabbits against the respective phosphorylated chemically synthesized peptide (H-CPRFF-phosphoS-phosphoS-KSKKDK-NH2) and validated by enzyme-linked immunosorbent assay (Eurogentec).
For immunoblot analysis of recombinant protein, GST–D6PK, GST–D6PK_SSAA and GST–D6PK_SSDD were purified as described previously. For λ-phosphatase treatments, GST–D6PK was treated with 400 U λ-phosphatase (New England Biolabs) for 20 min at room temperature with the protein bound to glutathione agarose before washing and elution. Equal protein amounts were loaded on an SDS–PAGE and blotted and probed with anti-aS310p/S311p (1:250) or anti-glutathione S-transferase (aGST, 27-4577-01; 1:2,000, Cytiva) primary antibodies and anti-rabbit horse radish peroxidase-conjugated (1:100,000; A9169; Sigma) or anti-rabbit alkaline phosphatase-conjugated secondary antibodies (1:1,000, A3937; Sigma). Immunoblots were imaged using a Fujifilm LAS 4000 mini (Fuji).
Immunohistochemistry
For immunostaining, 5-day-old light-grown seedlings were treated with 10 µM BFA or a corresponding mock solution for 30 min and fixed for 1 h at room temperature under vacuum in 4% (v/v) paraformaldehyde in PBS pH 7.4. Cell walls were partially digested for 30 min at 37 °C with 2% (w/v) Driselase (Sigma). Plasma membranes were permeabilized for 1 h at room temperature with 3% (v/v) Nonidet P-40 (AppliChem) in 10% (v/v) DMSO/PBS. The samples were blocked for 1 h at room temperature with 4% (w/v) BSA in PBS before anti-aS310p/S311p (1:100), diluted in blocking solution, was added for 4 h at 37 °C. Following washes with 0.1% Triton X-100 (AppliChem) in PBS, 4 h incubation at 37 °C with a Cy3-conjugated anti-rabbit antibody (1:600; Dianova) was done for primary antibody detection. Following renewed washes, the immunostained seedling roots were examined in an Olympus FV1000 confocal laser scanning microscope (Olympus). For the quantification of co-localization, YFP (YFP–D6PK) and Cy3 (aS310p/S311p) signals of immunostained seedlings were determined using the Fiji JaCoP plug-in and by calculating the Pearson’s correlation coefficient (r).
Mass spectrometry
For phosphoproteomics analysis of in vitro phosphorylated GST–D6PK, in-gel trypsin digestion was performed according to standard procedures46. Briefly, the samples were run on a NuPAGE 4–12% Bis–Tris protein gel (ThermoFisher Scientific) for 5 min. Subsequently, the proteins were separated on a short SDS–PAGE gel and, for identification of phosphosites, separated on a long SDS–PAGE gel. The protein gel slice was excised and reduced with 50 mM 1,4 dithiothreitol, alkylated with 55 mm chloroacetamide and digested overnight with trypsin. The tryptic peptides were eluted and dried in a vacuum concentrator and dissolved in 0.1% (v/v) formic acid in high-performance liquid chromatography (HPLC)-grade water before liquid chromatography–mass spectrometry analysis.
