
Abstract
A summit held March 2023 in Scottsdale, Arizona (USA) focused on the intronic hexanucleotide expansion in the C9ORF72 gene and its relevance in frontotemporal dementia (FTD) and amyotrophic lateral sclerosis (ALS; C9ORF72-FTD/ALS). The goal of this summit was to connect basic scientists, clinical researchers, drug developers, and individuals affected by C9ORF72-FTD/ALS to evaluate how collaborative efforts across the FTD-ALS disease spectrum might break down existing disease silos. Presentations and discussions covered recent discoveries in C9ORF72-FTD/ALS disease mechanisms, availability of disease biomarkers and recent advances in therapeutic development, and clinical trial design for prevention and treatment for individuals affected by C9ORF72-FTD/ALS and asymptomatic pathological expansion carriers. The C9ORF72-associated hexanucleotide repeat expansion is an important locus for both ALS and FTD. C9ORF72-FTD/ALS may be characterized by loss of function of the C9ORF72 protein and toxic gain of functions caused by both dipeptide repeat (DPR) proteins and hexanucleotide repeat RNA. C9ORF72-FTD/ALS therapeutic strategies discussed at the summit included the use of antisense oligonucleotides, adeno-associated virus (AAV)-mediated gene silencing and gene delivery, and engineered small molecules targeting RNA structures associated with the C9ORF72 expansion. Neurofilament light chain, DPR proteins, and transactive response (TAR) DNA-binding protein 43 (TDP-43)–associated molecular changes were presented as biomarker candidates. Similarly, brain imaging modalities (i.e., magnetic resonance imaging [MRI] and positron emission tomography [PET]) measuring structural, functional, and metabolic changes were discussed as important tools to monitor individuals affected with C9ORF72-FTD/ALS, at both pre-symptomatic and symptomatic disease stages. Finally, summit attendees evaluated current clinical trial designs available for FTD or ALS patients and concluded that therapeutics relevant to FTD/ALS patients, such as those specifically targeting C9ORF72, may need to be tested with composite endpoints covering clinical symptoms of both FTD and ALS. The latter will require novel clinical trial designs to be inclusive of all patient subgroups spanning the FTD/ALS spectrum.
Plain Language Summary
The C9ORF72 Summit was held in March 2023 in Scottsdale, Arizona (USA). Some people who have the disease frontotemporal dementia or the disease amyotrophic lateral sclerosis have a change in one of their genes; the name of the gene is C9ORF72. People who carry this genetic difference usually inherited it from a parent. Researchers are improving their understanding of how the change in the C9ORF72 gene affects people, and efforts are being made to use this knowledge to develop treatments for amyotrophic lateral sclerosis and frontotemporal dementia. In addition to studying the cellular and molecular mechanisms of how the C9ORF72 mutation leads to cellular dysfunction and frontotemporal dementia and amyotrophic lateral sclerosis clinical symptoms, a large effort of the research community is aimed at develo** measurements, called biomarkers, that could enhance therapy development efforts in multiple ways. Examples include monitoring of disease activity, identifying those at risk of develo** amyotrophic lateral sclerosis or frontotemporal dementia, predicting which people might benefit from a particular treatment, and showing that a drug has had a biological effect. Markers that identify healthy people who are at risk of develo** amyotrophic lateral sclerosis or frontotemporal dementia could be used to test treatments that would start before a person shows any symptoms and hopefully would delay or even prevent their onset.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Why was this summit held? |
This summit was held to foster a breakdown of frontotemporal dementia (FTD) and amyotrophic lateral sclerosis (ALS) disease silos. Areas included all levels of investigation, from basic research through biomarker development, therapeutic target discovery and therapeutics development, and clinical trial design. Additional aims were evaluate the most effective treatment and care of individuals affected by FTD/ALS, including the most prevalent familial form of FTD/ALS, C9ORF72-related FTD/ALS, discussions, and collaborative efforts. |
What were key takeaways from this summit? |
Critical information in our understanding of the biology around the C9ORF72 expansion is still lacking, and many questions remain unanswered: What molecular and cellular changes occur at pre-symptomatic stages and how early do they occur? Why are some individuals only affected with ALS-associated symptoms, while others show FTD-associated clinical symptoms, and yet others have symptoms of both diseases? How do non-neuronal cells respond to the C9ORF72 repeat expansion, how do they contribute to neuronal degeneration, and do these contributions differ between the frontal cortex and spinal cord? |
Biomarkers, based on both imaging and on molecular readouts, are crucial to improving disease modeling, predicting disease progression, predicting onset of symptoms spanning ALS and FTD, and determining which therapies are engaging their targets in all affected brain regions. |
Basket and/or platform trials are needed that include all patient subgroups, including mutation carriers and patients experiencing the FTD/ALS disease spectrum. |
Commentary
A mutation in the C9ORF72 gene is one of the most common causes of both frontotemporal dementia (FTD) and amyotrophic lateral sclerosis (ALS). To explore the convergence of these two diseases with a common genetic root, a summit was held. Participants in this summit, held March 2023 in Scottsdale, Arizona (USA), discussed the biology (genetics, RNA, proteins) of C9ORF72-related FTD and ALS (C9ORF72-FTD/ALS) and the potential relevance of this biology to biomarkers, therapeutic target identification, and the design of clinical trials in the affected and pre-symptomatic populations. The information presented and discussed at the summit is presented in this article.
The contents of this manuscript are based on discussions at the C9ORF72 Summit and did not entail any animal or human subjects research that required review/approval by the Institutional Animal Care & Use Committee (IACUC) or Institutional Review Board (IRB).
Translating Genetic Discoveries into Therapies
Researchers have identified genetic causes for 60–70% of familial ALS and familial FTD and about 10% of sporadic ALS and sporadic FTD [1, 2]. Interestingly, the C9ORF72 locus was identified as the most commonly linked genetic factor to both ALS and FTD [3]. A pathological intronic hexanucleotide repeat expansion (HRE) that is believed to have arisen around the fall of Rome is recognized as the source of a common founder, and Viking conquests have been postulated as the mechanism of dissemination of C9ORF72 [4,5,6]. The age range at symptom onset is broad for patients who carry the C9ORF72 repeat expansion [7, 8]. This expansion is known to have incomplete, but age-dependent, penetrance; while patients may occasionally develop ALS or FTD in their 20s, others survive into their ninth decade without develo** symptoms [7].
Screening for the C9ORF72 HRE and other genes associated with ALS and FTD became common with the advent of next-generation sequencing [9], and it has become standard practice in many places across the world for both familial and apparently sporadic cases. Genomic approaches are also beginning to unravel the mystery of the biology of ALS and FTD. Cellular pathways that have been linked to ALS and FTD based on genomic discoveries include neuronal projection, cytoskeleton alterations, membrane trafficking, and cyclic-nucleotide-mediated signaling [10, 11].
The discovery of the C9ORF72 gene as the most common genetic cause of ALS and FTD has yielded novel opportunities for therapeutic intervention (e.g., gene therapy trials [discussed below]). The failure of these trials, however, has illuminated the limits of the current understanding of the biology of C9ORF72-FTD/ALS, as well as the critical importance of improving this understanding to help ensure that future trials have a greater likelihood of success.
Overall, numerous novel therapies for ALS and FTD are emerging, but the belief is that these are likely be most effective when patients are treated as early as possible in their disease course. With advanced technologies in genome sequencing and gene therapy approaches, one could speculate that whole-genome sequencing early in life might become available to everyone and would pave the road for future precision medicine approaches for ALS and FTD patients as well as for gene carriers.
C9ORF72 Disease Biology
Mutations in the C9ORF72 gene are characterized by a GGGGCC (G4C2)n HRE in a non-coding region on chromosome 9p21; these mutations represent the most common genetic abnormality in FTD (10%-30%) and are also highly prevalent in ALS (20%-50%) and the spectrum disease of FTD-ALS (30%) [4, 12]. Extensive research over the past 10 years on the role of the (G4C2)n repeat expansion in C9ORF72 has led to the proposals of different disease-causing mechanisms for mutant C9ORF72: (1) protein loss of function; (2) toxic RNA gain of function; and (3) toxicity caused by repeat-associated non-ATG initiated (RAN) translation, which leads to the accumulation of dipeptide repeats (DPR) [13]. Ongoing studies are investigating the cellular consequences of these mechanistic pathways, including defects in nuclear-cytoplasmic trafficking of RNAs and proteins due to RNA toxicity and DPR formation, such as transactive response DNA-binding protein 43 (TDP-43) [14, 15], which further contribute to C9ORF72 disease pathogenesis.
