
Abstract
Cisplatin is a clinically advanced and highly effective anticancer drug used in the treatment of a wide variety of malignancies, such as head and neck, lung, testis, ovary, breast cancer, etc. However, it has only a limited use in clinical practice due to its severe adverse effects, particularly nephrotoxicity; 20%–35% of patients develop acute kidney injury (AKI) after cisplatin administration. The nephrotoxic effect of cisplatin is cumulative and dose dependent and often necessitates dose reduction or withdrawal. Recurrent episodes of AKI result in impaired renal tubular function and acute renal failure, chronic kidney disease, uremia, and hypertensive nephropathy. The pathophysiology of cisplatin-induced AKI involves proximal tubular injury, apoptosis, oxidative stress, inflammation, and vascular injury in the kidneys. At present, there are no effective drugs or methods for cisplatin-induced kidney injury. Recent in vitro and in vivo studies show that numerous natural products (flavonoids, saponins, alkaloids, polysaccharide, phenylpropanoids, etc.) have specific antioxidant, anti-inflammatory, and anti-apoptotic properties that regulate the pathways associated with cisplatin-induced kidney damage. In this review we describe the molecular mechanisms of cisplatin-induced nephrotoxicity and summarize recent findings in the field of natural products that undermine these mechanisms to protect against cisplatin-induced kidney damage and provide potential strategies for AKI treatment.
Similar content being viewed by others


Introduction
Cisplatin is a clinically advanced and highly effective anticancer drug that is used for the treatment of various solid tumors, such as lung cancer, stomach cancer, and ovarian cancer [1]. However, nephrotoxicity is the major side effect of cisplatin administration. Clinically, the risk of nephrotoxicity in patients taking cisplatin is between 20% and 35% and leads to death in acute kidney injury (AKI) patients [2, 3]. In addition, pediatric patients also develop nephrotoxicity when using cisplatin [4]. Patients with AKI are clinically characterized by impaired renal tubular function, acute renal failure, a reduction in whole blood cells, anemia, physical tremors, weight loss, gastrointestinal dysfunction, lethargy, and orbital tightening, which limit the antitumor use of cisplatin [5]. Cisplatin mediates nephrotoxicity via a number of different cytotoxic mechanisms. In addition to DNA damage, cisplatin also causes cytoplasmic organelle dysfunction, particularly in the endoplasmic reticulum and mitochondria, activates apoptotic pathways, and inflicts cellular damage via oxidative stress and inflammation [6].
Presently, there is no clinically effective drug to prevent or treat cisplatin-induced nephrotoxicity. Many high-efficacy and low-toxicity drugs from natural products have been developed to protect against cisplatin-induced AKI. For example, ginseng, curcumin, and pomegranate can act as antioxidants and anti-inflammatory agents and possibly protect against oxidative stress by restoring the levels of antioxidant enzymes [7]. In addition, pretreatment with vitamin supplements, such as vitamin E and riboflavin (vitamin B), significantly reduces serum urea and increases the expression levels of antioxidant enzymes in children with steroid-responsive nephrotic syndrome [8]. These natural products have potential antioxidant and anti-inflammatory properties and can be used as supplements to alleviate cisplatin-induced nephrotoxicity.
In this review, we first introduce the pathological manifestations of cisplatin-induced nephrotoxicity and clarify the molecular events of the underlying mechanisms. Finally, we summarize the roles of various kinds of natural products in protecting against cisplatin-induced AKI. This review focuses on the different mechanisms and protective effects of natural products, providing a comprehensive understanding of the prevention of cisplatin-induced nephrotoxicity and potential implications for drug combinations or natural supplements for AKI patients.
Pathological manifestations of cisplatin-induced nephrotoxicity
Clinically, different doses of cisplatin may lead to different degrees of nephrotoxicity. Patients who receive a single dose of cisplatin may suffer from reversible kidney injury, while large doses or multiple courses of treatment may cause irreversible renal failure [9]. Pharmacokinetic studies also show that nephrotoxicity is mainly due to the high volume of cisplatin distribution and long-term accumulation of cisplatin in the kidney [10]. In general, the pathological mechanisms of cisplatin-induced nephrotoxicity mainly manifest as decreases in renal blood flow and glomerular filtration rate [11] and ischemia or necrosis of proximal renal tubular epithelial cells [12].
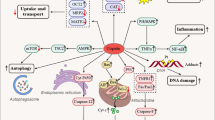
Histopathological changes in cisplatin-induced nephrotoxicity are positively correlated with the dose of cisplatin. First, cisplatin is passively absorbed into renal tubular cells via organic cation transporter 2 (OCT2) and forms hydrates with water molecules, leading to continuous accumulation in renal cells [13]. The formation of cisplatin hydrate is a reversible process, and cisplatin hydrate can be dissociated into cisplatin and water molecules and discharged from the cells [13]. Thus, the accumulation and retention of cisplatin in renal cells leads to DNA damage, oxidative stress, apoptosis, and autophagy (Fig. 1).

The normal epithelium is damaged by cisplatin, as characterized by the loss of brush borders, epithelial cell necrosis, sloughing and obstruction, and immune cell infiltration.
Cisplatin first causes shedding of the brush shape of renal tubular epithelial cells. With increasing cisplatin accumulation, epithelial cells undergo necrosis and are gradually shed, accompanied by the formation of proteinaceous casts [14]. Moreover, the proximal tubule basement membrane becomes thickened, and tubules become dilated [15]. Electron microscopy observation of epithelial cell ultrastructure shows swollen and vacuolated mitochondria, endoplasmic reticulum expansion, and increased numbers of lysosomes [16]. Taken together, these organelle malfunctions result in the destruction and sloughing of epithelial cells, as well as the formation of intratubular obstructions.
Damaged renal tubular epithelial cells recruit many immune cells, such as macrophages, dendritic cells, and T cells, which release a variety of inflammatory factors [17]. Moreover, cisplatin can cause reduced medullary blood flow and exacerbate tubular cell injury, leading to acute ischemic injury in the kidneys [18]. Instead of the typical self-regulatory renal vasodilation in ischemic kidneys, evident vasoconstriction occurs in cisplatin-induced AKI, leading to hypoxic injury and vascular injury in severe cases [19]. Some studies have shown that cisplatin forms a complex with reduced glutathione in the liver and then enters the kidney. Cisplatin is decomposed into a nephrotoxic metabolite due to the action of glutamyltransferase in the brush edge of the renal proximal tubule, causing renal cell apoptosis or necrosis [20].
Mechanisms of cisplatin-induced nephrotoxicity
The application of cisplatin chemotherapy is often limited by severe adverse effects, including nephrotoxicity, ototoxicity, neurotoxicity, and vomiting. Nephrotoxicity, which is the major limiting factor of cisplatin use, involves various mechanisms, such as oxidative stress, apoptosis, inflammation, and autophagy (Fig. 2). Understanding the underlying mechanism is important for investigating intervention strategies for nephrotoxicity.

The mechanisms mainly include the transport and metabolism of cisplatin, apoptosis, autophagy, DNA damage, oxidative stress, and inflammation, which work together to aggravate AKI induced by cisplatin.
Cellular uptake and transport
Cisplatin is mainly excreted through the kidneys. It becomes concentrated during excretion, and the concentration in renal tubular epithelial cells is much higher than that in the blood. In the kidney, cisplatin is absorbed by renal cells via passive diffusion. During excretion, cisplatin and its metabolites are secreted and reabsorbed in the renal tubules during glomerular filtration, leading to a high concentration of cisplatin in the kidneys.
Recent studies have shown that cisplatin is taken up by renal tubular cells via OCT2, copper ion transporter 1 (CTR1), and solute carrier family 22 member 2 [21]. In addition, cisplatin is secreted into the lumen by solute carrier family 47 member 1 and multidrug and toxin extrusion 1 [22]. Knockdown of the Oct2 gene can significantly reduce cisplatin-induced nephrotoxicity [23]. Consistently, patients with Oct2 mutations show low OCT2 expression and reduced cisplatin transport into renal tubular cells, resulting in decreased nephrotoxicity [24]. In addition, when CTR1 expression is downregulated, cisplatin uptake and the subsequent cytotoxicity decrease significantly [25]. Moreover, peroxiredoxin I (Prx I)-deficient mice have higher resistance to cisplatin-induced nephrotoxicity than wild-type mice due to increased cisplatin excretion via the high expression of the renal efflux transporters multidrug resistance-related protein 2 (MRP2) and MRP4 in Prx I-deficient mice [26].
DNA damage
Cisplatin mediates its cytotoxic effects by binding DNA to form adducts that cause DNA damage [27]. In an aqueous environment, the chloride ligand of cisplatin is replaced by water molecules to form a positively charged hydrated complex ion, which is transferred to the nucleus by DNA electrostatic attraction. Then, this complex binds to DNA to form an adduct, resulting in DNA cross-linking and preventing DNA synthesis and replication in rapidly proliferating cells [28]. This phenomenon is pronounced in cells with defective DNA repair.
However, cisplatin binds nonspecifically to nuclear DNA, and less than 1% of platinum binds to nuclear DNA [29]. Interestingly, mitochondrial DNA is more sensitive than nuclear DNA to cisplatin-mediated cytotoxicity [30]. The positively charged metabolites produced by the hydrolysis of cisplatin preferentially accumulate in mitochondria, which are negatively charged. Therefore, the sensitivity of cells to cisplatin depends on mitochondrial density and the mitochondrial membrane potential in cells [31]. Given that the renal proximal tubule contains sites of quite high mitochondrial density, it is the most highly sensitive site in the kidney to cisplatin [32].
Apoptosis
It has been reported that a low concentration (8 μM) of cisplatin causes renal tubular epithelial apoptosis, while a high concentration (800 μM) of cisplatin induces necrosis [33]. Cisplatin-induced apoptosis in renal tubular cells is primarily associated with mitochondria-mediated endogenous pathways, death receptor-mediated exogenous pathways, and endoplasmic reticulum stress (ERS) pathways.
Mitochondria-mediated endogenous pathways
Cisplatin-induced mitochondria-mediated apoptotic pathways mainly include caspase-dependent and -independent pathways. When cisplatin enters renal tubular epithelial cells, BAX translocates to mitochondria and activates caspase-2, resulting in the release of cytochrome c, second mitochondria-derived activator of caspase/direct inhibitor of apoptosis proteins binding protein with low Pi (isoelectric point) (SMAC/DIABLO), high temperature requirement A2 (HtrA2/Omi), and apoptosis-inducing factor (AIF) from mitochondria [34]. Then, caspase-9 is activated, which eventually leads to apoptosis [35]. Apart from the caspase-dependent pathway, cytoplasmic Omi/HtrA2 also promotes caspase-independent apoptosis by binding and cleaving inhibitors of apoptotic proteins after cisplatin-induced apoptotic stimulation [36].
AIF is an apoptosis-related protein located on the mitochondrial membrane, and poly (ADP-ribose) polymerase-1 (PARP-1) is a nuclear factor that participates in DNA repair and protein modification. Once cellular DNA is severely damaged by cisplatin, nuclear PARP-1 activity is increased, causing AIF activation and nuclear translocation, which induces apoptosis [37]. PARP-1 activation is a primary signal in the process of cisplatin-induced nephrotoxicity. Moreover, PARP-1 inhibition or deletion protects the kidneys from nephrotoxicity, providing a therapeutic strategy for cisplatin-induced nephrotoxicity [38].
The role of p53 in cisplatin-induced cytotoxicity mainly involves activation of the mitochondrial pathway. Upon exposure to cisplatin-induced cellular DNA damage, p53 is phosphorylated, and the proapoptotic protein BAX undergoes structural modifications and alters mitochondrial membrane integrity, causing the activation of p53 upregulated modulator of apoptosis-α and Ca2+-independent phospholipase A2. Then, the antiapoptotic proteins BCL-2 and BCL-XL are downregulated, triggering the mitochondrial apoptotic pathway [39].
