
Abstract
The recent progress in genome editing technology using the engineered zinc finger nucleases (ZFN), transcriptional activator-like effector nucleases (TALEN), and more recently, clustered regularly interspaced short palindromic repeat-CRISPR-associated protein 9 (CRISPR-Cas9) system have enabled the possibility of precisely modifying target sites in the genome. This technology brings hope of a cure for many genetic diseases. With this review, our goal is to discuss how targeted genome editing can be combined with hematopoietic stem cell transplantation and other approaches to be used for the treatment of a particular group of genetic diseases, the lysosomal storage disorders. We also highlight which diseases within this group would potentially benefit from this treatment and what are the main problems to be addressed to transform this promising technology into reality.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
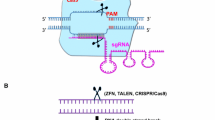
Human genetic diseases arise from mutations in genomic DNA that abrogate normal function of genes. The emergence of recombinant DNA technology, which could enable the delivery of a “new gene” into the cells of patients, has brought hope of a cure for these diseases, and represents the prototype of gene therapy. Different biological strategies for genome engineering have appeared, but most of them have shown limitations related to low efficiency in gene delivery or in maintaining its expression or present significant risks related to the random insertion of the therapeutic gene into the genome [1, 2]. Recently developed protocols for genomic editing through the use of nucleases have provided a much simpler and safer way for targeted gene modification. The engineered proteins zinc finger nucleases (ZFNs) and transcriptional activator-like effector nucleases (TALENs) are based on DNA-binding proteins, whereas the clustered regularly interspaced short palindromic repeat-CRISPR associated protein 9 (CRISPR-Cas9) system is based on RNA-guided DNA recognition [3]. They can all be programmed to generate targeted double-strand breaks in genomic DNA. The break activates repair through error-prone non-homologous end joining or homology-directed repair. If non-homologous end joining is activated, the result is insertions and/or deletions (indels) that disrupt the target locus. In the presence of a donor template with homology to the targeted locus, the homology-directed repair pathway operates allowing for precise mutations to be made or corrected, or for DNA sequences to be inserted at much higher frequencies than conventional gene targeting. Double-strand break generation can increase rates of homologous recombination of ssDNA and dsDNA donors by 5-fold and 130-fold, respectively [4]. Several studies in cells and animal models using this technology as a form of gene therapy have been published, demonstrating its advantages over conventional gene therapy protocols (Table 1). With this review, our goal is to discuss how targeted genomic editing can be used for the treatment of a particular group of genetic diseases, the lysosomal storage disorders (LSD), which diseases within this group would benefit from this technology, and what are the main problems to be addressed to transform this promising technology into reality.
Lysosomal Storage Disorders
Lysosomal storage disorders (LSD) are a group of approximately 50 genetically inherited diseases, characterized by total or partial deficiency of one specific enzyme involved in the degradation pathways of macromolecules in the lysosome [14]. They are monogenic and for most of them, a large number of mutations have been described. Some mutations cause complete loss of enzyme activity, while others only reduce this activity. Storage of undergraded or partially degraded material, usually the substrate of the defective enzyme, occurs in the lysosome. Conventionally, LSD are grouped based on the chemical nature of the non-degraded substrates that accumulate, including mucopolysaccharidoses, lipidoses, glycogenoses, and oligosaccharidoses [1].
Despite the great heterogeneity of symptoms, most of these diseases are characterized by its progressive course with high morbidity and increased mortality, although there are significant variations between different diseases and among patients with the same disease [15]. Generally, these diseases are multisystemic, and clinical features include organomegaly, central nervous system dysfunction, and coarse hair and faces. Most patients are asymptomatic at birth and present the onset during childhood. Their frequency varies in different regions and populations, but although individually rare, the combined estimated prevalence ranges from 1:4000 to 1:9000 live births [14].