Liquid chromatography-tandem mass spectroscopy analysis was performed on Orbitrap mass spectrometer systems (Thermo Fisher Scientific) coupled on-line to a Dionex 3000 HPLC (Thermo Fisher Scientific) with a 75 μm × 2 cm trap column (Reprosil Pur ODS-3 5 μm particles (Dr. Maisch HPLC)) and a 75 μm × 40 cm analytical column (3 μm particles C18 Reprosil Gold 120 (Dr. Maisch HPLC)). Peptides were separated at a flow rate of 300 nl min−1 over a 50 min gradient from 4% to 32% acetonitrile in 5% dimethylsulfoxide, 0.1% formic acid, followed by a washing step (column temperature 50 °C). For the in vitro D6PK redox and kinase assay samples, full-scan mass spectra (m/z 360–1,300) were acquired in profile mode on an Orbitrap Fusion Lumos Tribrid Mass Spectrometer with 60,000 resolution, an automatic gain control target value of 5 × 105 or 4 × 105 and 10 or 50 ms maximum injection time, respectively. For the top 20 precursor ions, Orbitrap readout MS2 scans were performed, using higher-energy collisional dissociation (HCD) fragmentation with 28% or 30% normalized collision energy, 15,000 resolution, an automatic gain control target value of 2 × 105 or 5 × 104, 1.7 or 1.3 m/z isolation width and 50 ms, 10 ms or 22 ms maximum injection time, respectively. The minimum intensity threshold was set to 2 × 104 with a dynamic exclusion of 20 s. For the EDTA kinase assay samples, the dynamic exclusion was set to 10 ms for precursors from a customized inclusion mass list and 60 ms for all other precursors. Immunoprecipitates from the D6PK redox assay were measured with comparable settings on a Q Exactive HF (ThermoFisher). Here the MS1 automatic gain control target value was set to 3 × 106, the normalized collision energy was 25% and MS2 spectra were acquired at 30,000 resolution. To confirm and more precisely monitor phospho-modified peptides identified in the data-dependent acquisition analysis of the EDTA kinase assay samples, a targeted Parallel Reaction Monitoring (PRM) was set up on the Orbitrap Fusion Lumos Tribrid Mass Spectrometer (Thermo Fisher) using the same liquid chromatography gradient settings as described above. PRM measurements were performed with the acquisition method switching between experiments after one duty cycle. The first experiment consisted of a full-scan MS1 spectrum recorded in the orbitrap (360 to 1,300 m/z, 15,000 resolution, automatic gain control target value of 4 × 105, maximum injection time 10 ms), followed by a targeted MS2 PRM scan triggering MS2 scans based on a list containing retention time window, m/z and charge information from the previous data-dependent acquisition experiment. For the targeted mass spectrometry analysis 2 (tMS2) PRM scan, the scheduled precursors were isolated (isolation window 0.7 m/z), fragmented via HCD (normalized collision energy [NCE] 28%) and recorded in the Lumos orbitrap (120 to 2,000 m/z, 15,000 resolution, automatic gain control target 2 × 105, maximum injection time 100 ms).
Peptide and protein identification and quantification were performed with MaxQuant47 using standard settings (version 1.5.8.3 in the case of the redox assay, version 1.6.3.3 in the case of the kinase assays). Raw files were searched against the Araport11 database (Araport11_genes.201606.pep.fasta) at www.arabidopsis.org and common contaminants. An E. coli reference database (562_Escherichiacoli_NCBI. fasta) was added when recombinantly expressed proteins were analysed. The D6PK–PDK1 kinase experiment was searched against a custom database with the protein sequences of D6PK, PIP5K1 and PDK1. Cysteine modification with carbamidomethyl (+57.0214) or N-ethylmaleimide (+125.0476) as well as oxidation of methionine and N-terminal protein acetylation were set as variable modifications for the redox experiments. For the in vitro kinase assay samples, carbamidomethylation of cysteine was set as fixed, and phosphorylation of serine, threonine or tyrosine as variable modification. Trypsin/P was specified as the proteolytic enzyme, with up to two missed cleavage sites allowed. The match between run function was enabled. Results were filtered to 1% peptide spectra matched, protein and site false discovery rate. Mass spectra were displayed with MaxQuant viewer48. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD037885 (ref. 49). RAW files from the PRM measurement were imported into Skyline (64 bit)50 for data filtering and analysis. Peaks were integrated using the automatic peak-finding function followed by manual curation of all peak boundaries and transitions. The summed area under the fragment ion traces was exported for data visualization in Microsoft Excel (v. 16.51).
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All material, data, accession codes, unique identifiers or web links for publicly available datasets will be made available upon request, unless not already provided in the article. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD037885. Source data are provided with this paper.