Death receptor-mediated exogenous pathways
In the exogenous apoptotic pathways, cisplatin binds to death receptors such as tumor necrosis factor receptor 1 (TNFR1), TNFR2, and FAS on the cell membrane to activate caspase-8, which further activates caspase-3, ultimately leading to apoptosis [40]. Cisplatin upregulates the expression of tumor necrosis factor-α (TNF-α), promoting the interaction of TNF-α and TNF receptors, including TNFR1 and TNFR2. TNFR1 has a death domain and is able to directly trigger exogenous apoptosis. However, TNFR2 mainly regulates the inflammatory response to induce apoptosis because it has no death domain [41]. In addition, cisplatin can also activate the FAS/FAS-L system [42], and the FAS-associated death domain further interacts with FAS or TNFR1 to trigger apoptosis, but the detailed mechanisms have not been elucidated.
Endoplasmic reticulum stress pathways
Cisplatin can also activate the apoptotic pathway that is mediated by ERS. After cisplatin enters cells, it acts on the cytochrome P450 (CYP450) enzymatic system on the endoplasmic reticulum membrane to induce oxidative stress and activate caspase-12, which leads to apoptosis [43]. As expected, cisplatin-induced apoptosis is significantly reduced in cytochrome P450, family 2, subfamily E, polypeptide 1 (Cyp2e1)-knockout mice [44]. Similarly, another study showed that the expression of the ERS marker X-box-binding protein 1 was increased, and calpain and caspase-12 cleavage products were observed in rat kidneys after cisplatin treatment [45]. Furthermore, transfection with an anti-caspase-12 antibody significantly attenuated cisplatin-induced apoptosis in porcine kidney LLC-PK1 cells [46]. The ERS pathway is also involved in the activation of endoplasmic reticulum phospholipase A2, which limits downstream p53 and activates upstream caspase-3. The endoplasmic reticulum may be a link between p53 and caspase-3 in the absence of mitochondrial dysfunction [47].
Oxidative stress
In recent years, studies have shown that oxidative stress and nitrosative stress play vital roles in cisplatin-induced nephrotoxicity, which is characterized by increased malondialdehyde (MDA), 4-hydroxy, 8-hydroxydeoxyguanosine, and 3-nitrotyrosine, and decreased superoxide dismutase (SOD) and catalase (CAT) after cisplatin treatment. Thus, reactive oxygen species (ROS) scavengers and antioxidants show robust protective effects against nephrotoxicity [48].
After entering renal tubular cells, cisplatin can rapidly react with the thiol-containing antioxidants glutathione and metallothionein to degrade or inactivate them. Moreover, some antioxidant enzymes, such as glutathione peroxidase, SOD, and glutathione reductase, are also inhibited, leading to increased ROS levels [49]. ROS affect the activity of mitochondrial complex enzymes I–IV, thereby inhibiting the normal transmission of the oxidative respiratory chain and leading to adenosine triphosphate depletion [40]. Then, increased ROS results in lipid peroxidation, changing membrane structure and permeability, which further affect cellular function [50]. Finally, ROS impair amino acids, proteins, and carbohydrates, thus promoting DNA damage and apoptosis. In addition, increased ROS can induce increased expression of FAS-L, FAS, TNFR1, and TNF-α, eventually resulting in apoptosis [45].
Inflammation
Cisplatin-induced nephrotoxicity is associated with the inflammatory response. Renal TNF-α expression is increased in a cisplatin-induced nephrotoxic mouse, and cisplatin-induced renal insufficiency and injury can be significantly alleviated by TNF-α inhibition or knockout, indicating that increased TNF-α expression plays an important role in cisplatin-induced nephrotoxicity [51]. Interestingly, after cisplatin administration, TNF-α in the circulation and urine may be derived from renal epithelial cells rather than immune cells. Moreover, TNF-α induces the production of ROS, further activating the transcription factor, nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB), which in turn induces the production of proinflammatory cytokines such as TNF-α [52]. The inhibition of NF-κB transcriptional activity by JSH-23 (a kind of NF-κB inhibitor) improves kidney function in mice [53].
TNF-α activates proinflammatory cytokines and chemokines to trigger oxidative stress, ultimately exacerbating kidney damage. Hydroxyl free radicals produced by cisplatin are involved in the phosphorylation of p38 mitogen-activated protein kinase (p38 MAPK) and the regulation of TNF-α synthesis, ultimately inducing the activation of NF-κB. Therefore, the hydroxyl radical scavenger dimethyl thiourea inhibits p38 MAPK activation and TNF-α mRNA expression in murine kidneys. The inhibition of p38 MAPK reduces the production of TNF-α, thereby effectively protecting against cisplatin-induced kidney damage [54]. Other cytokines, such as transforming growth factor-β, monocyte chemoattractant protein-1 (MCP-1), intercellular adhesion molecule, and heme oxygenase-1 (HO-1), are also associated with cisplatin-induced nephrotoxicity [55]. N-Acetylcysteine (NAC), an antioxidative agent, effectively inhibits inflammation and activation of the complement system to exert renal protection [56]. Mitochondrial dysfunction leads to the formation of O2–, while the inflammatory response induced by cisplatin involves the upregulation of TNF-α, nicotinamide adenine dinucleotide phosphate oxidase, and inducible nitric oxide synthase (iNOS), which directly leads to NO– formation. NO– and O2– produce ONOO–, which has strong oxidation and nitration properties, further inducing apoptosis and necrosis [57].
Autophagy
Autophagy plays an important role in maintaining cellular homeostasis and surviving cisplatin-induced nephrotoxicity. In NRK-52E cells treated with cisplatin, the increases in autophagy and apoptosis were both inhibited after beclin-1 knockdown, indicating that autophagy mediates cell damage [58]. However, another study showed that autophagy inhibition accelerated apoptosis, demonstrating the protective effect of autophagy in cisplatin-induced kidney injury [59]. Moreover, autophagy can prevent AKI and proximal tubule apoptosis caused by cisplatin [60].
Studies have reported that the suppression of mammalian target of the rapamycin (mTOR) activity alleviates the inhibitory phosphorylation of Unc-51-like autophagy activating kinase 1, which leads to the activation of autophagy [61]. Pretreatment with rapamycin, an mTOR inhibitor, induces autophagy to improve renal function in rats with ischemia/reperfusion [62]. Interestingly, NAD(P)H quinone dehydrogenase 1 deletion (an oxidative stress barrier) enhances the effect of rapamycin and leads to increased tuberous sclerosis complex 2 phosphorylation, indicating that autophagy may be activated to counter the increased stress and protect against AKI [63].
Current treatment of cisplatin-induced nephrotoxicity
Various treatments have been applied to address the different mechanisms of cisplatin-induced nephrotoxicity (Table 1). For example, cimetidine acts as an OCT2 inhibitor that inhibits the transportation of cisplatin in the kidney to protect against AKI [64], carvedilol works as an antioxidant against the oxidative stress process [65], cilastatin inhibits the apoptotic pathway [66], and rosiglitazone reduces inflammation [67].
At present, although several kinds of drugs are applied clinically in response to kidney damage caused by cisplatin, these drugs exhibit different degrees of inadequacy. For example, hydration and diuresis in the clinic enhance cisplatin excretion and reduce renal exposure [68]. However, the disadvantage is that a large amount of hydration is required before and after cisplatin administration [69]. Moreover, adverse reactions such as osmotic pressure changes may occur during chemoprevention. In addition, metabolic waste in the body can be excreted through hemodialysis, which is often accompanied by hypophosphatemia and heart rate disorders. Amifostine is a broad-spectrum cytoprotective agent approved by the FDA as a kidney protectant for cisplatin chemotherapy in patients with advanced ovarian cancer; however, its application in other tumors is limited due to blood pressure drops and hypocalcemia [70].
Protective effects of natural products that prevent cisplatin-induced nephrotoxicity
Traditional and complementary medicines, including a variety of natural products, such as herbs, vitamins, minerals, trace elements, and nutritional supplements, have been widely used in most countries [71]. Adopting natural products in healthcare can improve the physical fitness of patients. To better understand the roles of natural products in AKI, we summarized the protective effects of various classes of natural products on cisplatin-induced nephrotoxicity (Fig. 3 and Tables 2 and 3).

Potential natural product treatments for cisplatin-induced nephrotoxicity classified by chemical structures.
Flavonoids
Studies have shown that formononetin can effectively reduce OCT2 expression and increase MRP expression, resulting in decreased accumulation of cisplatin in renal tubular cells [72]. Similarly, puerarin protects against cisplatin-induced nephrotoxicity and promotes the antitumor activity of cisplatin in COLO205 and HeLa tumor cells in a dose-dependent manner [73]. Interestingly, naringin can alleviate cisplatin-induced renal dysfunction by inhibiting the inflammatory response and reducing apoptosis [74]. Flavonoids with multiple activities, such as icariin, breviscapine, epicatechin and epicatechin gallate, sappanone A, morin and its hydrate, quercetin, silymarin, daidzein, and xanthohumol, can reduce cisplatin-induced oxidative and nitrosative stress and decrease creatinine (Cre) and blood urea nitrogen (BUN) levels to improve renal function, thereby alleviating cisplatin-induced nephrotoxicity [75,76,77,78,79,80,81,82,83,84]. In addition, wogonin markedly inhibits receptor-interacting protein kinase 1-mediated necrosis and the canonical WNT pathway (WNT/β-catenin pathway) to protect against cisplatin-induced nephrotoxicity [85]. Further studies demonstrated that baicalein and apigenin ameliorated cisplatin-induced renal damage through the upregulation of antioxidant pathways and downregulation of the MAPK and NF-κB signaling pathways [86].
Interestingly, Scutellaria baicalensis Georgi not only enhances the therapeutic efficacy of cisplatin but also attenuates chemotherapy-induced AKI [87]. Glycyrrhizic acid, 18β-glycyrrhetinic acid, hypericin, and eriodictyol reduce AKI by inhibiting the cisplatin-induced phosphorylation of NF-κB and upregulating the expression of nuclear factor erythroid 2 (NFE2)-related factor 2 (NRF2) and HO-1 [88,89,90]. D-Pinitol and mangiferin attenuate inflammatory infiltration, DNA damage, and renal dysfunction in rats by modulating the MAPK pathway [91]. Furthermore, cisplatin-induced oxidative stress is mitigated by hesperidin and hesperetin by reducing MDA/Myeloperoxidase (MPO) levels and increasing SOD/Glutathione (GSH) levels. Galangin and the isoflavonoid biochanin A exhibit renoprotective effects in mice by targeting the inflammatory response and p53-mediated apoptosis. Importantly, luteolin significantly reduces histological and biochemical changes induced by cisplatin by blocking platinum accumulation and inflammation [92]. Genistein and naringin inhibit the NF-κB and iNOS pathways and p53 activation to improve HK-2 cell viability and kidney morphology in the presence of cisplatin and have become a potential effective treatment strategy for AKI [93]. A recent study demonstrated that scutellarin and anthocyanin from the fruits of Panax ginseng attenuate cisplatin-induced nephrotoxicity by inhibiting TNF-α [94]. In summary, flavonoids exhibit great potential as dietary supplements to ameliorate cisplatin-induced nephrotoxicity.
It is worth noting that the flavonoid phloretin is a robust toxicant (LC50 = 362 μM) that potentiates H2O2-induced toxicity, which is consistent with the previously noted cytotoxicity of phloretin and other hydroxychalcones. This toxicity is due to the oxidative activities of these polyphenols and the possible induction of mitochondrial toxicity [95].