Loss of C9ORF72 Function in C9ORF72-FTD/ALS
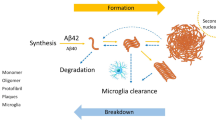
The repeat expansion causes C9ORF72 haploinsufficiency in ALS/FTD through the downregulation of C9ORF72 transcripts and reduced C9ORF72 protein expression (Fig. 1) [4, 12, 16,17,18,19,72, 80,81,82,83]. Monitoring CSF or blood neurofilaments in these individuals could inform impending phenoconversion. However, because pre-symptomatic increases in NfL are not seen as consistently among C9ORF72 repeat expansion carriers as is observed for rapidly progressive forms of ALS linked to mutations in the superoxide dismutase 1 gene (SOD1-ALS), alternate biomarkers or a multimodal approach will likely be needed to predict phenoconversion in C9ORF72 expansion carriers [84].
Both the prognostic utility of neurofilaments and their use as response biomarkers could improve clinical trial design and inform interpretation of clinical trial data. The latter would additionally benefit from the use of DPR proteins, which have potential pharmacodynamic information on upstream target engagement. Indeed, early investigations to identify approaches to neutralize or degrade C9ORF72 repeat RNA enabled the monitoring of the intended effects of putative therapeutics, such as a lowering of DPR proteins [85,86,87,88,89,90,91]. While assays to measure poly(GP) DPR proteins in human CSF were developed first, poly(GP), poly(GA), and poly(GR) have since been detected in CSF from asymptomatic and symptomatic C9ORF72 repeat expansion carriers, and these two groups have levels of DPR proteins that are generally similar [78, 92]. Despite the fact that CSF DPR proteins in symptomatic C9ORF72 expansion carriers do not associate with clinical traits such as age at disease onset, revised Amyotrophic Lateral Sclerosis Functional Rating Scale (ALSFRS-R) score, rate of decline of ALSFRS-R score, or survival, data from preclinical models and human studies support the use of CSF DPR proteins as pharmacodynamic biomarkers.[78, 85, 86, 90, 92,93,94,95] In fact, measures of DPR proteins have been used as an exploratory endpoint in clinical trials of investigational agents, such as antisense oligonucleotides (ASOs), for C9ORF72-FTD/ALS.
Biomarkers of C9ORF72-FTD/ALS Based on TDP-43–Associated Pathology
Loss of splicing repression by the TDP-43 is linked to ALS and FTD pathology [96]. TDP-43 has been explored extensively as a fluid biomarker for ALS and FTD, including in CSF, plasma, circulating lymphomonocytes, and platelets, but no robust measure of pathological TDP-43 protein itself has been developed as of yet.
In brains of patients with ALS or FTD, TDP-43 is compromised in its function of repressing the splicing of cryptic exons [97]. The stathmin-2 (STMN2) cryptic exon is a biomarker for both ALS and FTD [98,99,100]. Additionally, the cryptic exon splicing event in the UNC13A locus is a biomarker that results from loss of TDP-43 [101, 102]. Researchers used a monoclonal antibody specific to a TDP-43–dependent cryptic epitope to show that C9ORF72-associated ALS, including in pre-symptomatic mutation carriers, involves losing TDP-43 splicing repression [103]. With the use of mass spectrometry, TDP-43–dependent cryptic peptides can also be detected in C9ORF72-associated ALS [104]. These findings have two major implications. First, therapeutic strategies could overcome the loss of TDP-43 splicing repression for C9ORF72-related and sporadic ALS and FTD. Second, develo** TDP-43 cryptic peptides as functional biomarkers could lead to early-stage diagnosis of ALS and FTD and could aid in monitoring the biological response of therapeutic treatment in clinical trials.
Neuroimaging of the C9ORF72 FTD/ALS Syndrome
Advances in neuroimaging sequences and computation have allowed the non-invasive quantification of cerebral structural (MRI, grey matter volumetry, and white matter tractography), functional (blood oxygen level-dependent [BOLD] MRI), and metabolic (fluorodeoxyglucose [FDG] PET) changes that have been applied to the study of the C9ORF72-related syndrome (comprehensively reviewed in [105]).
Symptomatic C9ORF72-related FTD and ALS has been consistently associated with more widespread cortical and subcortical grey and white matter changes—more so than has non-C9ORF72 FTD or ALS [106]—with longitudinal changes detectable in some patients over months [107]. Consistent thalamic involvement, particularly of the pulvinar sub-region, occurs in both FTD and ALS [108], with an overlap** regional hypometabolic thalamic signature also seen in ALS cases using FDG PET [109]. Alterations in cerebral networks, as demonstrated in resting state BOLD MRI, have been associated with both ALS and FTD phenotypes [110, 111].
In asymptomatic C9ORF72 expansion carriers, substantial MRI changes have been detected, including grey and white matter loss [112, 113], gyrification abnormalities [114], hypothalamic atrophy [115], changes in resting-state network connectivity [116], and reduced cerebral perfusion [117]. FDG PET has demonstrated regional hypometabolism encompassing the cingulate gyrus, frontal and temporal neocortices, and thalami [118, 119]. Future studies might usefully exploit the unrivaled sensitivity of magnetoencephalography (MEG) to subtle changes in cortical motor neurophysiology, as noted in a small study of asymptomatic carriers of varied ALS-causing genotypes [120]. In relation to understanding the different phenotypic outcomes, a study of the cervical spinal cord showed that a sub-group of asymptomatic C9ORF72 expansion carriers aged over 40 years who were relatives of those affected by ALS (rather than FTD) showed evidence of early pathology of the white matter of the corticospinal tract [121].
The differing clinical needs of patients tend to result in the dichotomization of symptomatic neuroimaging cohorts into either ALS or FTD. Through the increasing partnership with families affected by C9ORF72 and international data-sharing platforms, the opportunity to study larger numbers of younger asymptomatic expansion carriers (including those aged < 18 years) will deepen the understanding of a syndrome that may have a broader neurodevelopmental dimension than previously realized [122].
Summary of the Working Group Discussions on C9ORF72 Biomarkers
Table 2 summarizes the highlights of the C9ORF72 Biomarker working group.
C9ORF72-FTD/ALS Gene Therapy and Small Molecule Approaches
Gene therapy is a potential treatment approach for C9ORF72-FTD/ALS with the primary goal of targeting the underlying disease-causing genetic mutation. Specifically, gene therapy strategies aim to reduce the abnormal HRE within the C9ORF72 gene. One approach involves using small RNA molecules, known as ASOs, to target and bind to the expanded repeat sequences. Parallel approaches explore the design of small molecule compounds that can inhibit the activity of the abnormal repeat expansion in the C9ORF72 gene. Finally, small molecule compounds targeting the underlying mechanisms and pathological processes associated with C9ORF72 repeat expansion are also being investigated as therapeutic avenues for C9ORF72-FTD/ALS.
Gene Therapy for C9ORF72-FTD/ALS: Challenges and Opportunities
The entire field of gene therapy has been very active recently, with many clinical trials underway in an array of different diseases. The approaches being investigated for C9ORF72-FTD/ALS include ASOs, adeno-associated virus (AAV)–mediated gene silencing using CRISPR or RNA interference (RNAi) technology, and AAV-mediated gene delivery of therapeutic proteins [123, 124]. ASO treatments are a great way to explore the possibility of modifying mutant gene expression in ALS using a reversible system whereas AAV methods using constitutive promoter systems are permanent and cannot be switched off if problems are encountered. However, while ASOs require constant delivery through clinical visits and a lumbar puncture, AAV infusions are “one and done,” which is more practical for patients. Newer technologies to allow regulated expression of therapeutic genes are an exciting area that may allow more flexible use of AAV constructs in the clinic. Using gene therapy to deliver therapeutic proteins to the brain is another exciting area—either directly or “ex vivo” using cells as a delivery vehicle.