Although several of these diseases have no specific therapy so far, for some LSD, hematopoietic stem cell transplantation (HSCT) and enzyme replacement therapy (ERT) are available, and other approaches, such as gene therapy, are being developed. These treatments are based on the fact that most lysosomal enzymes can be internalized by deficient cells via mannose-6-phosphate receptors. After endocytic internalization, the enzyme is transported by endosomes to the lysosome, resulting in the breakdown of accumulated storage material [16]. However, these treatments are not available to all LSDs, many of them are in the experimental phase, and they are not fully effective and curative.
In HSCT, stem cells from bone marrow or umbilical cord blood from healthy donors are transplanted. Evidence shows that its efficacy relies not only on the migration of donor cells into bone marrow and reconstitution of the blood lineage, but to the subsequent migration of engrafted cells into many disease target organs, including the brain, where they replace the resident enzyme-deficient population; thus becoming a local and steady source of the functional enzyme [17]. When successful, HSCT may prolong the patient’s life, preserve neurocognition and enhance somatic changes. Major drawbacks of the HSCT include the significant risks associated with this procedure, such as the possibility of develo** graft-versus-host disease, the difficulty of finding HLA-compatible donors and development of chimerism [18•]. So, its use in many countries has been deferred in favor of ERT whenever it is available.
In ERT, the deficient recombinant enzyme is administered to the patient by means of repeated intravenous injections. Despite being an effective and safe treatment option for various LSD, ERT also has important limitations. Among them are the adverse reactions presented by some patients, the high cost of treatment, the life-long dependence on weekly 4–5-h-long infusions and the limited ability to correct neurological and skeletal pathology [19].
Given the limitations presented by existing therapies in the treatment of LSD, investigation of new therapies aiming at increasing treatment effectiveness becomes necessary. Many studies have been developed in this direction, each using a different aspect of the disease to the development of different approaches, among them gene therapy. Gene therapy may overcome some of these problems, as it may allow constant delivery of the enzyme direct to target organs and eliminates the need for weekly infusions. Also, correction of a few cells could lead to the enzyme being secreted into the circulation and taken up by their neighboring cells, resulting in widespread correction of the biochemical defects. So, the number of cells that must be modified with a gene transfer vector is relatively low [15]. Moreover, precise transcriptional regulation is probably not necessary as over-expression of lysosomal enzymes does not appear to be detrimental and as little as 5–10 % normal levels of enzyme can be therapeutic for several LSD [20].
Genetic modification can be performed either ex vivo or in vivo. The ex vivo strategy is based on modification of cells in culture and transplantation of the modified cell into patient [1]. Cells that are most commonly considered therapeutic targets for monogenic diseases are stem cells. Advances in collection and isolation of these cells from a variety of sources have promoted autologous gene therapy as a viable option for LSD. In mouse models of LSDs, genetically modified neural stem cells encoding for enzyme genes effectively decreased lysosomal storage, reduced pathology, and extended life span of animals [21]. Mesenchymal stem cells and inducible pluripotent stem cells (iPSC) are also being used with this purpose [6, 7, 22]. However, as already mentioned, conventional gene therapy protocols have important limitations; among them are safety issues related to immune response and the possibility of insertional mutagenesis in the case of viral vectors, and low efficiency with non-viral vectors.
Genome Editing Combined with HSCT: Potential to Treat LSD
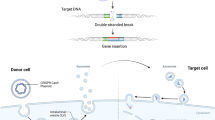
The use of endonucleases for targeted genome editing can solve the limitations presented by the usual gene therapy protocols. These enzymes are custom molecular scissors, allowing cutting DNA into well-defined, perfectly specified pieces, in virtually all cell types. Moreover, they can be delivered to the cells by plasmids that transiently express the nucleases, or by transcribed RNA, avoiding the use of viruses [23]. This technology, combined with HSCT, may represent a milestone for treatment of LSD. Hematopoietic stem cells extracted from a patient could be transfected with vectors encoding endonucleases designed to cleave at sites near the specific mutation and with a donor vector. The donor vector contains a region homologous to the mutated region, however, with the correct nucleotide sequence, and would serve as a template for repair of DNA damage after the double-strand break. Then, the cells which internalize the two vectors, in which the cleavage and homologous recombination occur, would have the correct gene sequence, and may be selected and implanted back into the patient (Fig. 1). Combining autologous HSCT with nuclease-mediated genome editing would have the advantages of lower risk of infection during the patient’s treatment, since the recovery of the immune function is rapid. Also, it would avoid the development of rejection (graft-versus-host disease), since the donor and the recipient are the same individual [24].