References
Hammes, U. Z., Murphy, A. S. & Schwechheimer, C. Auxin transporters—a biochemical view. Cold Spring Harb. Perspect. Biol. 14, a039875 (2022).
Hajny, J., Tan, S. & Friml, J. Auxin canalization: from speculative models toward molecular players. Curr. Opin. Plant Biol. 65, 102174 (2022).
Verma, S., Attuluri, V.P.S. & Robert, H.S. An essential function for auxin in embryo development. Cold Spring Harb. Perspect. Biol. (2021).
Han, H., Adamowski, M., Qi, L., Alotaibi, S. S. & Friml, J. PIN-mediated polar auxin transport regulations in plant tropic responses. New Phytol. 232, 510–522 (2021).
Zourelidou, M. et al. Auxin efflux by PIN-FORMED proteins is activated by two different protein kinases, D6 PROTEIN KINASE and PINOID. Elife 3, eLife.02860 (2014).
Bogre, L., Okresz, L., Henriques, R. & Anthony, R. G. Growth signalling pathways in Arabidopsis and the AGC protein kinases. Trends Plant Sci. 8, 424–431 (2003).
Bassukas, A. E. L., **ao, Y. & Schwechheimer, C. Phosphorylation control of PIN auxin transporters. Curr. Opin. Plant Biol. 65, 102146 (2022).
Zourelidou, M. et al. The polarly localized D6 PROTEIN KINASE is required for efficient auxin transport in Arabidopsis thaliana. Development 136, 627–636 (2009).
Willige, B. C. et al. D6PK AGCVIII kinases are required for auxin transport and phototropic hypocotyl bending in Arabidopsis. Plant Cell 25, 1674–1688 (2013).
Barbosa, I. C., Zourelidou, M., Willige, B. C., Weller, B. & Schwechheimer, C. D6 PROTEIN KINASE activates auxin transport-dependent growth and PIN-FORMED phosphorylation at the plasma membrane. Dev. Cell 29, 674–685 (2014).
Marhava, P. et al. A molecular rheostat adjusts auxin flux to promote root protophloem differentiation. Nature 558, 297–300 (2018).
Mergner, J. et al. Mass-spectrometry-based draft of the Arabidopsis proteome. Nature 579, 409–414 (2020).
Barbosa, I. C. et al. Phospholipid composition and a polybasic motif determine D6 PROTEIN KINASE polar association with the plasma membrane and tropic responses. Development 143, 4687–4700 (2016).
Weller, B. et al. Dynamic PIN-FORMED auxin efflux carrier phosphorylation at the plasma membrane controls auxin efflux-dependent growth. Proc. Natl Acad. Sci. USA 114, E887–E896 (2017).
Tan, S. et al. The lipid code-dependent phosphoswitch PDK1-D6PK activates PIN-mediated auxin efflux in Arabidopsis. Nat. Plants 6, 556–569 (2020).
**ao, Y. & Offringa, R. PDK1 regulates auxin transport and Arabidopsis vascular development through AGC1 kinase PAX. Nat. Plants 6, 544–555 (2020).
Galvan-Ampudia, C. S. & Offringa, R. Plant evolution: AGC kinases tell the auxin tale. Trends Plant Sci. 12, 541–547 (2007).
Simon, M. L. et al. A PtdIns(4)P-driven electrostatic field controls cell membrane identity and signalling in plants. Nat. Plants 2, 16089 (2016).
Greaves, J. & Chamberlain, L. H. Dual role of the cysteine-string domain in membrane binding and palmitoylation-dependent sorting of the molecular chaperone cysteine-string protein. Mol. Biol. Cell 17, 4748–4759 (2006).
Wolven, A., Okamura, H., Rosenblatt, Y. & Resh, M. D. Palmitoylation of p59fyn is reversible and sufficient for plasma membrane association. Mol. Biol. Cell 8, 1159–1173 (1997).