The major challenge for using gene therapy to modify C9ORF72 toxicity in patients is the complexity of how the HREs cause cell death, as discussed throughout this article. As there is both a loss of function due to reduced C9ORF72 protein levels and a gain of function due to toxic products driven by the repeats, any gene therapy approach has to take both of these processes into consideration. As of the writing of this article, there have been three clinical trials in this area. In the first trial, mutant C9ORF72 expression was suppressed by a mixed backbone ASO targeting the antisense strand in an N-of-one study, which resulted in a drop in the DPR protein poly(GP), but the patient’s ALS functional rating score was unchanged and neurofilament levels actually increased [86]. A larger study led by Biogen Inc. (Cambridge, MA, USA) used an ASO against C9ORF72 (BIIB078) in a phase 1 study; however, patients on the higher dose of the drug trended towards greater decline, so the clinical development was halted. Wave Life Sciences (Cambridge, MA, USA) used a similar ASO-targeting strategy, and this phase 1b/2 trial was also halted (ClinicalTrials.gov: NCT04931862). While there are no current trials using AAV and gene editing techniques to correct the C9ORF72 repeat expansion, many studies are under development, as described extensively in two reviews [123, 124].
In addition to gene editing approaches to treat C9ORF72, other gene therapy approaches are also applicable to all forms of ALS, as they focus on replacing cells expressing the mutation while at the same time being engineered to release powerful growth factors. One recent study showed that this approach is safe in patients with sporadic ALS [125] and opens up other opportunities for similar approaches in C9ORF72 patients.
Small Molecule Approaches to Degrade the HRE in C9ORF72-FTD/ALS
Another way to target the HRE in C9ORF72 is via a small molecule approach, which would have the advantage of being delivered orally as a pill. A particular small molecule, known as an RNase recruiting chimera (RIBOTAC), was engineered to eliminate (G4C2)n through the use of a heterobifunctional compound that selectively binds the target with one end and depletes it with the other end by activating ribonuclease L that can cleave RNA when it is in close proximity [126]. This approach decreased the abundance of (G4C2)n in vitro by being selective for the very thermodynamically stable hairpin RNA structures. After being delivered intracerebroventricularly in mice, the RIBOTAC displayed prolonged activity and decreased pathological aggregates in the cortex.
An additional small molecule approach used a nuclear RNA exosome to degrade RNA and was designed to be a blood–brain penetrant [127]. This RNA-targeted small molecule is selective in induced pluripotent stem cells and bioactive in multiple mouse models of C9ORF72-FTD/ALS upon delivery by injection. Multiple mechanisms are available to affect the decay of (G4C2)n in cells and mouse models, and dual-functioning compounds can affect the RNA in both the nucleus and cytoplasm.
Small molecules can affect RNA biology in many ways, including affecting the degradation of RNA. Known drug collections can be used to find ligands that bind RNA and that can be optimized, including being designed to be brain penetrant.
Clinical Trial Design for C9ORF72-FTD/ALS
Experiences from Alzheimer’s disease (AD) and spinal muscular atrophy suggest that treating FTD and ALS will be most successful if treatment is initiated early—ideally in pre-symptomatic stages. Such a prevention approach has been implemented in the DIAN-TU AD trial [128]. However, unlike AD, in FTD, little is known about the progression of biomarker and clinical changes that could be used to determine the criteria for enrollment or to identify the best clinical trial endpoints at different disease stages. Similar difficulties are found in ALS. The onset of genetic FTD or ALS, particularly for C9ORF72-associated FTD and ALS, is difficult to predict [8]. Further, the number of eligible patients, especially for C9ORF72-FTD, is limited, so it is challenging to power trials for efficacy.
Clinical Trials for C9ORF72—Current Landscape and Hopes for the Future
Current clinical trials for C9ORF72 ALS include both trials that enroll gene-targeted and gene-enriched clinical cohorts of patients affected by C9ORF72, respectively (Table 3) [129]. As highlighted above, the gene-targeted trials are focused on using ASOs to reduce the repeat-containing toxic RNA transcripts and DPR, while RNAi using AAV vectors for degrading the C9ORF72 repeat-containing RNAs is in pre-clinical development [129]. A recently halted trial of the ASO known as WVE-004 was selective against the RNA V1 and V3 repeat-containing transcript variants [129]. The discontinued trial of the ASO known as BIIB078 in adults with C9ORF72-associated ALS showed reduced concentrations of DPRs (poly[GP] and poly[GA]) in CSF, indicating target engagement, but results showed increased concentrations of CSF NfL compared with concentrations in the placebo group and poorer trends in measures of clinical functional outcomes [129]. Cohorts of patients with C9ORF72-ALS have been the focus of trials targeting downstream pathways of autophagy or neuroinflammation via drugs such as the oral PIKFYVE inhibitor apilimod (LAM-002a, also known as AIT-101) [69], the LINE-1 retrotransposon inhibitor TPN-101 [129], and metformin [129].
Currently, nearly all C9ORF72 ALS clinical trials are in phase 1 or 2a testing, and there are no late-phase clinical efficacy trials yet. As more C9ORF72-targeted experimental compounds move into later phases of clinical trials, it is critical to develop innovative clinical trial designs that efficiently utilize the relatively small and available pool of C9ORF72-FTD and -ALS trial participants for all phases of clinical trial testing and for all concurrently available experimental therapies. This could be achieved by more innovative and efficient early clinical phase trial designs that have small sample sizes and short treatment durations yet are adequately powered for biomarker-driven primary outcomes and enriched by trial cohorts of clinically homogenous individuals with early FTD and ALS. Such trials could aid in efficient screening of experimental compounds and assist in making sound go/no-go decisions based on in vivo human biological efficacy, CNS target engagement, pharmacokinetic signals, and safety signals. Subsequently, successful compounds could be brought forward for late-phase testing in large, traditional, randomized, placebo-controlled, clinical efficacy trials.
Preparing for FTD/ALS Prevention Trials
We have already entered an era of ALS prevention trials with the initiation of the ATLAS study in the SOD1-ALS population [130]. This trial builds on the observation that a pre-symptomatic rise in NfL predicts imminent phenoconversion to clinically manifest ALS, at least in a subset of people with highly penetrant SOD1 mutations associated with rapidly progressive disease [80]. While additional biomarkers will be needed to empower similar disease prevention trials among pre-symptomatic C9ORF72 mutation carriers, the ATLAS study has forged a path that will shed important light on future FTD/ALS prevention trials.
The FTD Prevention Initiative (www.thefpi.org) seeks to bring together global FTD cohorts to prepare for clinical trials. The majority (approx. 80%) of C9ORF72 mutation carriers in this worldwide network of approximately 1000 mutation carriers do not meet diagnostic criteria for ALS or motor neuron disease (MND), suggesting that ALS may not be the most common phenotype for C9ORF72 mutation carriers. Some individuals meet diagnostic criteria for a FTD subtype, while others have intermediate FTD-ALS phenotype and may not fully meet either diagnostic criterion. The approach to modeling disease progression jointly includes both clinical endpoints and biomarkers, determines latent “disease age” as the years to/since clinical onset, and uses Bayesian priors derived from estimates of years since onset based on clinician report of symptoms or age relative to the mutation’s mean for pre-symptomatic patients; the controls use disease age, which is based on the mutation that their family carries, such as C9ORF72, GRN, or MAPT [84]. The baseline sample population included 796 patients who are carriers of genes for FTD: 347 carriers of C9ORF72, 281 carriers of GRN, and 168 carriers of MAPT. MRI-measured NfL chain and brain atrophy were observed to diverge 20–30 years before the disease had clinical impact on carriers of C9ORF72, and the dynamics differ between the genetic groups. Disease progression and selection of endpoints are specific to the mutation that carriers have.
Prevention trials may require informative biomarkers to serve as surrogate endpoints, whereas clinical endpoints are sufficient for trials of treatments in fully symptomatic individuals. Whether someone will develop ALS, FTD, a movement disorder, or some mixed phenotype because of a C9ORF72 expansion is currently not predictable. Therefore, prevention trials that begin treatment prior to symptom onset will need to consider the potential development of multiple C9ORF72 phenotypes. The sample size of a prevention trial can be reduced by using disease age as an inclusion criterion. Clinical trial power may be further increased by Bayesian efficacy analyses and model-derived endpoints, which may be necessary for addressing rare mutations such as MAPT and GRN.