Schematic representation of the proposed treatment. Hematopoietic stem cells extracted from a patient could be transfected with vectors encoding the tailored endonuclease and with a donor vector to guide the homologous recombination. Then, the corrected cells may be selected ex vivo and implanted back into the patient. HSC hematopoietic stem cells, DBS double strand break
However, the clinical experience obtained so far with conventional HSCT suggests that the success of this procedure will probably be limited to some LSD. Factors predicting the success of the treatment are generally disease-specific. The main LSD with good evidence of transplant efficacy are Hurler syndrome (MPS I), Hunter syndrome (MPS II), Maroteaux-Lamy syndrome (MPS VI), Sly syndrome (MPS VII), and α-mannosidosis [25]. These diseases would probably be also the ones with greater chance to benefit from the combined treatment HSCT + genome editing. Moreover, other LSDs with evidence from limited series that suggest that transplantation may be efficacious, or for which HSCT has not been widely applied, but it may be expected to have some advantage, the combined treatment may have advantages over the HSCT and become an option. In this group, diseases with milder phenotypes, for which HSCT was not formerly offered because of the perceived risk, can be included.
The HSCT is more effective in younger patients [18•]. Therefore, it is important to consider the time it would take to have the final diagnosis and identification of the mutation, the construction of vectors for site-specific cleavage and homologous recombination, and finally, cell repair and transplantation. Therefore, patients who have common mutations in certain populations could benefit, since the vectors are mutation-specific, and pre-clinical tests would be preferentially performed in these mutations, allowing the acquisition of information regarding correction rates and safety issues, for example. Table 2 shows the LSDs most likely to benefit from treatment with HSCT combined with genome editing, since they are treatable by HSCT and have reported common mutations in some populations.
The proposed strategy has been studied experimentally in other genetic disease models and the results are promising. Genovese et al. used ZFN genome editing in a model of X-linked severe combined immunodeficiency (SCID-X1). They showed evidence of targeted integration in human hematopoietic stem cells by long-term multilineage repopulation of transplanted mice. They also demonstrated the therapeutic potential of the strategy by targeting a corrective complementary DNA into the IL2RG gene of HSCs from healthy donors and a subject with SCID-X1. Gene-edited HSCs sustained normal hematopoiesis and gave rise to functional lymphoid cells that possess a selective growth advantage over those carrying disruptive IL2RG mutations [5•]. The more recent technology of the CRISPR-Cas9 system seems to be even more effective than ZFN and has been used in proof of principle treatment for genetic diseases. This system was used to correct a mutation that causes cystic fibrosis in both patient-derived primary cultured small intestinal cells and large intestinal stem cells. When assayed in organoid culture, disease-associated defects were rescued in these engineered cells [33].
Another approach that could be used for LDS is correcting other types of stem cells. Mesenchymal stem cells could be a good option for treating specific organs affected by the diseases. These cells can be easily obtained from bone marrow or adipose tissue. After editing their genome, the cells could be expanded in culture and reinserted into the patient to produce differentiated cells with corrected functions. Attempts with other gene therapy strategies have been made to use these cells to treat LSD. Meyerrose et al. (2008) transduced human bone marrow-derived mesenchymal stem cells with a lentiviral vector expressing β-glucuronidase and transplanted them into the xenotransplant model of MPS VII. Transduced cells persisted in the animals that underwent transplantation for the length of the study (4 months), expressed high levels of enzyme and reduced lysosomal storage in several critical tissues [34]. Moreover, studies using inducible pluripotent stem cells (iPSC) demonstrated that it is possible to generate mutations in these cells with the CRISPR-Cas9 system after inducing differentiation, allowing for studies of tissue-specific effects [7]. Such studies suggest that, in the future, it may be possible to generate iPSC from patients, correct the causative mutation, differentiate the cells and reintroduce them back into the patient.