Robinson, L. J. & Michel, T. Mutagenesis of palmitoylation sites in endothelial nitric oxide synthase identifies a novel motif for dual acylation and subcellular targeting. Proc. Natl Acad. Sci. USA 92, 11776–11780 (1995).
Giles, N. M. et al. Metal and redox modulation of cysteine protein function. Chem. Biol. 10, 677–693 (2003).
Marino, S. M. & Gladyshev, V. N. Cysteine function governs its conservation and degeneration and restricts its utilization on protein surfaces. J. Mol. Biol. 404, 902–916 (2010).
Ji, Y. et al. Direct detection of S-palmitoylation by mass spectrometry. Anal. Chem. 85, 11952–11959 (2013).
Abrami, L., Kunz, B., Iacovache, I. & van der Goot, F. G. Palmitoylation and ubiquitination regulate exit of the Wnt signaling protein LRP6 from the endoplasmic reticulum. Proc. Natl Acad. Sci. USA 105, 5384–5389 (2008).
Batistic, O., Sorek, N., Schultke, S., Yalovsky, S. & Kudla, J. Dual fatty acyl modification determines the localization and plasma membrane targeting of CBL/CIPK Ca2+ signaling complexes in Arabidopsis. Plant Cell 20, 1346–1362 (2008).
Hemsley, P. A., Taylor, L. & Grierson, C. S. Assaying protein palmitoylation in plants. Plant Methods 4, 2 (2008).
Wan, J., Roth, A. F., Bailey, A. O. & Davis, N. G. Palmitoylated proteins: purification and identification. Nat. Protoc. 2, 1573–1584 (2007).
Zhang, Y. L. et al. Protein palmitoylation is critical for the polar growth of root hairs in Arabidopsis. BMC Plant Biol. 15, 50 (2015).
Abrami, L. et al. Palmitoylated acyl protein thioesterase APT2 deforms membranes to extract substrate acyl chains. Nat. Chem. Biol. 17, 438–447 (2021).
Menz, J., Li, Z., Schulze, W. X. & Ludewig, U. Early nitrogen-deprivation responses in Arabidopsis roots reveal distinct differences on transcriptome and (phospho-) proteome levels between nitrate and ammonium nutrition. Plant J. 88, 717–734 (2016).
Kumar, M., Carr, P. & Turner, S. R. An atlas of Arabidopsis protein S-acylation reveals its widespread role in plant cell organization and function. Nat. Plants 8, 670–681 (2022).
Van Bel, M. et al. PLAZA 4.0: an integrative resource for functional, evolutionary and comparative plant genomics. Nucleic Acids Res. 46, D1190–D1196 (2018).
Sawano, A. & Miyawaki, A. Directed evolution of green fluorescent protein by a new versatile PCR strategy for site-directed and semi-random mutagenesis. Nucleic Acids Res. 28, E78 (2000).
Hansson, M. D., Rzeznicka, K., Rosenback, M., Hansson, M. & Sirijovski, N. PCR-mediated deletion of plasmid DNA. Anal. Biochem. 375, 373–375 (2008).
Katsiarimpa, A. et al. The Arabidopsis deubiquitinating enzyme AMSH3 interacts with ESCRT-III subunits and regulates their localization. Plant Cell 23, 3026–3040 (2011).
Clough, S. J. & Bent, A. F. Floral dip: a simplified method for Agrobacterium-mediated transformation of Arabidopsis thaliana. Plant J. 16, 735–743 (1998).
Schindelin, J. et al. Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676–682 (2012).
Bolte, S. & Cordelieres, F. P. A guided tour into subcellular colocalization analysis in light microscopy. J. Microsc. 224, 213–232 (2006).
Koulouras, G. et al. EasyFRAP-web: a web-based tool for the analysis of fluorescence recovery after photobleaching data. Nucleic Acids Res. 46, W467–W472 (2018).
Geiger, D. et al. Stomatal closure by fast abscisic acid signaling is mediated by the guard cell anion channel SLAH3 and the receptor RCAR1. Sci. Signal. 4, ra32 (2011).