Summary of the Working Group Discussions on C9ORF72 Therapeutics and Clinical Trial Design
The highlights of the C9ORF72 Therapeutics and Clinical Trials working group are summarized in Table 4.
Conclusion
Families who live with C9ORF72 typically understand all too well that ALS and FTD exist on a spectrum. Genetic carriers are at risk of develo** ALS, FTD, or a combination thereof, with no ability at this time to predict which disease may emerge in which person. Basic scientists working on C9ORF72 recognize that their research has equal applicability to both ALS and FTD, with little ability—to date—to identify which laboratory models are relevant to which clinical disease. However, medical care and clinical research are often siloed with ALS and FTD patients seen by different clinical care centers with distinct training.
Working in silos has helped clinics to develop the specialization needed to tend to each of these highly complex diseases. However, the full spectrum of symptoms can go unrecognized, something that can be particularly painful to families dealing with unrecognized behavioral FTD symptoms in loved ones with ALS, and vice versa.
The clinical research silos of FTD versus ALS also create a number of specific problems. Both fields are actively working to detect the earliest manifestation of clinical disease in genetic carriers, a critical step to design clinical trials to test the ability of disease-modifying therapies that might protect against the development of disease. Collaboration across the two fields of expertise is critical—including an awareness of the respective bodies of literature in the two fields—with a willingness of researchers to learn from each other. This is especially important given that it is not currently known which disease will emerge in these genetic carriers. Sponsors seeking to test the effectiveness of a given treatment for C9ORF72 or shared causes of FTD/ALS must usually select one or the other condition. Two phase 2 basket trials are underway in 2023 for C9ORF72 FTD and C9ORF72 ALS, but the lack of a unified outcome measure of efficacy means that one or the other condition must be selected for phase 3 trials. Biorepositories are often built for either ALS or FTD, with different core data elements, tissue processing protocols, and participant profiles, and those differences challenge the ability to leverage resources in rare diseases where every sample and every data point are of critical importance.
The ALS and FTD communities can work together to understand genetics, pathophysiology, and disease progression. Families who carry C9ORF72 require holistic support. Specific tools may help to overcome these challenges; these include: (1) the development and use of tools to promote linkage and collaboration of care and research resources across ALS and FTD, such as minimal dataset or usage of the same tokenization tools; (2) collaboration on the inclusion criteria and outcome measures for prodromal prevention clinical trials in genetic carriers at risk of ALS and FTD; (3) increased collaboration across FTD and ALS in the support of families and individuals at risk of disease; and (4) development of training and communication resources to make it easier for ALS clinics and families to understand and support FTD-related symptoms.
References
Grassano M, Calvo A, Moglia C, Sbaiz L, Brunetti M, Barberis M, et al. Systematic evaluation of genetic mutations in ALS: a population-based study. J Neurol Neurosurg Psychiatry. 2022;93(11):1190–3.
Kirola L, Mukherjee A, Mutsuddi M. Recent updates on the genetics of amyotrophic lateral sclerosis and frontotemporal dementia. Mol Neurobiol. 2022;59(9):5673–94.
Laaksovirta H, Peuralinna T, Schymick JC, Scholz SW, Lai SL, Myllykangas L, et al. Chromosome 9p21 in amyotrophic lateral sclerosis in Finland: a genome-wide association study. Lancet Neurol. 2010;9(10):978–85.
Renton AE, Majounie E, Waite A, Simon-Sanchez J, Rollinson S, Gibbs JR, et al. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron. 2011;72(2):257–68.
Majounie E, Renton AE, Mok K, Dopper EG, Waite A, Rollinson S, et al. Frequency of the C9orf72 hexanucleotide repeat expansion in patients with amyotrophic lateral sclerosis and frontotemporal dementia: a cross-sectional study. Lancet Neurol. 2012;11(4):323–30.
Pliner HA, Mann DM, Traynor BJ. Searching for Grendel: origin and global spread of the C9ORF72 repeat expansion. Acta Neuropathol. 2014;127(3):391–6.
Murphy NA, Arthur KC, Tienari PJ, Houlden H, Chiò A, Traynor BJ, et al. Age-related penetrance of the C9orf72 repeat expansion. Sci Rep. 2017;7(1):2116.
Moore KM, Nicholas J, Grossman M, McMillan CT, Irwin DJ, Massimo L, et al. Age at symptom onset and death and disease duration in genetic frontotemporal dementia: an international retrospective cohort study. Lancet Neurol. 2020;19(2):145–56.
Arthur KC, Doyle C, Chiò A, Traynor BJ. Use of genetic testing in amyotrophic lateral sclerosis by neurologists. JAMA Neurol. 2017;74(1):125–6.
Saez-Atienzar S, Bandres-Ciga S, Langston RG, Kim JJ, Choi SW, Reynolds RH, et al. Genetic analysis of amyotrophic lateral sclerosis identifies contributing pathways and cell types. Sci Adv. 2021;7(3):eabd9036. https://doi.org/10.1126/sciadv.abd9036.
Podda MV, Grassi C. New perspectives in cyclic nucleotide-mediated functions in the CNS: the emerging role of cyclic nucleotide-gated (CNG) channels. Pflug Arch. 2014;466(7):1241–57.
DeJesus-Hernandez M, Mackenzie IR, Boeve BF, Boxer AL, Baker M, Rutherford NJ, et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron. 2011;72(2):245–56.
Ranganathan R, Haque S, Coley K, Shepheard S, Cooper-Knock J, Kirby J. Multifaceted genes in amyotrophic lateral sclerosis-frontotemporal dementia. Front Neurosci. 2020;14:684.
Boeynaems S, Bogaert E, Van Damme P, Van Den Bosch L. Inside out: the role of nucleocytoplasmic transport in ALS and FTLD. Acta Neuropathol. 2016;132(2):159-73. https://doi.org/10.1007/s00401-016-1586-5.
Moore S, Rabichow BE, Sattler R. The Hitchhiker’s guide to nucleocytoplasmic trafficking in neurodegeneration. Neurochem Res. 2020;45(6):1306–27.
Gijselinck I, Van Langenhove T, van der Zee J, Sleegers K, Philtjens S, Kleinberger G, et al. A C9orf72 promoter repeat expansion in a Flanders-Belgian cohort with disorders of the frontotemporal lobar degeneration-amyotrophic lateral sclerosis spectrum: a gene identification study. Lancet Neurol. 2012;11(1):54–65.
van Blitterswijk M, Gendron TF, Baker MC, DeJesus-Hernandez M, Finch NA, Brown PH, et al. Novel clinical associations with specific C9ORF72 transcripts in patients with repeat expansions in C9ORF72. Acta Neuropathol. 2015;130(6):863–76.
Frick P, Sellier C, Mackenzie IRA, Cheng CY, Tahraoui-Bories J, Martinat C, et al. Novel antibodies reveal presynaptic localization of C9orf72 protein and reduced protein levels in C9orf72 mutation carriers. Acta Neuropathol Commun. 2018;6(1):72.
Waite AJ, Bäumer D, East S, Neal J, Morris HR, Ansorge O, et al. Reduced C9orf72 protein levels in frontal cortex of amyotrophic lateral sclerosis and frontotemporal degeneration brain with the C9ORF72 hexanucleotide repeat expansion. Neurobiol Aging. 2014;35(7):1779.e5-1779.e13.
**ao S, MacNair L, McGoldrick P, McKeever PM, McLean JR, Zhang M, et al. Isoform-specific antibodies reveal distinct subcellular localizations of C9orf72 in amyotrophic lateral sclerosis. Ann Neurol. 2015;78(4):568–83.
** Z, Zhang M, Bruni AC, Maletta RG, Colao R, Fratta P, et al. The C9orf72 repeat expansion itself is methylated in ALS and FTLD patients. Acta Neuropathol. 2015;129(5):715–27.