Pitfalls to be Addressed
The potential of combining autologous HSCT with genome editing in gene therapy for LSD is evident. However, before this technology can be used as treatment for these diseases, some obstacles must be overcome. Some of them are related to hematopoietic stem cells, and are common to other forms of gene therapy on stem cells. After modification, it would be ideal that the selected cells could be expanded in culture before being reimplanted into the patient. Several protocols for expansion of these cells, the majority using cytokines in the culture medium, are being studied and showed good results in mouse cells [35]. However, when tested in larger animals or humans, the same protocols do not seem to work well [36, 37]. Thus, appropriate clinical expansion protocols are still lacking.
Regarding HSCT, some LSD, including MPS III (Sanfilippo syndrome), Batten disease, and late infantile ceroid lipofuscinosis (LINCL) do not respond to this treatment [25]. In these cases, it would be necessary to try to modify other cell types. Also, the extent of myeloablation and the number of modified cells that would be transplanted must be well studied.
Even though genome editing with nucleases has been applied in many model organisms, there are still fundamental attributes that are unclear and need further investigation. These include molecular aspects of the systems. For example, the ratio of Cas9 to gRNA in the CRISPR-Cas9 system greatly affects mutagenesis efficiency [38, 39]. Theoretically, the more complexes are formed, the higher editing efficiency is expected. However, excessive cleavage efficiency can result in off-target breaks, one of the main problems related to nucleases. Unspecific cleavages were reported in several studies, not only using CRISPR-Cas but also for ZFN and TALEN [40–42]. Several methods have been studied to detect these off-target sites and thus improve the selection of cells before use. However, it is important to develop methods to optimize the systems by controlling component expression, improving target selection criteria, and engineering the nucleases to provide higher specificity.
Another area that needs optimization is the delivery of the genetic material to the cells. In the case of hematopoietic cells, for which the protocols for in vitro expansion do not work well, it is important that a large fraction of cells taken from the patient is properly modified. This largely depends on the efficiency of transfection. There are different ways of delivery systems for generating active nuclease components (e.g., plasmid-, DNA fragment-, and RNA-based delivery), and the best way must be determined for each cell type. Optimized electroporation and lipofection protocols for each cell type are also crucial to the success of the strategy. Besides being used directly for genome editing [43], viral vectors are being used in combination with ZFN, TALEN, and CRISPR-Cas9. For transfection-resistant cell types, the use of viral vectors, such as integrase-deficient lentiviral vectors, adeno-associated virus-derived vectors, or adenoviral vectors, instead of plasmids, can be an option. An integrase-deficient lentivirus vector was successfully used for delivering ZFN into a panel of human cell lines and stem cells [44]. However, for TALEN delivery, this method does not seem to be so effective. For this system, adeno-associated virus vectors are indicated [45]. Viral vectors are also being used for delivering CRISPR-Cas9 system components. Maggio et al. transduced cells with this combination and observed rates of targeted mutagenesis similar to those achieved by isogenic adenovirus encoding TALEN targeting the same chromosomal region [46]. Moreover, factors affecting recombination efficiency, including the characteristics of the mutations, sizes and positions of the homologous flanking arms, and the stability of the given templates before HDR occurs remain to be evaluated.