Perez, V. I. et al. Detection and quantification of protein disulfides in biological tissues a fluorescence-based proteomic approach. Methods Enzymol. 473, 161–177 (2010).
Winther, J. R. & Thorpe, C. Quantification of thiols and disulfides. Biochim. Biophys. Acta 1840, 838–846 (2014).
Hemsley, P. A. Assaying protein S-acylation in plants. Methods Mol. Biol. 1043, 141–146 (2013).
Forrester, M. T. et al. Site-specific analysis of protein S-acylation by resin-assisted capture. J. Lipid Res. 52, 393–398 (2011).
Shevchenko, A., Tomas, H., Havlis, J., Olsen, J. V. & Mann, M. In-gel digestion for mass spectrometric characterization of proteins and proteomes. Nat. Protoc. 1, 2856–2860 (2006).
Cox, J. & Mann, M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 26, 1367–1372 (2008).
Tyanova, S. et al. Visualization of LC-MS/MS proteomics data in MaxQuant. Proteomics 15, 1453–1456 (2015).
Perez-Riverol, Y. et al. The PRIDE database and related tools and resources in 2019: improving support for quantification data. Nucleic Acids Res. 47, D442–D450 (2019).
MacLean, B. et al. Skyline: an open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics 26, 966–968 (2010).
Acknowledgements
This work was supported by grants from the Deutsche Forschungsgemeinschaft to C.S. (SCHW751/12-2, SCHW751/14-1, SCHW751/15-1, SCHW751/16-1) and a postdoctoral fellowship from the Alexander-von-Humboldt foundation (NLD 1216207 HFST-P) to Y.X. The authors are grateful to U. Z. Hammes and P. Jewaria (Technical University of Munich) for their critical and thorough comments on the manuscript, and they acknowledge J. Sala and E. Grill (both Technical University of Munich) for the gifts of GST–GNC and GST–CPK21, respectively. U. Krauss and E. Knieps-Grünhagen (Forschungszentrum Jülich, Germany) are thanked for performing biochemical analyses that ultimately did not become part of this study. We further acknowledge support by the Center for Advanced Light Microscopy (CALM) of the TUM School of Life Sciences.
Funding
Open access funding provided by Technische Universität München.
Author information
Authors and Affiliations
Contributions
A.G., A.E.L.B., Y.X., I.C.R.B. and C.S. conceived and designed the analysis; A.G. collected the data; J.M., P.G., B.M. and B.K. contributed data or analysis tools; A.G., A.E.L.B., I.C.R.B., J.M. and P.G. performed the analyses; C.S. and A.G. wrote the paper; all authors edited and approved the paper.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Plants thanks Remko Offringa, Tatsuya Sakai and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 CXX(X)P motif repeats are a highly specific feature of AGC1 kinases.
Muscle alignment of the middle domain sequences of Arabidopsis thaliana AGC1 – AGC4 kinases with cysteine (C) and proline (P) residues of CXX(X)P motifs, as well as adjacent serine (S) residues and polybasic motifs (K/R) highlighted. Three residues of the flanking kinase subdomains VII (DFD) and VIII (SMS sequence of the activation loop) are included in the alignment to better delineate the middle domain.
Extended Data Fig. 2 CXX(X)P motif cysteines but not prolines are important for plasma membrane association and auxin transport but do not impact D6PK kinase activity.
A and B. Representative confocal images of transiently transformed protoplasts overexpressing wild type YFP-D6PK from 35Sp::YFP-D6PK or its mutant variants with individual or all five cysteines (C) mutated to serines (S) or alanine (A) (panel A) or with individual or all four prolines (P) mutated to glycines (G) (panel B). Scale bars = 10 µm. C. Result from in vitro kinase assays with recombinant wild type and mutant GST-D6PK as specified using a cytoplasmic loop fragment of PIN1 in GST-PIN1 as trans-phosphorylation substrate. AR, autoradiography; CBB, Coomassie Brilliant Blue-stained gel as loading control. This experiment was repeated three times with similar results. D. Representative images of the apical hook region of three-days old dark-grown seedlings with the specified genotypes. Scale bar = 0.5 mm.