Many flavonoids show strong protective effects against cisplatin-induced AKI. To date, researchers have found that many kinds of flavonoids activate NRF2/HO-1 signaling and inhibit NF-κB activity to alleviate kidney injury. More interestingly, some flavonoids not only protect against cisplatin-induced kidney injury but also synergistically inhibit the growth of tumors, enhancing the efficacy of cisplatin in tumor-bearing mice [74]. These results suggest that flavonoids may be used in the comprehensive treatment of cancer patients. Although flavonoids exhibit strong protection against kidney injury, there are some challenges in the clinical application of flavonoids. For example, monomers of flavonoid compounds are difficult to extract and have poor lipid solubility and low bioavailability, limiting their clinical applications [96]. If researchers can overcome these challenges, flavonoids will become promising drugs for AKI treatment.
Saponins
Oxidative stress and inflammation are important mechanisms involved in the pathogenesis of AKI. Some studies have shown that saikosaponin D can increase the survival rate of HK-2 cells and maintain the normal morphology of the nucleus. Saikosaponin D can inhibit the activation of the NF-κB-P38-JNK-MAPK signaling cascade, thereby reducing cisplatin-induced apoptosis [97]. Red ginseng, ginsenoside Rg5, and Platycodon grandiflorum saponins can inhibit inflammation by reducing the expression of cyclooxygenase-2 and iNOS to inhibit acute tubular necrosis and apoptosis [98, 99]. Renal oxidative stress, as evidenced by increased MDA levels and declines in GSH and SOD activities, is significantly reduced by saponins from Terminalia arjuna [100].
In addition, some saponin components mainly regulate autophagy and apoptosis to exert protective effects against kidney injury. Ginsenoside 20(S)-Rg3 and ginsenoside Rb3 can inhibit autophagy to improve renal injury by blocking the JNK-P53-caspase-3 signaling cascade [101, 153,154,155,156]. Recent reports show that pretreatment with curcumin can ameliorate cisplatin-induced kidney damage by suppressing inflammation and apoptosis [157].
The mechanisms of cisplatin-induced kidney damage involve various pathways, such as inflammatory mediators, oxidative stress, necrosis and apoptosis, and autophagy. To date, researchers have not found that these mechanisms are involved in cisplatin-induced nephrotoxicity, starting with excess ROS generation, which leads to oxidative stress, triggering inflammatory and autophagy pathways that damage DNA and induce apoptosis in the kidney. It is still unclear how the various pathways integrate and ultimately lead to kidney damage. In recent years, many natural products have been discovered by different mechanisms. A natural compound may have multiple active targets rather than only one unique target. Therefore, a natural product may play multiple roles and exhibit wide use and may have increased potential toxicity or side effects. Since some pathways of cisplatin-induced kidney injury are also involved in the antitumor effects of cisplatin, natural products may also affect cisplatin-mediated antitumor effects. While most compounds have anti-inflammatory and antioxidant properties, NAC and vitamin E have been reported to act as antioxidants and contribute to the development of lung cancer [158]. Therefore, it is unclear whether natural compounds with antioxidant activity interfere with the development of tumors while protecting against kidney injury. In this case, in addition to check the protective role against AKI, it is necessary to further study if natural products have effects on tumor growth, which may help to break through the limited use of cisplatin in clinic.
It is worth noting that natural products that have robust therapeutic effects on cisplatin-induced AKI also alleviate kidney diseases caused by other factors. Further research is needed to verify the beneficial effects of certain products on humans and other animals with kidney diseases to elucidate the detailed mechanisms of the renoprotective effects. To achieve the desired protective effect against nephrotoxicity, researchers should take all aspects of the relevant mechanisms into account and consider comprehensive measures or combinations of drugs. In addition, although certain natural products are excellent in protecting against kidney damage in vitro and in vivo, it is necessary to study the optimal dose for protecting against different tumors and different cisplatin strengths.
Furthermore, the development of molecular biology technology has led to the research of targeted therapy using cisplatin and natural products or derivatives that are highly selective for the kidney or tumor as carriers, and chemically coupling these factors into biological treatments. Direct delivery of cisplatin to the tumor site rather than the kidney can not only reduce the amount of cisplatin needed but also improve the efficacy and reduce adverse reactions. This opens up new ideas for the study of protective measures against cisplatin-induced nephrotoxicity.
Change history
19 October 2022
A Correction to this paper has been published: https://doi.org/10.1038/s41401-022-01012-3
References
Dasari S, Tchounwou PB. Cisplatin in cancer therapy: molecular mechanisms of action. Eur J Pharmacol. 2014;740:364–78.
Gonzales-Vitale JC, Hayes DM, Cvitkovic E, Sternberg SS. The renal pathology in clinical trials of cis-platinum (II) diamminedichloride. Cancer. 1977;39:1362–71.
Pierson-Marchandise M, Gras V, Moragny J, Micallef J, Gaboriau L, Picard S, et al. The drugs that mostly frequently induce acute kidney injury: a case-noncase study of a pharmacovigilance database. Br J Clin Pharmacol. 2017;83:1341–9.
Barton CD, Pizer B, Jones C, Oni L, Pirmohamed M, Hawcutt DB. Identifying cisplatin-induced kidney damage in paediatric oncology patients. Pediatr Nephrol. 2018;33:1467–74.
Effects and side-effects of cisplatin. Lancet. 1982;1:682.
Pabla N, Dong Z. Cisplatin nephrotoxicity: mechanisms and renoprotective strategies. Kidney Int. 2008;73:994–1007.
Ridzuan NRA, Rashid NA, Othman F, Budin SB, Hussan F, Teoh SL. Protective role of natural products in cisplatin-induced nephrotoxicity. Mini Rev Med Chem. 2019;19:1134–43.
Mathew JL, Kabi BC, Rath B. Anti-oxidant vitamins and steroid responsive nephrotic syndrome in Indian children. J Paediatr Child Health. 2002;38:450–37.
Cornelison TL, Reed E. Nephrotoxicity and hydration management for cisplatin, carboplatin, and ormaplatin. Gynecol Oncol. 1993;50:147–58.
Ibrahim ME, Chang C, Hu Y, Hogan SL, Mercke N, Gomez M, et al. Pharmacokinetic determinants of cisplatin-induced subclinical kidney injury in oncology patients. Eur J Clin Pharmacol. 2019;75:51–7.
Li Q, Bowmer CJ, Yates MS. The protective effect of glycine in cisplatin nephrotoxicity: inhibition with NG-nitro-L-arginine methyl ester. J Pharm Pharmacol. 1994;46:346–51.
Safirstein R, Winston J, Moel D, Dikman S, Guttenplan J. Cisplatin nephrotoxicity: insights into mechanism. Int J Androl. 1987;10:325–46.
Eljack ND, Ma HY, Drucker J, Shen C, Hambley TW, New EJ, et al. Mechanisms of cell uptake and toxicity of the anticancer drug cisplatin. Metallomics. 2014;6:2126–33.
Ozkok A, Edelstein CL. Pathophysiology of cisplatin-induced acute kidney injury. Biomed Res Int. 2014;2014:967826.
Yang Y, Yu X, Zhang Y, Ding G, Zhu C, Huang S, et al. Hypoxia-inducible factor prolyl hydroxylase inhibitor roxadustat (FG-4592) protects against cisplatin-induced acute kidney injury. Clin Sci (Lond). 2018;132:825–38.
Prasad SB, Rosangkima G, Kharbangar A. Structural and biochemical changes in mitochondria after cisplatin treatment of Dalton’s lymphoma-bearing mice. Mitochondrion. 2010;10:38–45.
Salei N, Rambichler S, Salvermoser J, Papaioannou NE, Schuchert R, Pakalniskyte D, et al. The kidney contains ontogenetically distinct dendritic cell and macrophage subtypes throughout development that differ in their inflammatory properties. J Am Soc Nephrol. 2020;31:257–78.
Winston JA, Safirstein R. Reduced renal blood flow in early cisplatin-induced acute renal failure in the rat. Am J Physiol. 1985;249:F490–6.
Bonventre JV, Zuk A. Ischemic acute renal failure: an inflammatory disease? Kidney Int. 2004;66:480–5.
Wainford RD, Weaver RJ, Stewart KN, Brown P, Hawksworth GM. Cisplatin nephrotoxicity is mediated by gamma glutamyltranspeptidase, not via a C-S lyase governed biotransformation pathway. Toxicology. 2008;249:184–93.
Yonezawa A, Masuda S, Nishihara K, Yano I, Katsura T, Inui K. Association between tubular toxicity of cisplatin and expression of organic cation transporter rOCT2 (Slc22a2) in the rat. Biochem Pharmacol. 2005;70:1823–31.
Iwata K, Aizawa K, Kamitsu S, **gami S, Fukunaga E, Yoshida M, et al. Effects of genetic variants in SLC22A2 organic cation transporter 2 and SLC47A1 multidrug and toxin extrusion 1 transporter on cisplatin-induced adverse events. Clin Exp Nephrol. 2012;16:843–51.
Filipski KK, Mathijssen RH, Mikkelsen TS, Schinkel AH, Sparreboom A. Contribution of organic cation transporter 2 (OCT2) to cisplatin-induced nephrotoxicity. Clin Pharmacol Ther. 2009;86:396–402.
Ciarimboli G. Membrane transporters as mediators of cisplatin side-effects. Anticancer Res. 2014;34:547–50.
Pabla N, Murphy RF, Liu K, Dong Z. The copper transporter Ctr1 contributes to cisplatin uptake by renal tubular cells during cisplatin nephrotoxicity. Am J Physiol Ren Physiol. 2009;296:F505–11.
Okada K, Ma D, Warabi E, Morito N, Akiyama K, Murata Y, et al. Amelioration of cisplatin-induced nephrotoxicity in peroxiredoxin I-deficient mice. Cancer Chemother Pharmacol. 2013;71:503–9.
Wang D, Lippard SJ. Cellular processing of platinum anticancer drugs. Nat Rev Drug Discov. 2005;4:307–20.
Fujikawa Y, Kawanishi M, Kuraoka I, Yagi T. Frequencies of mutagenic translesion DNA synthesis over cisplatin-guanine intra-strand crosslinks in lacZ plasmids propagated in human cells. Mutat Res Genet Toxicol Environ Mutagen. 2014;770:23–8.
Miller RP, Tadagavadi RK, Ramesh G, Reeves WB. Mechanisms of cisplatin nephrotoxicity. Toxins (Basel). 2010;2:2490–518.
Yimit A, Adebali O, Sancar A, Jiang Y. Differential damage and repair of DNA-adducts induced by anti-cancer drug cisplatin across mouse organs. Nat Commun. 2019;10:309.
Hirama M, Isonishi S, Yasuda M, Ishikawa H. Characterization of mitochondria in cisplatin-resistant human ovarian carcinoma cells. Oncol Rep. 2006;16:997–1002.
Brady HR, Kone BC, Stromski ME, Zeidel ML, Giebisch G, Gullans SR. Mitochondrial injury: an early event in cisplatin toxicity to renal proximal tubules. Am J Physiol. 1990;258:F1181–7.
Chirino YI, Pedraza-Chaverri J. Role of oxidative and nitrosative stress in cisplatin-induced nephrotoxicity. Exp Toxicol Pathol. 2009;61:223–42.
Green DR, Reed JC. Mitochondria and apoptosis. Science. 1998;281:1309–12.