Conclusions
The need for new therapies aimed at increasing treatment effectiveness for LSDs is evident, since the few therapeutic options available have important limitations. Combining targeted genome editing and HSCT seems to be a promising strategy to treat some of these diseases, especially those that are responsive to HSCT and have common mutations described. ZFN, TALEN, and CRISPR-Cas9 systems have the potential to correct mutations on specific sites, via double-strand break followed by homologous recombination, and have proven to be efficient in treating other genetic diseases. However, since these technologies are very recent, some obstacles must be addressed. Additional methodological advances both in HSCT and nuclease-mediated genome editing will undoubtedly further enhance the use of these techniques, and make them a possible option for curing LSD.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance
Matte U, Baldo G, Giugliani R. Non viral gene transfer approaches for lysosomal storage disorders. In: Yuan X, editor. Non-Viral Gene Therapy [Internet]. InTech; 2011 [cited 2014 Nov 15]. Available from: http://www.intechopen.com/books/non-viral-gene-therapy/non-viral-gene-transfer-approaches-for-lysosomal-storage-disorders.
Baldo G, Giugliani R, Matte U. Gene delivery strategies for the treatment of mucopolysaccheridoses. Expert Opin Drug Deliv. 2014;11(3):449–59.
Wang D, Gao G. State-of-the-Art human gene therapy: part II. Gene therapy strategies and clinical applications. Discov Med. 2014;18(98):151–61.
DiCarlo JE, Norville JE, Mali P, Rios X, Aach J, Church GM. Genome engineering in Saccharomyces cerevisiae using CRISPR-Cas systems. Nucleic Acids Res. 2013; gkt135.
Genovese P, Schiroli G, Escobar G, Di Tomaso T, Firrito C, Calabria A, et al. Targeted genome editing in human repopulating haematopoietic stem cells. Nature. 2014;510(7504):235–40. This study showed that ZFN gene-edited hematopoietic stem cells sustained normal haematopoiesis and gave rise to functional cells that possess a selective growth advantage over the mutant cells.
Yusa K, Rashid ST, Strick-Marchand H, Varela I, Liu P-Q, Paschon DE, et al. Targeted gene correction of α1-antitrypsin deficiency in induced pluripotent stem cells. Nature. 2011;478(7369):391–4.
Sanders LH, Laganière J, Cooper O, Mak SK, Vu BJ, Huang YA, et al. LRRK2 mutations cause mitochondrial DNA damage in iPSC-derived neural cells from Parkinson’s disease patients: reversal by gene correction. Neurobiol Dis. 2014;62:381–6.
Sun N, Zhao H. Seamless correction of the sickle cell disease mutation of the HBB gene in human induced pluripotent stem cells using TALENs. Biotechnol Bioeng. 2014;111(5):1048–53.
Matsubara Y, Chiba T, Kashimada K, Morio T, Takada S, Mizutani S, et al. Transcription activator-like effector nuclease-mediated transduction of exogenous gene into IL2RG locus. Sci Rep. 2014;4:5043.
Low BE, Krebs MP, Joung JK, Tsai SQ, Nishina PM, Wiles MV. Correction of the Crb1rd8 allele and retinal phenotype in C57BL/6N mice via TALEN-mediated homology-directed repair. Invest Ophthalmol Vis Sci. 2014;55(1):387–95.
**e F, Ye L, Chang JC, Beyer AI, Wang J, Muench MO, et al. Seamless gene correction of β-thalassemia mutations in patient-specific iPSCs using CRISPR/Cas9 and piggyBac. Genome Res. 2014;24(9):1526–33.
Yin H, Xue W, Chen S, Bogorad RL, Benedetti E, Grompe M, et al. Genome editing with Cas9 in adult mice corrects a disease mutation and phenotype. Nat Biotechnol. 2014;32(6):551–3.
Long C, McAnally JR, Shelton JM, Mireault AA, Bassel-Duby R, Olson EN. Prevention of muscular dystrophy in mice by CRISPR/Cas9-mediated editing of germline DNA. Science. 2014;345(6201):1184–8.
Giugliani R. Newborn screening for lysosomal diseases: current status and potential interface with population medical genetics in Latin America. J Inherit Metab Dis. 2012;35(5):871–7.
Matte U, Lagranha VL, de Carvalho TG, Mayer FQ, Giugliani R. Cell microencapsulation: a potential tool for the treatment of neuronopathic lysosomal storage diseases. J Inherit Metab Dis. 2011;34(5):983–90.