Extended Data Fig. 3 Profile blots confirm the reduced plasma membrane polarity of YFP-D6PK_C4S, C5S and C1-5S.
Representative confocal images as shown in Fig. 2A (left panels) and profile blots (right panel) over a 20 µm line, as shown in the confocal images, confirm the reduction in relative plasma membrane protein abundance of YFP-D6PK_C4S, C5S and C1-5S. Images are representative of results from at least three independent experiments, imaging at least five roots each time.
Extended Data Fig. 4 2-BP treatments lead to increased YFP-D6PK transcript abundance.
A. Bar charts displaying the means and standard deviations of a qRT-PCR experiment testing for the transcription of YFP-D6PK after mock and 2-BP (2-bromo palmitate) treatment, as specified, from three biological replicates displayed as individual data points. Expression levels were compared using a One-way ANOVA F(4, 10) = 18.85, p = 0.0001. B - D. Representative confocal images of root tips from three days-old transgenic seedlings expressing D6PKp::YFP-D6PK and treated with 2-BP and CHX (cycloheximide) as specified. Scale bars = 50 µm. E. Immunoblots with anti-GFP (aGFP) ad anti-UGP (aUGP) antibodies on total protein extracts treated with 2-BP and CHX (cycloheximide) as specified. The CHX chase experiment, including the imaging and immunoblot, was performed once.
Extended Data Fig. 5 Mass spectrometric analysis of middle domain phosphosites.
Muscle alignment of D6PK and D6PKL middle domain sequences with the 31 amino acids deleted in YFP-D6PK∆SAN marked by a line. Serine residues in the middle region are highlighted in red, serines detected in their phosphorylated form in in vitro phosphorylation experiments are shown in bold letters together with their respective phosphopeptide intensity blots (upper panels) for the specified tryptic peptide fragments. The heatmap shows the relative abundance of the phosphorylated peptide in the different samples allowing to differentiate auto- and transphosphorylation events. The peptide corresponding to S310/S311, FFSSKSK, was recovered very rarely and only in its phosphorylated form, rendering it impossible to establish the corresponding correlations.
Extended Data Fig. 6 D6PK_SSAA mutations impair D6PK polar distribution and compromise basipetal auxin transport.
A Representative confocal images of transiently transformed protoplasts overexpressing wild type YFP-D6PK from 35Sp::YFP-D6PK or its mutant variants with SSAA and SSDD mutations. Scale bar = 10 µm. Transformations were performed twice and multiple protoplasts were imaged each time yielding comparable results. B. Result from in vitro kinase assays with recombinant wild type and mutant GST-D6PK as specified using a cytoplasmic loop fragment of PIN1 in GST-PIN1 as trans-phosphorylation substrate. AR, autoradiography; CBB, Coomassie Brilliant Blue-stained gel as loading control. Kinase assays were performed twice and similar results were obtained. C. Representative confocal images of the apical hook region of transgenic seedlings expressing the transgenes as specified in the d6pk d6pkl1 background. D and E. Bar charts displaying the means and standard deviations of the relative cytoplasmic fractions (D) and the polarity indices (E) as determined from confocal images as shown in (C). The three data points represent the means of five measurements from one root, representing a biological replicate. F. Representative images of three-days old dark-grown seedlings with the specified genotypes. Scale bar = 0.5 mm. G. Graph displaying the mean and standard deviation of basipetal auxin transport as measured in four-days old dark-grown seedlings with the specified genotypes. Each data point represents a measurement taken from five seedlings. In (D), (E) and (G), the statistically significant difference between groups was determined by One-way ANOVA and means were compared using a Tukey’s test (D, F(2, 36) = 18.80, p = 0.0000026; E, F(2, 36) = 15. 79, p = 0.0000119; G, F(4,25) = 7.997, p = 0.00027). Different letters indicate a statistically significant difference.