Guerrero-Beltran CE, Calderon-Oliver M, Martinez-Abundis E, Tapia E, Zarco-Marquez G, Zazueta C, et al. Protective effect of sulforaphane against cisplatin-induced mitochondrial alterations and impairment in the activity of NAD(P)H: quinone oxidoreductase 1 and gamma glutamyl cysteine ligase: studies in mitochondria isolated from rat kidney and in LLC-PK1 cells. Toxicol Lett. 2010;199:80–92.
Lau AH. Apoptosis induced by cisplatin nephrotoxic injury. Kidney Int. 1999;56:1295–8.
Rodrigues MA, Rodrigues JL, Martins NM, Barbosa F, Curti C, Santos NA, et al. Carvedilol protects against cisplatin-induced oxidative stress, redox state unbalance and apoptosis in rat kidney mitochondria. Chem Biol Interact. 2011;189:45–51.
Chirino YI, Trujillo J, Sanchez-Gonzalez DJ, Martinez-Martinez CM, Cruz C, Bobadilla NA, et al. Selective iNOS inhibition reduces renal damage induced by cisplatin. Toxicol Lett. 2008;176:48–57.
Servais H, Ortiz A, Devuyst O, Denamur S, Tulkens PM, Mingeot-Leclercq MP. Renal cell apoptosis induced by nephrotoxic drugs: cellular and molecular mechanisms and potential approaches to modulation. Apoptosis. 2008;13:11–32.
Tsuruya K, Ninomiya T, Tokumoto M, Hirakawa M, Masutani K, Taniguchi M, et al. Direct involvement of the receptor-mediated apoptotic pathways in cisplatin-induced renal tubular cell death. Kidney Int. 2003;63:72–82.
Hong SJ, Dawson TM, Dawson VL. Nuclear and mitochondrial conversations in cell death: PARP-1 and AIF signaling. Trends Pharmacol Sci. 2004;25:259–64.
Razzaque MS, Koji T, Kumatori A, Taguchi T. Cisplatin-induced apoptosis in human proximal tubular epithelial cells is associated with the activation of the Fas Fas ligand system. Histochem Cell Biol. 1999;111:359–65.
Boyce M, Yuan J. Cellular response to endoplasmic reticulum stress: a matter of life or death. Cell Death Differ. 2006;13:363–73.
Liu H, Baliga R. Cytochrome P450 2E1 null mice provide novel protection against cisplatin-induced nephrotoxicity and apoptosis. Kidney Int. 2003;63:1687–96.
Peyrou M, Hanna PE, Cribb AE. Cisplatin, gentamicin, and p-aminophenol induce markers of endoplasmic reticulum stress in the rat kidneys. Toxicol Sci. 2007;99:346–53.
Liu H, Baliga R. Endoplasmic reticulum stress-associated caspase 12 mediates cisplatin-induced LLC-PK1 cell apoptosis. J Am Soc Nephrol. 2005;16:1985–92.
Cilenti L, Kyriazis GA, Soundarapandian MM, Stratico V, Yerkes A, Park KM, et al. Omi/HtrA2 protease mediates cisplatin-induced cell death in renal cells. Am J Physiol Ren Physiol. 2005;288:F371–9.
Jiang M, Wang CY, Huang S, Yang T, Dong Z. Cisplatin-induced apoptosis in p53-deficient renal cells via the intrinsic mitochondrial pathway. Am J Physiol Ren Physiol. 2009;296:F983–93.
Jesse CR, Bortolatto CF, Wilhelm EA, Roman SS, Prigol M, Nogueira CW. The peroxisome proliferator-activated receptor-γ agonist pioglitazone protects against cisplatin-induced renal damage in mice. J Appl Toxicol. 2014;34:25–32.
Yadav DK, Kumar S, Choi EH, Chaudhary S, Kim MH. Molecular dynamic simulations of oxidized skin lipid bilayer and permeability of reactive oxygen species. Sci Rep. 2019;9:4496.
Ramesh G, Reeves WB. Salicylate reduces cisplatin nephrotoxicity by inhibition of tumor necrosis factor-alpha. Kidney Int. 2004;65:490–9.
Zhang B, Ramesh G, Norbury CC, Reeves WB. Cisplatin-induced nephrotoxicity is mediated by tumor necrosis factor-alpha produced by renal parenchymal cells. Kidney Int. 2007;72:37–44.
Ozkok A, Ravichandran K, Wang Q, Ljubanovic D, Edelstein CL. NF-kappa B transcriptional inhibition ameliorates cisplatin-induced acute kidney injury (AKI). Toxicol Lett. 2016;240:105–13.
Ramesh G, Reeves WB. p38 MAP kinase inhibition ameliorates cisplatin nephrotoxicity in mice. Am J Physiol Ren Physiol. 2005;289:F166–74.
dos Santos NA, Carvalho Rodrigues MA, Martins NM, dos Santos AC. Cisplatin-induced nephrotoxicity and targets of nephroprotection: an update. Arch Toxicol. 2012;86:1233–50.
Huang S, You J, Wang K, Li Y, Zhang Y, Wei H, et al. N-acetylcysteine attenuates cisplatin-induced acute kidney injury by inhibiting the C5a receptor. Biomed Res Int. 2019;2019:4805853.
Cummings BS, McHowat J, Schnellmann RG. Role of an endoplasmic reticulum Ca2+-independent phospholipase A2 in cisplatin-induced renal cell apoptosis. J Pharmacol Exp Ther. 2004;308:921–8.
Inoue K, Kuwana H, Shimamura Y, Ogata K, Taniguchi Y, Kagawa T, et al. Cisplatin-induced macroautophagy occurs prior to apoptosis in proximal tubules in vivo. Clin Exp Nephrol. 2010;14:112–22.
Jiang M, Wei Q, Dong G, Komatsu M, Su Y, Dong Z. Autophagy in proximal tubules protects against acute kidney injury. Kidney Int. 2012;82:1271–83.
Kaushal GP. Autophagy protects proximal tubular cells from injury and apoptosis. Kidney Int. 2012;82:1250–3.
Gong L, Pan Q, Yang N. Autophagy and inflammation regulation in acute kidney injury. Front Physiol. 2020;11:576463.
Su Y, Lu J, Gong P, Chen X, Liang C, Zhang J. Rapamycin induces autophagy to alleviate acute kidney injury following cerebral ischemia and reperfusion via the mTORC1/ATG13/ULK1 signaling pathway. Mol Med Rep. 2018;18:5445–54.
Kim TW, Kim YJ, Kim HT, Park SR, Lee MY, Park YD, et al. NQO1 deficiency leads enhanced autophagy in cisplatin-induced acute kidney injury through the AMPK/TSC2/mTOR signaling pathway. Antioxid Redox Signal. 2016;24:867–83.
Katsuda H, Yamashita M, Katsura H, Yu J, Waki Y, Nagata N, et al. Protecting cisplatin-induced nephrotoxicity with cimetidine does not affect antitumor activity. Biol Pharm Bull. 2010;33:1867–71.
Carvalho Rodrigues MA, Gobe G, Santos NA, Santos AC. Carvedilol protects against apoptotic cell death induced by cisplatin in renal tubular epithelial cells. J Toxicol Environ Health A. 2012;75:981–90.
Ozkaya O, Yavuz O, Can B, Dilek M, Savli E, Acikgoz Y, et al. Effect of rosiglitazone on cisplatin-induced nephrotoxicity. Ren Fail. 2010;32:368–71.
Humanes B, Lazaro A, Camano S, Moreno-Gordaliza E, Lazaro JA, Blanco-Codesido M, et al. Cilastatin protects against cisplatin-induced nephrotoxicity without compromising its anticancer efficiency in rats. Kidney Int. 2012;82:652–63.
Duffy EA, Fitzgerald W, Boyle K, Rohatgi R. Nephrotoxicity: evidence in patients receiving cisplatin therapy. Clin J Oncol Nurs. 2018;22:175–83.
Shord SS, Thompson DM, Krempl GA, Hanigan MH. Effect of concurrent medications on cisplatin-induced nephrotoxicity in patients with head and neck cancer. Anticancer Drugs. 2006;17:207–15.
Capizzi RL. Amifostine reduces the incidence of cumulative nephrotoxicity from cisplatin: laboratory and clinical aspects. Semin Oncol. 1999;26:72–81.
Cohen M, Hunter J. Complementary medicine products: interpreting the evidence base. Intern Med J. 2017;47:992–8.
Huang D, Wang C, Duan Y, Meng Q, Liu Z, Huo X, et al. Targeting Oct2 and P53: formononetin prevents cisplatin-induced acute kidney injury. Toxicol Appl Pharmacol. 2017;326:15–24.
Ma X, Yan L, Zhu Q, Shao F. Puerarin attenuates cisplatin-induced rat nephrotoxicity: the involvement of TLR4/NF-kappaB signaling pathway. PLoS One. 2017;12:e0171612.
Athira KV, Madhana RM, Lahkar M. Flavonoids, the emerging dietary supplement against cisplatin-induced nephrotoxicity. Chem Biol Interact. 2016;248:18–20.
Ma P, Zhang S, Su X, Qiu G, Wu Z. Protective effects of icariin on cisplatin-induced acute renal injury in mice. Am J Transl Res. 2015;7:2105–14.
Lou XY, Cheng JL, Zhang B. Therapeutic effect and mechanism of breviscapine on cisplatin-induced nephrotoxicity in mice. Asian Pac J Trop Med. 2015;8:873–7.
Sahin K, Tuzcu M, Gencoglu H, Dogukan A, Timurkan M, Sahin N, et al. Epigallocatechin-3-gallate activates Nrf2/HO-1 signaling pathway in cisplatin-induced nephrotoxicity in rats. Life Sci. 2010;87:240–5.
Kanlaya R, Thongboonkerd V. Protective effects of epigallocatechin-3-gallate from green tea in various kidney diseases. Adv Nutr. 2019;10:112–21.
Kang L, Zhao H, Chen C, Zhang X, Xu M, Duan H. Sappanone A protects mice against cisplatin-induced kidney injury. Int Immunopharmacol. 2016;38:246–51.
Wei Z, He X, Kou J, Wang J, Chen L, Yao M, et al. Renoprotective mechanisms of morin in cisplatin-induced kidney injury. Int Immunopharmacol. 2015;28:500–6.
Tan RZ, Wang C, Deng C, Zhong X, Yan Y, Luo Y, et al. Quercetin protects against cisplatin-induced acute kidney injury by inhibiting Mincle/Syk/NF-kappaB signaling maintained macrophage inflammation. Phytother Res. 2020;34:139–52.
Ninsontia C, Pongjit K, Chaotham C, Chanvorachote P. Silymarin selectively protects human renal cells from cisplatin-induced cell death. Pharm Biol. 2011;49:1082–90.
Tomar A, Kaushik S, Khan SI, Bisht K, Nag TC, Arya DS, et al. The dietary isoflavone daidzein mitigates oxidative stress, apoptosis, and inflammation in CDDP-induced kidney injury in rats: impact of the MAPK signaling pathway. J Biochem Mol Toxicol. 2020;34:e22431.
Li F, Yao YY, Huang H, Hao H, Ying MZ. Xanthohumol attenuates cisplatin-induced nephrotoxicity through inhibiting NF-kappa B and activating Nrf2 signaling pathways. Int Immunopharmacol. 2018;61:277–82.
Badawy AM, El-Naga RN, Gad AM, Tadros MG, Fawzy HM. Wogonin pre-treatment attenuates cisplatin-induced nephrotoxicity in rats: Impact on PPAR-gamma, inflammation, apoptosis and Wnt/beta-catenin pathway. Chem Biol Interact. 2019;308:137–46.
He X, Li C, Wei Z, Wang J, Kou J, Liu W, et al. Protective role of apigenin in cisplatin-induced renal injury. Eur J Pharmacol. 2016;789:215–21.