Hollak CEM, Wijburg FA. Treatment of lysosomal storage disorders: successes and challenges. J Inherit Metab Dis. 2014;37(4):587–98.
Wilkinson FL, Sergijenko A, Langford-Smith KJ, Malinowska M, Wynn RF, Bigger BW. Busulfan conditioning enhances engraftment of hematopoietic donor-derived cells in the brain compared with irradiation. Mol Ther. 2013;21(4):868–76.
Lund TC. Hematopoietic stem cell transplant for lysosomal storage diseases. Pediatr Endocrinol Rev PER. 2013;11 Suppl 1:91–8. This review discusses what is currently known of HSCT related outcomes in the treatment of many LSDs.
Baldo G, Mayer FQ, Martinelli BZ, de Carvalho TG, Meyer FS, de Oliveira PG, et al. Enzyme replacement therapy started at birth improves outcome in difficult-to-treat organs in mucopolysaccharidosis I mice. Mol Genet Metab. 2013;109(1):33–40.
Hawkins-Salsbury JA, Reddy AS, Sands MS. Combination therapies for lysosomal storage disease: is the whole greater than the sum of its parts? Hum Mol Genet. 2011;20(R1):R54–60.
Shihabuddin LS, Cheng SH. Neural stem cell transplantation as a therapeutic approach for treating lysosomal storage diseases. Neurotherapeutics. 2011;8(4):659–67.
Reiser J, Zhang X-Y, Hemenway CS, Mondal D, Pradhan L, La Russa VF. Potential of mesenchymal stem cells in gene therapy approaches for inherited and acquired diseases. Expert Opin Biol Ther. 2005;5(12):1571–84.
Sander JD, Joung JK. CRISPR-Cas systems for genome editing, regulation and targeting. Nat Biotechnol. 2014;32(4):347–55.
Montiel-Equihua CA, Thrasher AJ, Gaspar HB. Gene therapy for severe combined immunodeficiency due to adenosine deaminase deficiency. Curr Gene Ther. 2012;12(1):57–65.
Wynn R. Stem cell transplantation in inherited metabolic disorders. ASH Educ Program Book. 2011;2011(1):285–91.
Valkonen S, Hietala M, Savontaus ML, Aula P. Origin of Finnish mutations causing aspartylglucosaminuria. Hereditas. 1999;131(3):191–5.
Giraldo P, Alfonso P, Irún P, Gort L, Chabás A, Vilageliu L, et al. Map** the genetic and clinical characteristics of Gaucher disease in the Iberian Peninsula. Orphanet J Rare Dis. 2012;7(1):17.
Tappino B, Biancheri R, Mort M, Regis S, Corsolini F, Rossi A, et al. Identification and characterization of 15 novel GALC gene mutations causing Frabbe disease. Hum Mutat. 2010;31(12):E1894–915.
Bernstein DL, Hülkova H, Bialer MG, Desnick RJ. Cholesteryl ester storage disease: review of the findings in 135 reported patients with an underdiagnosed disease. J Hepatol. 2013;58(6):1230–43.
Riise Stensland HMF, Klenow HB, Nguyen LV, Hansen GM, Malm D, Nilssen Ø. Identification of 83 novel alpha-mannosidosis-associated sequence variants: functional analysis of MAN2B1 missense mutations. Hum Mutat. 2012;33(3):511–20.
Berger J, Löschl B, Bernheimer H, Lugowska A, Tylki-Szymanska A, Gieselmann V, et al. Occurrence, distribution, and phenotype of arylsulfatase a mutations in patients with metachromatic leukodystrophy. Am J Med Genet. 1997;69(3):335–40.
Terlato NJ, Cox GF. Can mucopolysaccharidosis type I disease severity be predicted based on a patient’s genotype? A comprehensive review of the literature. Genet Med. 2003;5(4):286–94.
Schwank G, Koo B-K, Sasselli V, Dekkers JF, Heo I, Demircan T, et al. Functional repair of CFTR by CRISPR/Cas9 in intestinal stem cell organoids of cystic fibrosis patients. Cell Stem Cell. 2013;13(6):653–8.