Belzil VV, Bauer PO, Prudencio M, Gendron TF, Stetler CT, Yan IK, et al. Reduced C9orf72 gene expression in c9FTD/ALS is caused by histone trimethylation, an epigenetic event detectable in blood. Acta Neuropathol. 2013;126(6):895–905.
Haeusler AR, Donnelly CJ, Periz G, Simko EA, Shaw PG, Kim MS, et al. C9orf72 nucleotide repeat structures initiate molecular cascades of disease. Nature. 2014;507(7491):195–200.
Rizzu P, Blauwendraat C, Heetveld S, Lynes EM, Castillo-Lizardo M, Dhingra A, et al. C9orf72 is differentially expressed in the central nervous system and myeloid cells and consistently reduced in C9orf72, MAPT and GRN mutation carriers. Acta Neuropathol Commun. 2016;4(1):37.
**ao S, MacNair L, McLean J, McGoldrick P, McKeever P, Soleimani S, et al. C9orf72 isoforms in amyotrophic lateral sclerosis and frontotemporal lobar degeneration. Brain Res. 2016;1647:43–9.
Burberry A, Suzuki N, Wang JY, Moccia R, Mordes DA, Stewart MH, et al. Loss-of-function mutations in the C9ORF72 mouse ortholog cause fatal autoimmune disease. Sci Transl Med. 2016;8(347):347ra93.
McCauley ME, O'Rourke JG, Yáñez A, Markman JL, Ho R, Wang X, et al. C9orf72 in myeloid cells suppresses STING-induced inflammation. Nature. 2020;585(7823):96–101.
O'Rourke JG, Bogdanik L, Yanez A, Lall D, Wolf AJ, Muhammad AK, et al. C9orf72 is required for proper macrophage and microglial function in mice. Science. 2016;351(6279):1324–9.
Burberry A, Wells MF, Limone F, Couto A, Smith KS, Keaney J, et al. C9orf72 suppresses systemic and neural inflammation induced by gut bacteria. Nature. 2020;582(7810):89–94.
Atanasio A, Decman V, White D, Ramos M, Ikiz B, Lee HC, et al. C9orf72 ablation causes immune dysregulation characterized by leukocyte expansion, autoantibody production, and glomerulonephropathy in mice. Sci Rep. 2016;6:23204.
Jiang J, Cleveland DW. Bidirectional transcriptional inhibition as therapy for ALS/FTD caused by repeat expansion in C9orf72. Neuron. 2016;92(6):1160–3.
Lopez-Herdoiza MB, Bauché S, Wilmet B, Le Duigou C, Roussel D, Frah M, et al. C9ORF72 knockdown triggers FTD-like symptoms and cell pathology in mice. Front Cell Neurosci. 2023;17:1155929.
Lall D, Lorenzini I, Mota TA, Bell S, Mahan TE, Ulrich JD, et al. C9orf72 deficiency promotes microglial-mediated synaptic loss in aging and amyloid accumulation. Neuron. 2021;109(14):2275-91.e8.
Dane TL, et al. Reduced C9orf72 expression exacerbates polyGR toxicity in patient iPSC-derived motor neurons and a Type I protein arginine methyltransferase inhibitor reduces that toxicity. Front Cell Neurosci. 2023;17:1134090.
Shi Y, Lin S, Staats KA, Li Y, Chang WH, Hung ST, et al. Haploinsufficiency leads to neurodegeneration in C9ORF72 ALS/FTD human induced motor neurons. Nat Med. 2018;24(3):313–25.
Boivin M, Pfister V, Gaucherot A, Ruffenach F, Negroni L, Sellier C, et al. Reduced autophagy upon C9ORF72 loss synergizes with dipeptide repeat protein toxicity in G4C2 repeat expansion disorders. EMBO J. 2020;39(4):e100574.
Shao Q, Liang C, Chang Q, Zhang W, Yang M, Chen JF. C9orf72 deficiency promotes motor deficits of a C9ALS/FTD mouse model in a dose-dependent manner. Acta Neuropathol Commun. 2019;7(1):32.
Zhu Q, Jiang J, Gendron TF, McAlonis-Downes M, Jiang L, Taylor A, et al. Reduced C9ORF72 function exacerbates gain of toxicity from ALS/FTD-causing repeat expansion in C9orf72. Nat Neurosci. 2020;23(5):615–24.
Dong W, Zhang L, Sun C, Gao X, Guan F, Li J, et al. Knock in of a hexanucleotide repeat expansion in the C9orf72 gene induces ALS in rats. Anim Model Exp Med. 2020;3(3):237–44.
Pang W, Hu F. Cellular and physiological functions of C9ORF72 and implications for ALS/FTD. J Neurochem. 2021;157(3):334–50.
Bauer CS, Cohen RN, Sironi F, Livesey MR, Gillingwater TH, Highley JR, et al. An interaction between synapsin and C9orf72 regulates excitatory synapses and is impaired in ALS/FTD. Acta Neuropathol. 2022;144(3):437–64.
Ho WY, Navakkode S, Liu F, Soong TW, Ling SC. Deregulated expression of a longevity gene, Klotho, in the C9orf72 deletion mice with impaired synaptic plasticity and adult hippocampal neurogenesis. Acta Neuropathol Commun. 2020;8(1):155.
**ao S, McKeever PM, Lau A, Robertson J. Synaptic localization of C9orf72 regulates post-synaptic glutamate receptor 1 levels. Acta Neuropathol Commun. 2019;7(1):161.
McGoldrick P, Lau A, You Z, Durcan TM, Robertson J. Loss of C9orf72 perturbs the Ran-GTPase gradient and nucleocytoplasmic transport, generating compositionally diverse Importin β-1 granules. Cell Rep. 2023;42(3): 112134.
Sumitomo A, Tomoda T. Autophagy in neuronal physiology and disease. Curr Opin Pharmacol. 2021;60:133–40.
Wang SM, Wu HE, Yasui Y, Geva M, Hayden M, Maurice T, et al. Nucleoporin POM121 signals TFEB-mediated autophagy via activation of SIGMAR1/sigma-1 receptor chaperone by pridopidine. Autophagy. 2023;19(1):126–51.
Sivadasan R, Hornburg D, Drepper C, Frank N, Jablonka S, Hansel A, et al. C9ORF72 interaction with cofilin modulates actin dynamics in motor neurons. Nat Neurosci. 2016;19(12):1610–8.
Gu J, Lee CW, Fan Y, Komlos D, Tang X, Sun C, et al. ADF/cofilin-mediated actin dynamics regulate AMPA receptor trafficking during synaptic plasticity. Nat Neurosci. 2010;13(10):1208–15.
Giampetruzzi A, Danielson EW, Gumina V, Jeon M, Boopathy S, Brown RH, et al. Modulation of actin polymerization affects nucleocytoplasmic transport in multiple forms of amyotrophic lateral sclerosis. Nat Commun. 2019;10(1):3827.
Wurz AI, Schulz AM, O'Bryant CT, Sharp JF, Hughes RM. Cytoskeletal dysregulation and neurodegenerative disease: Formation, monitoring, and inhibition of cofilin-actin rods. Front Cell Neurosci. 2022;16:982074.
Braems E, Swinnen B, Van Den Bosch L. C9orf72 loss-of-function: a trivial, stand-alone or additive mechanism in C9 ALS/FTD? Acta Neuropathol. 2020;140(5):625–43.
Mori K, Arzberger T, Grasser FA, Gijselinck I, May S, Rentzsch K, et al. Bidirectional transcripts of the expanded C9orf72 hexanucleotide repeat are translated into aggregating dipeptide repeat proteins. Acta Neuropathol. 2013;126(6):881–93.
Mackenzie IR, Frick P, Grässer FA, Gendron TF, Petrucelli L, Cashman NR, et al. Quantitative analysis and clinico-pathological correlations of different dipeptide repeat protein pathologies in C9ORF72 mutation carriers. Acta Neuropathol. 2015;130(6):845–61.
Swinnen B, Robberecht W, Van Den Bosch L. RNA toxicity in non-coding repeat expansion disorders. EMBO J. 2020;39(1):e101112.