Huang TH, Wu TH, Guo YH, Li TL, Chan YL, Wu CJ. The concurrent treatment of Scutellaria baicalensis Georgi enhances the therapeutic efficacy of cisplatin but also attenuates chemotherapy-induced cachexia and acute kidney injury. J Ethnopharmacol. 2019;243:112075.
Wu CH, Chen AZ, Yen GC. Protective effects of glycyrrhizic acid and 18beta-Glycyrrhetinic acid against cisplatin-induced nephrotoxicity in BALB/c mice. J Agric Food Chem. 2015;63:1200–9.
Chao CS, Tsai CS, Chang YP, Chen JM, Chin HK, Yang SC. Hyperin inhibits nuclear factor kappa B and activates nuclear factor E2-related factor-2 signaling pathways in cisplatin-induced acute kidney injury in mice. Int Immunopharmacol. 2016;40:517–23.
Li CZ, ** HH, Sun HX, Zhang ZZ, Zheng JX, Li SH, et al. Eriodictyol attenuates cisplatin-induced kidney injury by inhibiting oxidative stress and inflammation. Eur J Pharmacol. 2016;772:124–30.
Sadhukhan P, Saha S, Dutta S, Sil PC. Mangiferin ameliorates cisplatin induced acute kidney injury by upregulating Nrf-2 via the activation of PI3K and exhibits synergistic anticancer activity with cisplatin. Front Pharmacol. 2018;9:638.
Domitrovic R, Cvijanovic O, Pugel EP, Zagorac GB, Mahmutefendic H, Skoda M. Luteolin ameliorates cisplatin-induced nephrotoxicity in mice through inhibition of platinum accumulation, inflammation and apoptosis in the kidney. Toxicology. 2013;310:115–23.
Sung MJ, Kim DH, Jung YJ, Kang KP, Lee AS, Lee S, et al. Genistein protects the kidney from cisplatin-induced injury. Kidney Int. 2008;74:1538–47.
Sun CY, Nie J, Zheng ZL, Zhao J, Wu LM, Zhu Y, et al. Renoprotective effect of scutellarin on cisplatin-induced renal injury in mice: Impact on inflammation, apoptosis, and autophagy. Biomed Pharmacother. 2019;112:108647.
Halliwell B. Are polyphenols antioxidants or pro-oxidants? What do we learn from cell culture and in vivo studies? Arch Biochem Biophys. 2008;476:107–12.
Rawat DS, Thakur BK, Semalty M, Semalty A, Badoni P, Rawat MS. Baicalein-phospholipid complex: a novel drug delivery technology for phytotherapeutics. Curr Drug Discov Technol. 2013;10:224–32.
Ma X, Dang C, Kang H, Dai Z, Lin S, Guan H, et al. Saikosaponin-D reduces cisplatin-induced nephrotoxicity by repressing ROS-mediated activation of MAPK and NF-kappaB signalling pathways. Int Immunopharmacol. 2015;28:399–408.
Li W, Yan MH, Liu Y, Liu Z, Wang Z, Chen C, et al. Ginsenoside Rg5 ameliorates cisplatin-induced nephrotoxicity in mice through inhibition of inflammation, oxidative stress, and apoptosis. Nutrients. 2016;8:566.
Zhang W, Hou J, Yan X, Leng J, Li R, Zhang J, et al. Platycodon grandiflorum saponins ameliorate cisplatin-induced acute nephrotoxicity through the NF-kappaB-mediated inflammation and PI3K/Akt/apoptosis signaling pathways. Nutrients. 2018;10:1328.
Sherif IO. Amelioration of cisplatin-induced nephrotoxicity in rats by triterpenoid saponin of Terminalia arjuna. Clin Exp Nephrol. 2015;19:591–7.
Han MS, Han IH, Lee D, An JM, Kim SN, Shin MS, et al. Beneficial effects of fermented black ginseng and its ginsenoside 20(S)-Rg3 against cisplatin-induced nephrotoxicity in LLC-PK1 cells. J Ginseng Res. 2016;40:135–40.
**ng JJ, Hou JG, Ma ZN, Wang Z, Ren S, Wang YP, et al. Ginsenoside Rb3 provides protective effects against cisplatin-induced nephrotoxicity via regulation of AMPK-/mTOR-mediated autophagy and inhibition of apoptosis in vitro and in vivo. Cell Prolif. 2019;52:e12627.
Zhang Y, Tao X, Yin L, Xu L, Xu Y, Qi Y, et al. Protective effects of dioscin against cisplatin-induced nephrotoxicity via the microRNA-34a/sirtuin 1 signalling pathway. Br J Pharmacol. 2017;174:2512–27.
Kang HR, Lee D, Eom HJ, Lee SR, Lee KR, Kang KS, et al. Identification and mechanism of action of renoprotective constituents from peat moss Sphagnum palustre in cisplatin-induced nephrotoxicity. J Funct Foods. 2016;20:358–68.
Jung K, Lee D, Yu JS, Namgung H, Kang KS, Kim KH. Protective effect and mechanism of action of saponins isolated from the seeds of gac (Momordica cochinchinensis Spreng.) against cisplatin-induced damage in LLC-PK1 kidney cells. Bioorg Med Chem Lett. 2016;26:1466–70.
Adewale OO, Oduyemi OI, Ayokunle O. Oral administration of leaf extracts of Momordica charantia affect reproductive hormones of adult female Wistar rats. Asian Pac J Trop Biomed. 2014;4:S521–4.
Tam PP, Law LK, Yeung HW. Effects of alpha-momorcharin on preimplantation development in the mouse. J Reprod Fertil. 1984;71:33–8.
Patel JC, Dhirawani MK, Doshi JC. “Karella” in the treatment of diabetes mellitus. Indian J Med Sci. 1968;22:30–2.
Feng L, Ke N, Cheng F, Guo Y, Li S, Li Q, et al. The protective mechanism of ligustrazine against renal ischemia/reperfusion injury. J Surg Res. 2011;166:298–305.
Michel HE, Menze ET. Tetramethylpyrazine guards against cisplatin-induced nephrotoxicity in rats through inhibiting HMGB1/TLR4/NF-kappaB and activating Nrf2 and PPAR-gamma signaling pathways. Eur J Pharmacol. 2019;857:172422.
Yadav YC, Srivastava DN. Nephroprotective and curative effects of Ficus religiosa latex extract against cisplatin-induced acute renal failure. Pharm Biol. 2013;51:1480–5.
Domitrovic R, Cvijanovic O, Pernjak-Pugel E, Skoda M, Mikelic L, Crncevic-Orlic Z. Berberine exerts nephroprotective effect against cisplatin-induced kidney damage through inhibition of oxidative/nitrosative stress, inflammation, autophagy and apoptosis. Food Chem Toxicol. 2013;62:397–406.
Hagar H, Medany AE, Salam R, Medany GE, Nayal OA. Betaine supplementation mitigates cisplatin-induced nephrotoxicity by abrogation of oxidative/nitrosative stress and suppression of inflammation and apoptosis in rats. Exp Toxicol Pathol. 2015;67:133–41.
Chan TY. Aconite poisoning. Clin Toxicol (Philos). 2009;47:279–85.
Wu JJ, Guo ZZ, Zhu YF, Huang ZJ, Gong X, Li YH, et al. A systematic review of pharmacokinetic studies on herbal drug Fuzi: Implications for Fuzi as personalized medicine. Phytomedicine. 2018;44:187–203.
Stegelmeier BL, Colegate SM, Brown AW. Dehydropyrrolizidine alkaloid toxicity, cytotoxicity, and carcinogenicity. Toxins (Basel). 2016;8:356.
Chen Q, Peng HX, Dong L, Chen LJ, Ma XB, Peng YP, et al. Activation of the NRF2-ARE signalling pathway by the Lentinula edodes polysaccharose LNT alleviates ROS-mediated cisplatin nephrotoxicity. Int Immunopharmacol. 2016;36:1–8.
Rjeibi I, Feriani A, Ben Saad A, Sdayria J, Saidi I, Ncib S, et al. Lycium europaeum extract: a new potential antioxidant source against cisplatin-induced liver and kidney injuries in mice. Oxid Med Cell Longev. 2018;2018:1630751.
Rjeibi I, Feriani A, Ben Saad A, Ncib S, Sdayria J, Hfaiedh N, et al. Lycium europaeum Linn as a source of polysaccharide with in vitro antioxidant activities and in vivo anti-inflammatory and hepato-nephroprotective potentials. J Ethnopharmacol. 2018;225:116–27.
Liu Q, Song J, Li H, Dong L, Dai S. Schizandrin B inhibits the cisDDP-induced apoptosis of HK2 cells by activating ERK/NFkappaB signaling to regulate the expression of survivin. Int J Mol Med. 2018;41:2108–16.
Mundhe NA, Kumar P, Ahmed S, Jamdade V, Mundhe S, Lahkar M. Nordihydroguaiaretic acid ameliorates cisplatin induced nephrotoxicity and potentiates its anti-tumor activity in DMBA induced breast cancer in female Sprague-Dawley rats. Int Immunopharmacol. 2015;28:634–42.
** J, Li M, Zhao Z, Sun X, Li J, Wang W, et al. Protective effect of Wuzhi tablet (Schisandra sphenanthera extract) against cisplatin-induced nephrotoxicity via Nrf2-mediated defense response. Phytomedicine. 2015;22:528–35.
Biessels GJ, Vermeij FH, Leijten FS. [Epileptic seizure after a cup of tea: intoxication with Japanese star anise]. Ned Tijdschr Geneeskd. 2002;146:808–11.
Potocnjak I, Domitrovic R. Carvacrol attenuates acute kidney injury induced by cisplatin through suppression of ERK and PI3K/Akt activation. Food Chem Toxicol. 2016;98:251–61.
Zang H, Yang Q, Li J. Eleutheroside B Protects against acute kidney injury by activating IGF pathway. Molecules. 2019;24:3876.
Domitrovic R, Cvijanovic O, Susnic V, Katalinic N. Renoprotective mechanisms of chlorogenic acid in cisplatin-induced kidney injury. Toxicology. 2014;324:98–107.
Gao L, Wu WF, Dong L, Ren GL, Li HD, Yang Q, et al. Protocatechuic aldehyde attenuates cisplatin-induced acute kidney injury by suppressing Nox-mediated oxidative stress and renal inflammation. Front Pharmacol. 2016;7:479.
Eren H, Aydin HR, Tumkaya L, Kazaz IO, Kalkan Y, Kazaz SN, et al. Whortleberry protects kidney against the cisplatin-induced nephrotoxicity: an experimental study. Ren Fail. 2018;40:466–74.
Nho JH, Jung HK, Lee MJ, Jang JH, Sim MO, Jeong DE, et al. Beneficial effects of cynaroside on cisplatin-induced kidney injury in vitro and in vivo. Toxicol Res. 2018;34:133–41.
Zhang R, Yin L, Zhang B, Shi H, Sun Y, Ji C, et al. Resveratrol improves human umbilical cord-derived mesenchymal stem cells repair for cisplatin-induced acute kidney injury. Cell Death Dis. 2018;9:965.
Kim JS, Kim KS, Son JY, Kim HR, Park JH, Lee SH, et al. Protective effects of Dendropanax morbifera against cisplatin-induced nephrotoxicity without altering chemotherapeutic efficacy. Antioxidants (Basel). 2019;8:256.
Dehnamaki F, Karimi A, Pilevarian AA, Fatemi I, Hakimizadeh E, Kaeidi A, et al. Treatment with troxerutin protects against cisplatin-induced kidney injury in mice. Acta Chir Belg. 2019;119:31–7.