Meyerrose TE, Roberts M, Ohlemiller KK, Vogler CA, Wirthlin L, Nolta JA, et al. Lentiviral-transduced human mesenchymal stem cells persistently express therapeutic levels of enzyme in a xenotransplantation model of human disease. Stem Cells Dayt Ohio. 2008;26(7):1713–22.
Watts KL, Adair J, Kiem H-P. Hematopoietic stem cell expansion and gene therapy. Cytotherapy. 2011;13(10):1164–71.
De Lima M, McMannis J, Gee A, Komanduri K, Couriel D, Andersson BS, et al. Transplantation of ex vivo expanded cord blood cells using the copper chelator tetraethylenepentamine: a phase I/II clinical trial. Bone Marrow Transplant. 2008;41(9):771–8.
Tisdale JF, Hanazono Y, Sellers SE, Agricola BA, Metzger ME, Donahue RE, et al. Ex vivo expansion of genetically marked rhesus peripheral blood progenitor cells results in diminished long-term repopulating ability. Blood. 1998;92(4):1131–41.
Xu T, Li Y, Nostrand JDV, He Z, Zhou J. Cas9-based tools for targeted genome editing and transcriptional control. Appl Environ Microbiol. 2014;80(5):1544–52.
Wang H, Yang H, Shivalila CS, Dawlaty MM, Cheng AW, Zhang F, et al. One-step generation of mice carrying mutations in multiple genes by CRISPR/Cas-mediated genome engineering. Cell. 2013;153(4):910–8.
Guilinger JP, Pattanayak V, Reyon D, Tsai SQ, Sander JD, Joung JK, et al. Broad specificity profiling of TALENs results in engineered nucleases with improved DNA-cleavage specificity. Nat Methods. 2014;11(4):429–35.
Grau J, Boch J, Posch S. TALENoffer: genome-wide TALEN off-target prediction. Bioinforma Oxf Engl. 2013;29(22):2931–2.
Fine EJ, Cradick TJ, Bao G. Identification of off-target cleavage sites of zinc finger nucleases and TAL effector nucleases using predictive models. Methods Mol Biol Clifton NJ. 2014;1114:371–83.
Ian E. Alexander, David W. Russell. The potential of AAV-mediated gene targeting for gene and cell therapy applications. Curr Stem Cell Rep. 2015;in press.
Lombardo A, Genovese P, Beausejour CM, Colleoni S, Lee Y-L, Kim KA, et al. Gene editing in human stem cells using zinc finger nucleases and integrase-defective lentiviral vector delivery. Nat Biotechnol. 2007;25(11):1298–306.
Holkers M, Maggio I, Liu J, Janssen JM, Miselli F, Mussolino C, et al. Differential integrity of TALE nuclease genes following adenoviral and lentiviral vector gene transfer into human cells. Nucleic Acids Res. 2013;41(5):e63.
Maggio I, Holkers M, Liu J, Janssen JM, Chen X, Gonçalves MAFV. Adenoviral vector delivery of RNA-guided CRISPR/Cas9 nuclease complexes induces targeted mutagenesis in a diverse array of human cells. Sci Rep. 2014;4:5105.
Acknowledgments
The authors would like to thank CNPq for financial support.
Compliance with Ethics Guidelines
ᅟ
Conflict of Interest
TG de Carvalho, U Matte, R Giugliani, and G Baldo declare that they have no conflict of interest.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Author information
Authors and Affiliations
Corresponding author
Additional information
This article is part of the Topical Collection on Genome Editing
Rights and permissions
About this article

Cite this article
de Carvalho, T.G., da Silveira Matte, U., Giugliani, R. et al. Genome Editing: Potential Treatment for Lysosomal Storage Diseases. Curr Stem Cell Rep 1, 9–15 (2015). https://doi.org/10.1007/s40778-014-0007-8
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40778-014-0007-8