Swinnen B, Bento-Abreu A, Gendron TF, Boeynaems S, Bogaert E, Nuyts R, et al. A zebrafish model for C9orf72 ALS reveals RNA toxicity as a pathogenic mechanism. Acta Neuropathol. 2018;135(3):427–43.
Braems E, Bercier V, Van Schoor E, Heeren K, Beckers J, Fumagalli L, et al. HNRNPK alleviates RNA toxicity by counteracting DNA damage in C9orf72 ALS. Acta Neuropathol. 2022;144(3):465–88.
Wilson DM, 3rd, Cookson MR, Van Den Bosch L, Zetterberg H, Holtzman DM, Dewachter I. Hallmarks of neurodegenerative diseases. Cell. 2023;186(4):693–714.
Chua JP, De Calbiac H, Kabashi E, Barmada SJ. Autophagy and ALS: mechanistic insights and therapeutic implications. Autophagy. 2022;18(2):254–82.
Guo Q, Lehmer C, Martínez-Sánchez A, Rudack T, Beck F, Hartmann H, et al. In situ structure of neuronal C9orf72 Poly-GA aggregates reveals proteasome recruitment. Cell. 2018;172(4):696-705.e12.
Shao W, Todd TW, Wu Y, Jones CY, Tong J, Jansen-West K, et al. Two FTD-ALS genes converge on the endosomal pathway to induce TDP-43 pathology and degeneration. Science. 2022;378(6615):94–9.
Hayes LR, Duan L, Bowen K, Kalab P, Rothstein JD. C9orf72 arginine-rich dipeptide repeat proteins disrupt karyopherin-mediated nuclear import. Elife. 2020;9:e51685.
Cunningham KM, Maulding K, Ruan K, Senturk M, Grima JC, Sung H, et al. TFEB/Mitf links impaired nuclear import to autophagolysosomal dysfunction in C9-ALS. Elife. 2020;9:e59419.
Loveland AB, Svidritskiy E, Susorov D, Lee S, Park A, Zvornicanin S, et al. Ribosome inhibition by C9ORF72-ALS/FTD-associated poly-PR and poly-GR proteins revealed by cryo-EM. Nat Commun. 2022;13(1):2776.
Frottin F, Schueder F, Tiwary S, Gupta R, Körner R, Schlichthaerle T, et al. The nucleolus functions as a phase-separated protein quality control compartment. Science. 2019;365(6451):342–7.
Lopez-Gonzalez R, Lu Y, Gendron TF, Karydas A, Tran H, Yang D, et al. Poly(GR) in C9ORF72-related ALS/FTD compromises mitochondrial function and increases oxidative stress and DNA damage in iPSC-derived motor neurons. Neuron. 2016;92(2):383–91.
Andrade NS, Ramic M, Esanov R, Liu W, Rybin MJ, Gaidosh G, et al. Dipeptide repeat proteins inhibit homology-directed DNA double strand break repair in C9ORF72 ALS/FTD. Mol Neurodegener. 2020;15(1):13.
Safren N, Tank EM, Malik AM, Chua JP, Santoro N, Barmada SJ. Development of a specific live-cell assay for native autophagic flux. J Biol Chem. 2021;297(3):101003.
Chua JP, Bedi K, Paulsen MT, Ljungman M, Tank EMH, Kim ES, et al. Myotubularin-related phosphatase 5 is a critical determinant of autophagy in neurons. Curr Biol. 2022;32(12):2581-2595.e6.
Hung ST, Linares GR, Chang WH, Eoh Y, Krishnan G, Mendonca S, et al. PIKFYVE inhibition mitigates disease in models of diverse forms of ALS. Cell. 2023;186(4):786-802.e28.
Verde F, Otto M, Silani V. Neurofilament light chain as biomarker for amyotrophic lateral sclerosis and frontotemporal dementia. Front Neurosci. 2021;15: 679199.
Rohrer JD, Woollacott IO, Dick KM, Brotherhood E, Gordon E, Fellows A, et al. Serum neurofilament light chain protein is a measure of disease intensity in frontotemporal dementia. Neurology. 2016;87(13):1329–36.
Gendron TF, Heckman MG, White LJ, Veire AM, Pedraza O, Burch AR, et al. Comprehensive cross-sectional and longitudinal analyses of plasma neurofilament light across FTD spectrum disorders. Cell Rep Med. 2022;3(4): 100607.
Gendron TF, Daughrity LM, Heckman MG, Diehl NN, Wuu J, Miller TM, et al. Phosphorylated neurofilament heavy chain: a biomarker of survival for C9ORF72-associated amyotrophic lateral sclerosis. Ann Neurol. 2017;82(1):139–46.
Behzadi A, Pujol-Calderón F, Tjust AE, Wuolikainen A, Höglund K, Forsberg K, et al. Neurofilaments can differentiate ALS subgroups and ALS from common diagnostic mimics. Sci Rep. 2021;11(1):22128.
Gille B, De Schaepdryver M, Goossens J, Dedeene L, De Vocht J, Oldoni E, et al. Serum neurofilament light chain levels as a marker of upper motor neuron degeneration in patients with amyotrophic lateral sclerosis. Neuropathol Appl Neurobiol. 2019;45(3):291–304.
Ooi S, Patel SK, Eratne D, Kyndt C, Reidy N, Lewis C, et al. Plasma neurofilament light chain and clinical diagnosis in frontotemporal dementia syndromes. J Alzheimers Dis. 2022;89(4):1221–31.
Corrigendum to: recommendations to distinguish behavioural variant frontotemporal dementia from psychiatric disorders. Brain. 2020;143(7):e62.
Meeter LHH, Gendron TF, Sias AC, Jiskoot LC, Russo SP, Donker Kaat L, et al. Poly(GP), neurofilament and grey matter deficits in C9orf72 expansion carriers. Ann Clin Transl Neurol. 2018;5(5):583–97.
Cajanus A, Katisko K, Kontkanen A, Jääskeläinen O, Hartikainen P, Haapasalo A, et al. Serum neurofilament light chain in FTLD: association with C9orf72, clinical phenotype, and prognosis. Ann Clin Transl Neurol. 2020;7(6):903–10.
Benatar M, Wuu J, Andersen PM, Lombardi V, Malaspina A. Neurofilament light: a candidate biomarker of presymptomatic amyotrophic lateral sclerosis and phenoconversion. Ann Neurol. 2018;84(1):130–9.
Rojas JC, Wang P, Staffaroni AM, Heller C, Cobigo Y, Wolf A, et al. Plasma neurofilament light for prediction of disease progression in familial frontotemporal lobar degeneration. Neurology. 2021;96(18):e2296–312.
van der Ende EL, Meeter LH, Poos JM, Panman JL, Jiskoot LC, Dopper EGP, et al. Serum neurofilament light chain in genetic frontotemporal dementia: a longitudinal, multicentre cohort study. Lancet Neurol. 2019;18(12):1103–11.
Saracino D, Dorgham K, Camuzat A, Rinaldi D, Rametti-Lacroux A, Houot M, et al. Plasma NfL levels and longitudinal change rates in C9orf72 and GRN-associated diseases: from tailored references to clinical applications. J Neurol Neurosurg Psychiatry. 2021;92(12):1278–88.
Staffaroni AM, Quintana M, Wendelberger B, Heuer HW, Russell LL, Cobigo Y, et al. Temporal order of clinical and biomarker changes in familial frontotemporal dementia. Nat Med. 2022;28(10):2194–206.
Gendron TF, Chew J, Stankowski JN, Hayes LR, Zhang YJ, Prudencio M, et al. Poly(GP) proteins are a useful pharmacodynamic marker for C9ORF72-associated amyotrophic lateral sclerosis. Sci Transl Med. 2017;9(383):eaai7866.
Tran H, Moazami MP, Yang H, McKenna-Yasek D, Douthwright CL, Pinto C, et al. Suppression of mutant C9orf72 expression by a potent mixed backbone antisense oligonucleotide. Nat Med. 2022;28(1):117–24.
Lagier-Tourenne C, Baughn M, Rigo F, Sun S, Liu P, Li HR, et al. Targeted degradation of sense and antisense C9orf72 RNA foci as therapy for ALS and frontotemporal degeneration. Proc Natl Acad Sci USA. 2013;110(47):E4530–9.