Zhou P, Wu M, Ye C, Xu Q, Wang L. Meclofenamic acid promotes cisplatin-induced acute kidney injury by inhibiting fat mass and obesity-associated protein-mediated m(6)A abrogation in RNA. J Biol Chem. 2019;294:16908–17.
Sen Z, Jie M, **gzhi Y, Dongjie W, Dongming Z, **aoguang C. Total coumarins from Hydrangea paniculata protect against cisplatin-induced acute kidney damage in mice by suppressing renal inflammation and apoptosis. Evid Based Complement Altern Med. 2017;2017:5350161.
Yang C, Guo Y, Huang TS, Zhao J, Huang XJ, Tang HX, et al. Asiatic acid protects against cisplatin-induced acute kidney injury via anti-apoptosis and anti-inflammation. Biomed Pharmacother. 2018;107:1354–62.
Yu X, Meng X, Xu M, Zhang X, Zhang Y, Ding G, et al. Celastrol ameliorates cisplatin nephrotoxicity by inhibiting NF-kappaB and improving mitochondrial function. EBioMedicine. 2018;36:266–80.
Ansari MA. Sinapic acid modulates Nrf2/HO-1 signaling pathway in cisplatin-induced nephrotoxicity in rats. Biomed Pharmacother. 2017;93:646–53.
Zhang L, Gu Y, Li H, Cao H, Liu B, Zhang H, et al. Daphnetin protects against cisplatin-induced nephrotoxicity by inhibiting inflammatory and oxidative response. Int Immunopharmacol. 2018;65:402–7.
Kandemir FM, Yildirim S, Caglayan C, Kucukler S, Eser G. Protective effects of zingerone on cisplatin-induced nephrotoxicity in female rats. Environ Sci Pollut Res Int. 2019;26:22562–74.
Tilyek A, Chai C, Hou X, Zhou B, Zhang C, Cao Z, et al. The protective effects of Ribes diacanthum Pall on cisplatin-induced nephrotoxicity in mice. J Ethnopharmacol. 2016;178:297–306.
Yarijani ZM, Godini A, Madani SH, Najafi H. Reduction of cisplatin-induced renal and hepatic side effects in rat through antioxidative and anti-inflammatory properties of Malva sylvestris L. extract. Biomedicine Pharmacother. 2018;106:1767–74.
Gulec M, Iraz M, Yilmaz HR, Ozyurt H, Temel I. The effects of Ginkgo biloba extract on tissue adenosine deaminase, xanthine oxidase, myeloperoxidase, malondialdehyde, and nitric oxide in cisplatin-induced nephrotoxicity. Toxicol Ind Health. 2006;22:125–30.
Karwasra R, Kalra P, Gupta YK, Saini D, Kumar A, Singh S. Antioxidant and anti-inflammatory potential of pomegranate rind extract to ameliorate cisplatin-induced acute kidney injury. Food Funct. 2016;7:3091–101.
Jamshidzadeh A, Heidari R, Golzar T, Derakhshanfar A. Effect of Eisenia foetida extract against cisplatin-induced kidney injury in rats. J Diet Suppl. 2016;13:551–9.
Guo Y, Wang M, Mou J, Zhao Z, Yang J, Zhu F, et al. Pretreatment of Huaiqihuang extractum protects against cisplatin-induced nephrotoxicity. Sci Rep. 2018;8:7333.
Zhou L, Wei XH, Pan CS, Yan L, Gu YY, Sun K, et al. QiShenYiQi Pills, a compound Chinese medicine, prevented cisplatin induced acute kidney injury via regulating mitochondrial function. Front Physiol. 2017;8:1090.
Bratkov VM, Shkondrov AM, Zdraveva PK, Krasteva IN. Flavonoids from the Genus Astragalus: phytochemistry and biological activity. Pharmacogn Rev. 2016;10:11–32.
Sarker SD, Nahar L. An introduction to natural products isolation. Methods Mol Biol. 2012;864:1–25.
D’Orazio N, Gemello E, Gammone MA, de Girolamo M, Ficoneri C, Riccioni G. Fucoxantin: a treasure from the sea. Mar Drugs. 2012;10:604–16.
Shi MJ, McMillan KL, Wu JX, Gillings N, Flores B, Moe OW, et al. Cisplatin nephrotoxicity as a model of chronic kidney disease. Lab Invest. 2018;98:1105–21.
Sharp CN, Doll MA, Megyesi J, Oropilla GB, Beverly LJ, Siskind LJ. Subclinical kidney injury induced by repeated cisplatin administration results in progressive chronic kidney disease. Am J Physiol Ren Physiol. 2018;315:F161–72.
Landau SI, Guo X, Velazquez H, Torres R, Olson E, Garcia-Milian R, et al. Regulated necrosis and failed repair in cisplatin-induced chronic kidney disease. Kidney Int. 2019;95:797–814.
Mercantepe F, Mercantepe T, Topcu A, Yilmaz A, Tumkaya L. Protective effects of amifostine, curcumin, and melatonin against cisplatin-induced acute kidney injury. Naunyn Schmiedebergs Arch Pharmacol. 2018;391:915–31.
Ortega-Dominguez B, Aparicio-Trejo OE, Garcia-Arroyo FE, Leon-Contreras JC, Tapia E, Molina-Jijon E, et al. Curcumin prevents cisplatin-induced renal alterations in mitochondrial bioenergetics and dynamic. Food Chem Toxicol. 2017;107:373–85.
Topcu-Tarladacalisir Y, Sapmaz-Metin M, Karaca T. Curcumin counteracts cisplatin-induced nephrotoxicity by preventing renal tubular cell apoptosis. Ren Fail. 2016;38:1741–8.
Ugur S, Ulu R, Dogukan A, Gurel A, Yigit IP, Gozel N, et al. The renoprotective effect of curcumin in cisplatin-induced nephrotoxicity. Ren Fail. 2015;37:332–6.
Soetikno V, Sari SDP, Ul Maknun L, Sumbung NK, Rahmi DNI, Pandhita BAW, et al. Pre-treatment with Curcumin ameliorates cisplatin-induced kidney damage by suppressing kidney inflammation and apoptosis in rats. Drug Res (Stuttg). 2019;69:75–82.
Sayin VI, Ibrahim MX, Larsson E, Nilsson JA, Lindahl P, Bergo MO. Antioxidants accelerate lung cancer progression in mice. Sci Transl Med. 2014;6:221ra15.
Sawyer D, Conner CS, Scalley R. Cimetidine: adverse reactions and acute toxicity. Am J Hosp Pharm. 1981;38:188–97.
Tejedor A, Torres AM, Castilla M, Lazaro JA, de Lucas C, Caramelo C. Cilastatin protection against cyclosporin A-induced nephrotoxicity: clinical evidence. Curr Med Res Opin. 2007;23:505–13.
Crona DJ, Faso A, Nishijima TF, McGraw KA, Galsky MD, Milowsky MI. A systematic review of strategies to prevent cisplatin-induced nephrotoxicity. Oncologist. 2017;22:609–19.
Chtourou Y, Aouey B, Aroui S, Kebieche M, Fetoui H. Anti-apoptotic and anti-inflammatory effects of naringin on cisplatin-induced renal injury in the rat. Chem Biol Interact. 2016;243:1–9.
Amini N, Sarkaki A, Dianat M, Mard SA, Ahangarpour A, Badavi M. Protective effects of naringin and trimetazidine on remote effect of acute renal injury on oxidative stress and myocardial injury through Nrf-2 regulation. Pharmacol Rep. 2019;71:1059–66.
Pan H, Chen J, Shen K, Wang X, Wang P, Fu G, et al. Mitochondrial modulation by Epigallocatechin 3-Gallate ameliorates cisplatin induced renal injury through decreasing oxidative/nitrative stress, inflammation and NF-κB in mice. PLoS One. 2015;10:e0124775.
Fatima S, Al-Mohaimeed N, Al-Shaikh Y, Tyagi P, Banu N, Hasan S, et al. Combined treatment of epigallocatechin gallate and coenzyme Q10 attenuates cisplatin-induced nephrotoxicity via suppression of oxidative/nitrosative stress, inflammation and cellular damage. Food Chem Toxicol. 2016;94:213–20.
Singh MP, Chauhan AK, Kang SC. Morin hydrate ameliorates cisplatin-induced ER stress, inflammation and autophagy in HEK-293 cells and mice kidney via PARP-1 regulation. Int Immunopharmacol. 2018;56:156–67.
Lin CF, Kuo YT, Chen TY, Chien CT. Quercetin-rich guava (Psidium guajava) juice in combination with trehalose reduces autophagy, apoptosis and pyroptosis formation in the kidney and pancreas of type II diabetic rats. Molecules. 2016;21:334.
Wang C, Pan Y, Zhang QY, Wang FM, Kong LD. Quercetin and allopurinol ameliorate kidney injury in STZ-treated rats with regulation of renal NLRP3 inflammasome activation and lipid accumulation. PLoS One. 2012;7:e38285.
Al Humayed S, Al-Ani B, El Karib AO, Shatoor AS, Eid RA, Aziz S, et al. Suppression of acetaminophen-induced hepatocyte ultrastructural alterations in rats using a combination of resveratrol and quercetin. Ultrastruct Pathol. 2019;43:162–9.
Milton Prabu S, Shagirtha K, Renugadevi J. Quercetin in combination with vitamins (C and E) improves oxidative stress and renal injury in cadmium intoxicated rats. Eur Rev Med Pharm Sci. 2010;14:903–14.
Impellizzeri D, Bruschetta G, Ahmad A, Crupi R, Siracusa R, Di Paola R, et al. Effects of palmitoylethanolamide and silymarin combination treatment in an animal model of kidney ischemia and reperfusion. Eur J Pharmacol. 2015;762:136–49.
Meng XM, Li HD, Wu WF, Ming-Kuen Tang P, Ren GL, Gao L, et al. Wogonin protects against cisplatin-induced acute kidney injury by targeting RIPK1-mediated necroptosis. Lab Invest. 2018;98:79–94.
Shimizu T, Shibuya N, Narukawa Y, Oshima N, Hada N, Kiuchi F. Synergistic effect of baicalein, wogonin and oroxylin A mixture: multistep inhibition of the NF-kappaB signalling pathway contributes to an anti-inflammatory effect of Scutellaria root flavonoids. J Nat Med. 2018;72:181–91.
Sahu BD, Mahesh Kumar J, Sistla R. Baicalein, a bioflavonoid, prevents cisplatin-induced acute kidney injury by up-regulating antioxidant defenses and down-regulating the MAPKs and NF-kappaB pathways. PLoS One. 2015;10:e0134139.
Tang D, Chen K, Huang L, Li J. Pharmacokinetic properties and drug interactions of apigenin, a natural flavone. Expert Opin Drug Metab Toxicol. 2017;13:323–30.
Vasaikar N, Mahajan U, Patil KR, Suchal K, Patil CR, Ojha S, et al. D-pinitol attenuates cisplatin-induced nephrotoxicity in rats: Impact on pro-inflammatory cytokines. Chem Biol Interact. 2018;290:6–11.
Sahu AK, Verma VK, Mutneja E, Malik S, Nag TC, Dinda AK, et al. Mangiferin attenuates cisplatin-induced acute kidney injury in rats mediating modulation of MAPK pathway. Mol Cell Biochem. 2019;452:141–52.
Abd-Elhakim YM, Ghoneim MH, Ebraheim LLM, Imam TS. Taurine and hesperidin rescues carbon tetrachloride-triggered testicular and kidney damage in rats via modulating oxidative stress and inflammation. Life Sci. 2020;254:117782.
Chen X, Wei W, Li Y, Huang J, Ci X. Hesperetin relieves cisplatin-induced acute kidney injury by mitigating oxidative stress, inflammation and apoptosis. Chem Biol Interact. 2019;308:269–78.