Extended Data Fig. 7 D6PK phosphorylated at S310/S311 accumulates intracellularly.
A - L. Representative confocal images after immunostaining with aS310p/311p of four-days-old Arabidopsis thaliana seedlings overexpressing wild type YFP-D6PK (A -D) or YFP-D6PK_SSDD (I – L) under mock conditions and after BFA treatment. The d6pk d6pkl1 d6pkl2 (d6pk012) triple mutant is included for comparisons. Images shown in A – D are the same images as those shown in Fig. 5B - E. Scale bars = 50 µm (A, C, E, G, I, K) and 20 µm (B, D, F, H, J, L). Images are representative images taken from multiple roots imaged from at least two independent experiments.
Extended Data Fig. 8 The D6PK_ΔSAN mutation impairs D6PK polar distribution and compromises basipetal auxin transport.
A Representative confocal images of transiently transformed protoplasts overexpressing wild type YFP-D6PK from 35Sp::YFP-D6PK or its mutant variants with the ΔSAN deletion. Scale bar = 10 µm. B. Result from in vitro kinase assays with recombinant wild type and mutant GST-D6PK as specified using a cytoplasmic loop fragment of PIN1 in GST-PIN1 as trans-phosphorylation substrate. AR, autoradiography; CBB, Coomassie Brilliant Blue-stained gel as loading control. Localization and phosphorylation data are representative of two independent experiments. C. Representative images of three-days old dark-grown seedlings with the specified genotypes. Scale bar = 0.5 mm. D. Bar chart displaying the mean and standard deviation of relative basipetal auxin transport as measured in four-days old dark-grown seedlings with the specified genotypes. Each data point represents a measurement taken from five seedlings. In (D), the statistically significant difference between groups was determined by One-way ANOVA and means were compared using a Tukey’s test (D, F(3, 13) = 10.54, p = 0.0009).
Extended Data Fig. 9 A D6PK activation loop mimicking SMD mutation is sufficient to suppress the apical hook formation and, partially, also the auxin transport defect of pdk1 pdk2 mutants.
A. Representative images of three-days old dark-grown seedlings with the specified genotypes. Scale bar = 0.5 mm. B. Graph displaying the mean and standard deviation of relative basipetal auxin transport as measured in four-days old dark-grown seedlings with the specified genotypes. Each data point represents a measurement taken from five seedlings. In (B), the statistically significant difference between groups was determined by One-way ANOVA and means were compared using a Tukey’s test (B, F(4, 17) = 9.344, p = 0.0003). Different letters indicate a statistically significant difference.
Extended Data Fig. 10 Immunostaining in the pdk1 pdk2 complementation lines reveals D6PK activation loop phosphorylation after BFA treatment.
A - D. Representative confocal images of roots (A, C) and magnifications of root cortex and epidermal cells (B, D) from four-days-old seedlings expressing YFP-PDK1 and YFP-D6PK (negative control) from the PDK1 promoter (PDK1p) after BFA treatment. Shown are YFP fluorescence, fluorescence after immunostaining with the aS310p/S311 antibody, as well as a merge of the images (A, C) and their magnification (B, D). Scale bars = 50 µm (A, C) and 20 µm (B, D). Immunostainings were performed twice with comparable results.
Supplementary information
Supplementary Information
Supplementary Figs. 1–3, legends and Supplementary Table 1.
Source data
Source Data Fig. 3
Unprocessed western blots.
Source Data Fig. 3
Unprocessed western blots.
Source Data Extended Data Fig. 4
Unprocessed western blots.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Graf, A., Bassukas, A.E.L., **ao, Y. et al. D6PK plasma membrane polarity requires a repeated CXX(X)P motif and PDK1-dependent phosphorylation. Nat. Plants 10, 300–314 (2024). https://doi.org/10.1038/s41477-023-01615-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41477-023-01615-6
- Springer Nature Limited