Donnelly CJ, Zhang PW, Pham JT, Haeusler AR, Mistry NA, Vidensky S, et al. RNA toxicity from the ALS/FTD C9ORF72 expansion is mitigated by antisense intervention. Neuron. 2013;80(2):415–28.
Su Z, Zhang Y, Gendron TF, Bauer PO, Chew J, Yang WY, et al. Discovery of a biomarker and lead small molecules to target r(GGGGCC)-associated defects in c9FTD/ALS. Neuron. 2014;83(5):1043–50.
O’Rourke JG, Bogdanik L, Muhammad A, Gendron TF, Kim KJ, Austin A, et al. C9orf72 BAC transgenic mice display typical pathologic features of ALS/FTD. Neuron. 2015;88(5):892–901.
Lehmer C, Oeckl P, Weishaupt JH, Volk AE, Diehl-Schmid J, Schroeter ML, et al. Poly-GP in cerebrospinal fluid links C9orf72-associated dipeptide repeat expression to the asymptomatic phase of ALS/FTD. EMBO Mol Med. 2017;9(7):859–68.
Wilson KM, Katona E, Glaria I, Carcolé M, Swift IJ, Sogorb-Esteve A, et al. Development of a sensitive trial-ready poly(GP) CSF biomarker assay for C9orf72-associated frontotemporal dementia and amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry. 2022;93(7):761–71.
Jiang J, Zhu Q, Gendron TF, Saberi S, McAlonis-Downes M, Seelman A, et al. Gain of toxicity from ALS/FTD-linked repeat expansions in C9ORF72 is alleviated by antisense oligonucleotides targeting GGGGCC-containing RNAs. Neuron. 2016;90(3):535–50.
Krishnan G, Raitcheva D, Bartlett D, Prudencio M, McKenna-Yasek DM, Douthwright C, et al. Poly(GR) and poly(GA) in cerebrospinal fluid as potential biomarkers for C9ORF72-ALS/FTD. Nat Commun. 2022;13(1):2799.
Liu Y, Dodart JC, Tran H, Berkovitch S, Braun M, Byrne M, et al. Variant-selective stereopure oligonucleotides protect against pathologies associated with C9orf72-repeat expansion in preclinical models. Nat Commun. 2021;12(1):847.
Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, Chou TT, et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science. 2006;314(5796):130–3.
Ling JP, Pletnikova O, Troncoso JC, Wong PC. TDP-43 repression of nonconserved cryptic exons is compromised in ALS-FTD. Science. 2015;349(6248):650–5.
Klim JR, Williams LA, Limone F, Guerra San Juan I, Davis-Dusenbery BN, Mordes DA, et al. ALS-implicated protein TDP-43 sustains levels of STMN2, a mediator of motor neuron growth and repair. Nat Neurosci. 2019;22(2):167–79.
Prudencio M, Humphrey J, Pickles S, Brown AL, Hill SE, Kachergus JM, et al. Truncated stathmin-2 is a marker of TDP-43 pathology in frontotemporal dementia. J Clin Invest. 2020;130(11):6080–92.
Melamed Z, Lopez-Erauskin J, Baughn MW, Zhang O, Drenner K, Sun Y, et al. Premature polyadenylation-mediated loss of stathmin-2 is a hallmark of TDP-43-dependent neurodegeneration. Nat Neurosci. 2019;22(2):180–90.
Ma XR, Prudencio M, Koike Y, Vatsavayai SC, Kim G, Harbinski F, et al. TDP-43 represses cryptic exon inclusion in the FTD-ALS gene UNC13A. Nature. 2022;603(7899):124–30.
Brown AL, Wilkins OG, Keuss MJ, Hill SE, Zanovello M, Lee WC, et al. TDP-43 loss and ALS-risk SNPs drive mis-splicing and depletion of UNC13A. Nature. 2022;603(7899):131–7.
Irwin KE, Jasin P, Braunstein KE, Sinha I, Bowden KD, Moghekar A, et al. A fluid biomarker reveals loss of TDP-43 splicing repression in pre-symptomatic ALS. bioRxiv. 2023. https://doi.org/10.1101/2023.01.23.525202.
Seddighi S, Qi YA, Brown AL, Wilkins OG, Bereda C, Belair C, et al. Mis-spliced transcripts generate de novo proteins in TDP-43-related ALS/FTD. bioRxiv. 2023. https://doi.org/10.1101/2023.01.23.525149.
Li Hi Shing S, McKenna MC, Siah WF, Chipika RH, Hardiman O, Bede P. The imaging signature of C9orf72 hexanucleotide repeat expansions: implications for clinical trials and therapy development. Brain Imaging Behav. 2021;15(5):2693–719.
Floeter MK, Bageac D, Danielian LE, Braun LE, Traynor BJ, Kwan JY. Longitudinal imaging in C9orf72 mutation carriers: Relationship to phenotype. Neuroimage Clin. 2016;12:1035–43.
Floeter MK, Danielian LE, Braun LE, Wu T. Longitudinal diffusion imaging across the C9orf72 clinical spectrum. J Neurol Neurosurg Psychiatry. 2018;89(1):53–60.
Bocchetta M, Iglesias JE, Neason M, Cash DM, Warren JD, Rohrer JD. Thalamic nuclei in frontotemporal dementia: Mediodorsal nucleus involvement is universal but pulvinar atrophy is unique to C9orf72. Hum Brain Mapp. 2020;41(4):1006–16.
Van Laere K, Vanhee A, Verschueren J, De Coster L, Driesen A, Dupont P, et al. Value of 18fluorodeoxyglucose-positron-emission tomography in amyotrophic lateral sclerosis: a prospective study. JAMA Neurol. 2014;71(5):553–61.
Lee SE, Khazenzon AM, Trujillo AJ, Guo CC, Yokoyama JS, Sha SJ, et al. Altered network connectivity in frontotemporal dementia with C9orf72 hexanucleotide repeat expansion. Brain. 2014;137(Pt 11):3047–60.
Agosta F, Ferraro PM, Riva N, Spinelli EG, Domi T, Carrera P, et al. Structural and functional brain signatures of C9orf72 in motor neuron disease. Neurobiol Aging. 2017;57:206–19.
Bertrand A, Wen J, Rinaldi D, Houot M, Sayah S, Camuzat A, et al. Early cognitive, structural, and microstructural changes in presymptomatic c9orf72 carriers younger than 40 years. JAMA Neurol. 2018;75(2):236–45.
Rohrer JD, Nicholas JM, Cash DM, van Swieten J, Dopper E, Jiskoot L, et al. Presymptomatic cognitive and neuroanatomical changes in genetic frontotemporal dementia in the Genetic Frontotemporal dementia Initiative (GENFI) study: a cross-sectional analysis. Lancet Neurol. 2015;14(3):253–62.
Caverzasi E, Battistella G, Chu SA, Rosen H, Zanto TP, Karydas A, et al. Gyrification abnormalities in presymptomatic c9orf72 expansion carriers. J Neurol Neurosurg Psychiatry. 2019;90(9):1005–10.
Gorges M, Vercruysse P, Müller HP, Huppertz HJ, Rosenbohm A, Nagel G, et al. Hypothalamic atrophy is related to body mass index and age at onset in amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry. 2017;88(12):1033–41.
Lee SE, Sias AC, Mandelli ML, Brown JA, Brown AB, Khazenzon AM, et al. Network degeneration and dysfunction in presymptomatic C9ORF72 expansion carriers. Neuroimage Clin. 2017;14:286–97.
Mutsaerts H, Mirza SS, Petr J, Thomas DL, Cash DM, Bocchetta M, et al. Cerebral perfusion changes in presymptomatic genetic frontotemporal dementia: a GENFI study. Brain. 2019;142(4):1108–20.
Diehl-Schmid J, Licata A, Goldhardt O, Förstl H, Yakushew I, Otto M, et al. FDG-PET underscores the key role of the thalamus in frontotemporal lobar degeneration caused by C9ORF72 mutations. Transl Psychiatry. 2019;9(1):54.