Tomar A, Vasisth S, Khan SI, Malik S, Nag TC, Arya DS, et al. Galangin ameliorates cisplatin induced nephrotoxicity in vivo by modulation of oxidative stress, apoptosis and inflammation through interplay of MAPK signaling cascade. Phytomedicine. 2017;34:154–61.
Abdel-Daim MM, Abdellatief SA. Attenuating effects of caffeic acid phenethyl ester and betaine on abamectin-induced hepatotoxicity and nephrotoxicity. Environ Sci Pollut Res Int. 2018;25:15909–17.
Suliman FA, Khodeer DM, Ibrahiem A, Mehanna ET, El-Kherbetawy MK, Mohammad HMF, et al. Renoprotective effect of the isoflavonoid biochanin A against cisplatin induced acute kidney injury in mice: effect on inflammatory burden and p53 apoptosis. Int Immunopharmacol. 2018;61:8–19.
Alauddin ChaturvediS, Malik MY, Azmi L, Shukla I, Naseem Z, et al. Formononetin and biochanin A protects against ritonavir induced hepatotoxicity via modulation of NF kappa B/pAkt signaling molecules. Life Sci. 2018;213:174–82.
Yuan Y, Zha H, Rangarajan P, Ling EA, Wu C. Anti-inflammatory effects of Edaravone and Scutellarin in activated microglia in experimentally induced ischemia injury in rats and in BV-2 microglia. BMC Neurosci. 2014;15:125.
Qi ZL, Wang Z, Li W, Hou JG, Liu Y, Li XD, et al. Nephroprotective effects of anthocyanin from the fruits of Panax ginseng (GFA) on cisplatin-induced acute kidney injury in mice. Phytother Res. 2017;31:1400–9.
Gao S, Li L, Li L, Ni J, Guo R, Mao J, et al. Effects of the combination of tanshinone IIA and puerarin on cardiac function and inflammatory response in myocardial ischemia mice. J Mol Cell Cardiol. 2019;137:59–70.
Gao T, Zhao P, Yu XL, Cao SX, Zhang B, Dai M. Use of Saikosaponin D and JNK inhibitor SP600125, alone or in combination, inhibits malignant properties of human osteosarcoma U2 cells. Am J Transl Res. 2019;11:2070–80.
Zhao Q, Yang M, Deng YP, Yu HT, Wang LL, Teng FK, et al. The safety evaluation of salvianolic acid B and ginsenoside Rg1 combination on mice. Int J Mol Sci. 2015;16:29345–56.
Yokozawa T, Liu ZW. The role of ginsenoside-Rd in cisplatin-induced acute renal failure. Ren Fail. 2000;22:115–27.
Kim YJ, Lee MY, Son HY, Park BK, Ryu SY, Jung JY. Red ginseng ameliorates acute cisplatin-induced nephropathy. Planta Med. 2014;80:645–54.
Wang Z, Li YF, Han XY, Sun YS, Zhang LX, Liu W, et al. Kidney protection effect of ginsenoside Re and its underlying mechanisms on cisplatin-induced kidney injury. Cell Physiol Biochem. 2018;48:2219–29.
Qi Z, Li Z, Li W, Liu Y, Wang C, Lin H, et al. Pseudoginsengenin DQ exhibits therapeutic effects in cisplatin-induced acute kidney injury via Sirt1/NF-kappaB and caspase signaling pathway without compromising its antitumor activity in mice. Molecules. 2018;23:3038.
Qi Z, Li W, Tan J, Wang C, Lin H, Zhou B, et al. Effect of ginsenoside Rh2 on renal apoptosis in cisplatin-induced nephrotoxicity in vivo. Phytomedicine. 2019;61:152862.
Liang X, Yang Y, Huang Z, Zhou J, Li Y, Zhong X. Panax notoginseng saponins mitigate cisplatin induced nephrotoxicity by inducing mitophagy via HIF-1alpha. Oncotarget. 2017;8:102989–3003.
Tian Z, Pang H, Du S, Lu Y, Zhang L, Wu H, et al. Effect of Panax notoginseng saponins on the pharmacokinetics of aspirin in rats. J Chromatogr B Anal Technol Biomed Life Sci. 2017;1040:136–43.
Ma ZN, Liu Z, Wang Z, Ren S, Tang S, Wang YP, et al. Supplementation of American ginseng berry extract mitigated cisplatin-evoked nephrotoxicity by suppressing ROS-mediated activation of MAPK and NF-kappaB signaling pathways. Food Chem Toxicol. 2017;110:62–73.
Wang L, Meng Q, Wang C, Liu Q, Peng J, Huo X, et al. Dioscin restores the activity of the anticancer agent adriamycin in multidrug-resistant human leukemia K562/adriamycin cells by down-regulating MDR1 via a mechanism involving NF-kappaB signaling inhibition. J Nat Prod. 2013;76:909–14.
Chen L, Liu T, Wang Q, Liu J. Anti-inflammatory effect of combined tetramethylpyrazine, resveratrol and curcumin in vivo. BMC Complement Alter Med. 2017;17:233.
Zhang B, Lu C, Bai M, He X, Tan Y, Bian Y, et al. Tetramethylpyrazine identified by a network pharmacology approach ameliorates methotrexate-induced oxidative organ injury. J Ethnopharmacol. 2015;175:638–47.
Hussien NR, Al-Kuraishy HM, Al-Gareeb AI. Berberine and Pentoxifylline: a novel combination in amelioration of acute kidney injury. J Pak Med Assoc. 2019;69(Suppl 3):S98–S7.
Chen XY, Zhang Y, Zhu ZN, Liu HL, Guo HC, **ong C, et al. Protective effect of berberine on doxorubicin-induced acute hepatorenal toxicity in rats. Mol Med Rep. 2016;13:3953–60.
Sun M, Zhao W, **e Q, Zhan Y, Wu B. Lentinan reduces tumor progression by enhancing gemcitabine chemotherapy in urothelial bladder cancer. Surg Oncol. 2015;24:28–34.
Chen W, Cheng X, Chen J, Yi X, Nie D, Sun X, et al. Lycium barbarum polysaccharides prevent memory and neurogenesis impairments in scopolamine-treated rats. PLoS One. 2014;9:e88076.
Bunel V, Antoine MH, Nortier J, Duez P, Stevigny C. Protective effects of schizandrin and schizandrin B towards cisplatin nephrotoxicity in vitro. J Appl Toxicol. 2014;34:1311–9.
Li YJ, Liu HT, Xue CJ, **ng XQ, Dong ST, Wang LS, et al. The synergistic anti-tumor effect of schisandrin B and apatinib. J Asian Nat Prod Res. 2020;22:839–49.
Lee DW, Kwak IS, Lee SB, Song SH, Seong EY, Yang BY, et al. Post-treatment effects of erythropoietin and nordihydroguaiaretic acid on recovery from cisplatin-induced acute renal failure in the rat. J Korean Med Sci. 2009;24(Suppl):S170–5.
Li JL, Chen SY, Qin XL, Fu Q, Bi HC, Zhang Y, et al. Wuzhi tablet (Schisandra sphenanthera extract) is a promising tacrolimus-sparing agent for renal transplant recipients who are CYP3A5 expressers: a two-phase prospective study. Drug Metab Dispos. 2017;45:1114–9.
Qu X, Gao H, Tao L, Zhang Y, Zhai J, Sun J, et al. Astragaloside IV protects against cisplatin-induced liver and kidney injury via autophagy-mediated inhibition of NLRP3 in rats. J Toxicol Sci. 2019;44:167–75.
Bunel V, Antoine MH, Nortier J, Duez P, Stevigny C. Nephroprotective effects of ferulic acid, Z-ligustilide and E-ligustilide isolated from Angelica sinensis against cisplatin toxicity in vitro. Toxicol Vitr. 2015;29:458–67.
Masoumi F, Youssefi MR, Tabari MA. Combination of carvacrol and thymol against the poultry red mite (Dermanyssus gallinae). Parasitol Res. 2016;115:4239–43.
Diamond BJ, Shiflett SC, Feiwel N, Matheis RJ, Noskin O, Richards JA, et al. Ginkgo biloba extract: mechanisms and clinical indications. Arch Phys Med Rehabil. 2000;81:668–78.
Lee IC, Ko JW, Park SH, Shin NR, Shin IS, Kim YB, et al. Ameliorative effects of pine bark extract on cisplatin-induced acute kidney injury in rats. Ren Fail. 2017;39:363–71.
Potocnjak I, Broznic D, Kindl M, Kropek M, Vladimir-Knezevic S, Domitrovic R. Stevia and stevioside protect against cisplatin nephrotoxicity through inhibition of ERK1/2, STAT3, and NF-kappa B activation. Food Chem Toxicol. 2017;107:215–25.
Geyikoglu F, Emir M, Colak S, Koc K, Turkez H, Bakir M, et al. Effect of oleuropein against chemotherapy drug-induced histological changes, oxidative stress, and DNA damages in rat kidney injury. J Food Drug Anal. 2017;25:447–59.
Przychodzen P, Wyszkowska R, Gorzynik-Debicka M, Kostrzewa T, Kuban-Jankowska A, Gorska-Ponikowska M. Anticancer potential of oleuropein, the polyphenol of olive oil, with 2-methoxyestradiol, separately or in combination, in human osteosarcoma cells. Anticancer Res. 2019;39:1243–51.
Elseweidy MM, Askar ME, Elswefy SE, Shawky M. Vanillin as a new modulator candidate for renal injury induced by cisplatin in experimental rats. Cytokine. 2017;99:260–5.
Nasr S, Varshosaz J, Hajhashemi V. Ortho-vanillin nanoparticle-doped glucan microspheres exacerbate the anti-arthritic effects of methotrexate in adjuvant-induced arthritis in rats. Pharmacol Rep. 2020;72:680–91.
Ounjaijean S, Somsak V. Combination of zingerone and dihydroartemisinin presented synergistic antimalarial activity against Plasmodium berghei infection in BALB/c mice as in vivo model. Parasitol Int. 2020;76:102088.
Yan SL, Wang ZH, Mong MC, Yang YC, Yin MC. Combination of carnosine and asiatic acid provided greater anti-inflammatory protection for HUVE cells and diabetic mice than individual treatments of carnosine or asiatic acid alone. Food Chem Toxicol. 2019;126:192–8.
Yan YY, Bi H, Zhang W, Wen Q, Liu H, Li JX, et al. Downregulation and subcellular distribution of HER2 involved in MDA-MB-453 breast cancer cell apoptosis induced by lapatinib/celastrol combination. J Buon. 2017;22:644–51.
Zhang JQ, Yao ZT, Liang GK, Chen X, Wu HH, ** L, et al. [Combination of lapatinib with chlorogenic acid inhibits breast cancer metastasis by suppressing macrophage M2 polarization]. Zhejiang Da Xue Xue Bao Yi Xue Ban. 2015;44:493–9.
Potocnjak I, Simic L, Vukelic I, Domitrovic R. Oleanolic acid attenuates cisplatin-induced nephrotoxicity in mice and chemosensitizes human cervical cancer cells to cisplatin cytotoxicity. Food Chem Toxicol. 2019;132:110676.
Minich DM, Bland JS, Katke J, Darland G, Hall A, Lerman RH, et al. Clinical safety and efficacy of NG440: a novel combination of rho iso-alpha acids from hops, rosemary, and oleanolic acid for inflammatory conditions. Can J Physiol Pharmacol. 2007;85:872–83.
Khan SA, Priyamvada S, Khan W, Khan S, Farooq N, Yusufi AN. Studies on the protective effect of green tea against cisplatin induced nephrotoxicity. Pharmacol Res. 2009;60:382–91.