Popuri K, Beg MF, Lee H, Balachandar R, Wang L, Sossi V, et al. FDG-PET in presymptomatic C9orf72 mutation carriers. Neuroimage Clin. 2021;31: 102687.
Proudfoot M, Rohenkohl G, Quinn A, Colclough GL, Wuu J, Talbot K, et al. Altered cortical beta-band oscillations reflect motor system degeneration in amyotrophic lateral sclerosis. Hum Brain Mapp. 2017;38(1):237–54.
Querin G, Bede P, El Mendili MM, Li M, Pélégrini-Issac M, Rinaldi D, et al. Presymptomatic spinal cord pathology in c9orf72 mutation carriers: a longitudinal neuroimaging study. Ann Neurol. 2019;86(2):158–67.
van Veenhuijzen K, Westeneng HJ, Tan HHG, Nitert AD, van der Burgh HK, Gosselt I, et al. Longitudinal effects of asymptomatic C9orf72 carriership on brain morphology. Ann Neurol. 2023;93(4):668–80.
Amado DA, Davidson BL. Gene therapy for ALS: a review. Mol Ther. 2021;29(12):3345–58.
Meijboom KE, Brown RH. Approaches to gene modulation therapy for ALS. Neurotherapeutics. 2022;19(4):1159–79.
Baloh RH, Johnson JP, Avalos P, Allred P, Svendsen S, Gowing G, et al. Transplantation of human neural progenitor cells secreting GDNF into the spinal cord of patients with ALS: a phase 1/2a trial. Nat Med. 2022;28(9):1813–22.
Bush JA, Aikawa H, Fuerst R, Li Y, Ursu A, Meyer SM, et al. Ribonuclease recruitment using a small molecule reduced c9ALS/FTD r(G(4)C(2)) repeat expansion in vitro and in vivo ALS models. Sci Transl Med. 2021;13(617):eabd5991.
Bush JA, Meyer SM, Fuerst R, Tong Y, Li Y, Benhamou RI, et al. A blood-brain penetrant RNA-targeted small molecule triggers elimination of r(G(4)C(2))(exp) in c9ALS/FTD via the nuclear RNA exosome. Proc Natl Acad Sci USA. 2022;119(48):e2210532119.
Bateman RJ, Benzinger TL, Berry S, Clifford DB, Duggan C, Fagan AM, et al. The DIAN-TU next generation Alzheimer’s prevention trial: Adaptive design and disease progression model. Alzheimers Dement. 2017;13(1):8–19.
Garret M, Mehta K, Chen JY, Babu S. Rise of the genomic medicine era in amyotrophic lateral sclerosis. Pract Neurol. 2023;22(6):22–7.
Benatar M, et al. Design of a randomized, placebo-controlled, phase 3 trial of tofersen initiated in clinically presymptomatic SOD1 variant carriers: the ATLAS study. Neurotherapeutics. 2022;19(4):1248–58.
Acknowledgements
We would like to acknowledge the recent passing of Mr. Mark Nilsson who attended this summit with his wife and caretaker Ms. Paula Nilsson. We are honored to have met Mark and appreciated his perspectives as an individual living with ALS during our discussions. We would also like to thank David and Weezie Reese for their support and encouragement to host this summit, and Karis Anderson for her support towards the organization of this summit.
Medical Writing/Editorial Assistance
Kathy Boltz, PhD, of On Point Scientific, LLC, provided medical writing support, and Sharese Terrell Willis, PhD, of Doc’s Editing Shop, LLC, provided editorial support. This assistance was funded by the authors.
Funding
The summit was funded by the Arizona Community Foundation through a generous donation to the Barrow Neurological Foundation. This work was also supported by the Intramural Research Program of the NIH, National Institute on Aging (ZIAAG000933-15) to BJT. The journal’s fee was waived.
Author information
Authors and Affiliations
Consortia
Contributions
Rita Sattler: drafting, finalizing and submitting the manuscript. Bryan J. Traynor: drafting the manuscript. Janice Robertson: drafting the manuscript. Ludo Van Den Bosch: drafting the manuscript. Sami J. Barmada: drafting the manuscript. Clive Svendsen: drafting the manuscript. Matthew D. Disney: drafting the manuscript. Tania F. Gendron: drafting the manuscript. Philip C. Wong: drafting the manuscript. Martin R. Turner: drafting the manuscript. Adam Boxer: drafting the manuscript. Suma Babu: drafting the manuscript. Michael Benatar: drafting the manuscript. Michael Kurnellas: drafting the manuscript. Jonathan D. Rohrer: drafting the manuscript. Christopher J. Donnelly: drafting the manuscript. Lynette M. Bustos: generating figure. Kendall Van Keuren-Jensen: drafting the manuscript. Penny A. Dacks: drafting the manuscript. Marwan N. Sabbagh: drafting the manuscript.
Corresponding authors
Ethics declarations
Conflict of Interest
Janice Robertson, Sami J. Barmada, Clive Svendsen, Tania F. Gendron, Philip C. Wong, Martin R. Turner, Adam Boxer, Michael Kurnellas, Lynette M Bustos, Kendall Van Keuren-Jensen have nothing to disclose. Rita Sattler is a member of the Scientific Advisory Board of Spinogenix Inc, the LiveLikeLou Foundation, the Robert Packard Center and Northeast ALS consortium. Bryan J. Traynor holds patents on the diagnostic and therapeutic implications of the C9orf72 repeat expansion. Ludo Van Den Bosch is head of the Scientific Advisory Board of Augustine Therapeutics (Leuven, Belgium) and is part of the Investment Advisory Board of Droia Ventures (Meise, Belgium). Jonathan D Rohrer has provided consultancy or been on an advisory board for Novartis, Wave Life Sciences, Prevail, Alector, Aviado Bio, Arkuda Therapeutics, and Denali Therapeutics. Matthew D. Disney is a founder of Expansion Therapeutics. Christopher J Donnelly is cofounder and advisor for Confluence Therapeutics. Michael Benatar reports grants from the National Institutes of Health, the Muscular Dystrophy Association, and the ALS Association; and receives consulting fees for Alector, Alexion, Annexon, Arrowhead, Biogen, Cartesian, Denali, Eli Lilly, Horizon, Immunovant, Novartis, Roche, Sanofi, Takeda, UCB, and UniQure. The University of Miami has licensed intellectual property to Biogen Inc. to support the design of the ATLAS study. He has a provisional patent ‘Determining Onset of ALS’ (US Patent No. 17/941,117). He serves as a member of the Board of Trustees for the ALS Association. Suma Babu has received research funding from Biogen Inc. (including the BIIB078 ASO trial for C9ORF72 ALS), AI therapeutics, Novartis, Orion, Voyager Therapeutics, Medicinova, Ionis, American Academy of Neurology, AANEM Foundation, and Muscular Dystrophy Association. Penny A. Dacks is an employee of the Association for Frontotemporal Degeneration (AFTD), King of Prussia, PA, USA. Marwan N. Sabbagh is a consultant for Roche-Genentech, Eisai, Lilly, Synaptogenix, NeuroTherapia, T3D, Signant Health, Novo Nordisk, Corium, Prothena, KeiferRx; is on the board of directors at CervoMed; and receives stock options/owns stocks from Athira, Lighthouse Pharmaceuticals, Alzheon. Michael Kurnellas, new affiliation: Neuron23, Inc., South San Francisco, CA, USA. Lynette M Bustos, new affiliation: Ionis Pharmaceuticals, Inc., Carlsbad, CA, USA.
Ethical Approval
The contents of this manuscript are based on discussions at the C9orf72 Summit and did not entail any animal or human subjects research that required review/approval by the IACUC or IRB. All patients and pre-symptomatic gene mutation carriers who participated and/or disclosed their gene mutation status, did so of their own volition.
Additional information
The members of the Attendees of the inaugural C9ORF72 FTD/ALS Summit are listed in the online Supplementary material.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Sattler, R., Traynor, B.J., Robertson, J. et al. Roadmap for C9ORF72 in Frontotemporal Dementia and Amyotrophic Lateral Sclerosis: Report on the C9ORF72 FTD/ALS Summit. Neurol Ther 12, 1821–1843 (2023). https://doi.org/10.1007/s40120-023-00548-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40120-023-00548-8