Sahu BD, Rentam KK, Putcha UK, Kuncha M, Vegi GM, Sistla R. Carnosic acid attenuates renal injury in an experimental model of rat cisplatin-induced nephrotoxicity. Food Chem Toxicol. 2011;49:3090–7.
Shi B, Wang LF, Meng WS, Chen L, Meng ZL. Carnosic acid and fisetin combination therapy enhances inhibition of lung cancer through apoptosis induction. Int J Oncol. 2017;50:2123–35.
Liu H, Gu LB, Tu Y, Hu H, Huang YR, Sun W. Emodin ameliorates cisplatin-induced apoptosis of rat renal tubular cells in vitro by activating autophagy. Acta Pharmacol Sin. 2016;37:235–45.
Chen Y, Gan D, Huang Q, Luo X, Lin D, Hu J. Emodin and its combination with cytarabine induce apoptosis in resistant acute myeloid leukemia cells in vitro and in vivo. Cell Physiol Biochem. 2018;48:2061–73.
Hegazy MG, Emam MA. Ethanolic extract of Trigonella Foenum Graecum attenuates cisplatin-induced nephro- and hepatotoxicities in rats. Cell Mol Biol (Noisy-le-Gd). 2015;61:81–7.
Pradeep SR, Barman S, Srinivasan K. Attenuation of diabetic nephropathy by dietary fenugreek (Trigonella foenum-graecum) seeds and onion (Allium cepa) via suppression of glucose transporters and renin-angiotensin system. Nutrition. 2019;67-68:110543.
Jiang L, Liu Y, He P, Chen J, Liu S, Tan N. Geraniin ameliorates cisplatin-induced nephrotoxicity in mice. Free Radic Res. 2016;50:813–9.
Boakye-Gyasi E, Kasanga EA, Ameyaw EO, Abotsi WKM, Biney RP, Agyare C, et al. An isobolographic analysis of the anti-nociceptive effect of geraniin in combination with morphine or diclofenac. J Basic Clin Physiol Pharmacol. 2018;29:201–9.
Divya MK, Lincy L, Raghavamenon AC, Babu TD. Ameliorative effect of Apodytes dimidiata on cisplatin-induced nephrotoxicity in Wistar rats. Pharm Biol. 2016;54:2149–57.
Mahmoodnia L, Mohammadi K, Masumi R. Ameliorative effect of lycopene effect on cisplatin-induced nephropathy in patient. J Nephropathol. 2017;6:144–9.
Bayomy NA, Elbakary RH, Ibrahim MAA, Abdelaziz EZ. Effect of lycopene and rosmarinic acid on gentamicin induced renal cortical oxidative stress, apoptosis, and autophagy in adult male albino rat. Anat Rec (Hoboken). 2017;300:1137–49.
Mahgoub E, Kumaraswamy SM, Kader KH, Venkataraman B, Ojha S, Adeghate E, et al. Genipin attenuates cisplatin-induced nephrotoxicity by counteracting oxidative stress, inflammation, and apoptosis. Biomed Pharmacother. 2017;93:1083–97.
Kim BR, Jeong YA, Jo MJ, Park SH, Na YJ, Kim JL, et al. Genipin enhances the therapeutic effects of oxaliplatin by upregulating BIM in colorectal cancer. Mol Cancer Ther. 2019;18:751–61.
Huang H, Shen Z, Geng Q, Wu Z, Shi P, Miao X. Protective effect of Schisandra chinensis bee pollen extract on liver and kidney injury induced by cisplatin in rats. Biomed Pharmacother. 2017;95:1765–76.
Li YZ, Ren S, Yan XT, Li HP, Li W, Zheng B, et al. Improvement of cisplatin-induced renal dysfunction by Schisandra chinensis stems via anti-inflammation and anti-apoptosis effects. J Ethnopharmacol. 2018;217:228–37.
Katanic J, Matic S, Pferschy-Wenzig EM, Kretschmer N, Boroja T, Mihailovic V, et al. Filipendula ulmaria extracts attenuate cisplatin-induced liver and kidney oxidative stress in rats: In vivo investigation and LC-MS analysis. Food Chem Toxicol. 2017;99:86–102.
Hosseinian S, Khajavi Rad A, Hadjzadeh MA, Mohamadian Roshan N, Havakhah S, Shafiee S. The protective effect of Nigella sativa against cisplatin-induced nephrotoxicity in rats. Avicenna J Phytomed. 2016;6:44–54.
Mohebbati R, Shafei MN, Beheshti F, Soukhtanloo M, Roshan NM, Anaeigoudari A, et al. Mixed hydroalcoholic extracts of Nigella sativa and Curcuma longa improves adriamycin-induced renal injury in rat. Saudi J Kidney Dis Transpl. 2017;28:1270–81.
Farooqui Z, Ahmed F, Rizwan S, Shahid F, Khan AA, Khan F. Protective effect of Nigella sativa oil on cisplatin induced nephrotoxicity and oxidative damage in rat kidney. Biomed Pharmacother. 2017;85:7–15.
Al-Okbi SY, Mohamed DA, Hamed TE, Edris AE, Fouda K. Hepatic regeneration and reno-protection by fish oil, Nigella sativa Oil and combined fish oil/Nigella sativa volatiles in CCl4 treated rats. J Oleo Sci. 2018;67:345–53.
Cao SS, Yan M, Hou ZY, Chen Y, Jiang YS, Fan XR, et al. Danshen modulates Nrf2-mediated signaling pathway in cisplatin-induced renal injury. J Huazhong Univ Sci Technol Med Sci. 2017;37:761–5.
Guan Y, Wu XX, Duan JL, Yin Y, Guo C, Wei G, et al. Effects and mechanism of combination of rhein and danshensu in the treatment of chronic kidney disease. Am J Chin Med. 2015;43:1381–400.
Parhizgar S, Hosseinian S, Hadjzadeh MA, Soukhtanloo M, Ebrahimzadeh A, Mohebbati R, et al. Renoprotective effect of Plantago major against nephrotoxicity and oxidative stress induced by cisplatin. Iran J Kidney Dis. 2016;10:182–8.
Parhizgar S, Hosseinian S, Soukhtanloo M, Bideskan AE, Hadjzadeh MA, Shahraki S, et al. Plantago major protects against cisplatin-induced renal dysfunction and tissue damage in rats. Saudi J Kidney Dis Transpl. 2018;29:1057–64.
Adeyemi OO, Ishola IO, Ajani ID. Citrullus colocynthis Linn. fruit extract ameliorates cisplatin-induced hepato-renal toxicity in rats. J Complement Integr Med. 2017;15:jcim-2017-0086.
Adeoye BO, Asenuga ER, Oyagbemi AA, Omobowale TO, Adedapo AA. The protective effect of the ethanol leaf extract of Andrographis paniculata on cisplatin-induced acute kidney injury in rats through nrf2/KIM-1 signalling pathway. Drug Res (Stuttg). 2018;68:23–32.
Adeoye BO, Oyagbemi AA, Asenuga ER, Omobowale TO, Adedapo AA. The ethanol leaf extract of Andrographis paniculata blunts acute renal failure in cisplatin-induced injury in rats through inhibition of Kim-1 and upregulation of Nrf2 pathway. J Basic Clin Physiol Pharmacol. 2018;30:205–17.
Kim IH, Kwon MJ, Jung JH, Nam TJ. Protein extracted from Porphyra yezoensis prevents cisplatin-induced nephrotoxicity by downregulating the MAPK and NF-kappaB pathways. Int J Mol Med. 2018;41:511–20.
Arab HH, Mohamed WR, Barakat BM, Arafa el SA. Tangeretin attenuates cisplatin-induced renal injury in rats: Impact on the inflammatory cascade and oxidative perturbations. Chem Biol Interact. 2016;258:205–13.
Funaro A, Wu X, Song M, Zheng J, Guo S, Rakariyatham K, et al. Enhanced anti-inflammatory activities by the combination of luteolin and tangeretin. J Food Sci. 2016;81:H1320–7.
Do Amaral CL, Francescato HD, Coimbra TM, Costa RS, Darin JD, Antunes LM, et al. Resveratrol attenuates cisplatin-induced nephrotoxicity in rats. Arch Toxicol. 2008;82:363–70.
**an Y, Lin Y, Cao C, Li L, Wang J, Niu J, et al. Protective effect of umbilical cord mesenchymal stem cells combined with resveratrol against renal podocyte damage in NOD mice. Diabetes Res Clin Pract. 2019;156:107755.
Dallak M, Dawood AF, Haidara MA, Abdel Kader DH, Eid RA, Kamar SS, et al. Suppression of glomerular damage and apoptosis and biomarkers of acute kidney injury induced by acetaminophen toxicity using a combination of resveratrol and quercetin. Drug Chem Toxicol. 2020;1–7.
Xu GY, Tang XJ. Troxerutin (TXN) potentiated 5-Fluorouracil (5-Fu) treatment of human gastric cancer through suppressing STAT3/NF-kappa B and Bcl-2 signaling pathways. Biomed Pharmacother. 2017;92:95–107.
Sekine Y, Nakayama H, Miyazawa Y, Kato H, Furuya Y, Arai S, et al. Simvastatin in combination with meclofenamic acid inhibits the proliferation and migration of human prostate cancer PC-3 cells via an AKR1C3 mechanism. Oncol Lett. 2018;15:3167–72.
Tan RZ, Liu J, Zhang YY, Wang HL, Li JC, Liu YH, et al. Curcumin relieved cisplatin-induced kidney inflammation through inhibiting Mincle-maintained M1 macrophage phenotype. Phytomedicine. 2019;52:284–94.
Karahan MA, Yalcin S, Aydogan H, Buyukfirat E, Kucuk A, Kocarslan S, et al. Curcumin and dexmedetomidine prevents oxidative stress and renal injury in hind limb ischemia/reperfusion injury in a rat model. Ren Fail. 2016;38:693–8.
Cao L, Zhi D, Han J, Kumar Sah S, **e Y. Combinational effect of curcumin and metformin against gentamicin-induced nephrotoxicity: Involvement of antioxidative, anti-inflammatory and antiapoptotic pathway. J Food Biochem. 2019;43:e12836.
Acknowledgements
This work was supported by the State Key Program of National Natural Science Foundation of China (No. 81872878) and a grant from Zhejiang Provincial Natural Science Foundation (No. LGF20H030001).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
The original version of the article was unfortunately missing the Open Access License text and had an issue with the copyrightholder name.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Fang, Cy., Lou, Dy., Zhou, Lq. et al. Natural products: potential treatments for cisplatin-induced nephrotoxicity. Acta Pharmacol Sin 42, 1951–1969 (2021). https://doi.org/10.1038/s41401-021-00620-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41401-021-00620-9
- Springer Nature Singapore Pte Ltd.
Keywords
This article is cited by
-
JianPiYiShen formula prevents cisplatin-induced acute kidney injury in mice by improving necroptosis through MAPK pathway
BMC Complementary Medicine and Therapies (2024)
-
Contributing roles of mitochondrial dysfunction and hepatocyte apoptosis in liver diseases through oxidative stress, post-translational modifications, inflammation, and intestinal barrier dysfunction
Cellular and Molecular Life Sciences (2024)
-
Hot Melt Extrusion Technology as a Modern Strategy for Improving the Bioavailability of Flavonoids
Pharmaceutical Chemistry Journal (2024)
-
Protective and Therapeutic Effects of Gymnema sylvestre Leaf Extract on Cisplatin-Induced Nephrotoxicity
Proceedings of the National Academy of Sciences, India Section B: Biological Sciences (2024)
-
Long-term cisplatin nephrotoxicity after childhood cancer: a systematic review and meta-analysis
Pediatric Nephrology (2024)