
Abstract
Background
In recent years, the significance of the nervous system in the tumor microenvironment has gained increasing attention. The bidirectional communication between nerves and cancer cells plays a critical role in tumor initiation and progression. Perineural invasion (PNI) occurs when tumor cells invade the nerve sheath and/or encircle more than 33% of the nerve circumference. PNI is a common feature in various malignancies and is associated with tumor invasion, metastasis, cancer-related pain, and unfavorable clinical outcomes. The colon and rectum are highly innervated organs, and accumulating studies support PNI as a histopathologic feature of colorectal cancer (CRC). Therefore, it is essential to investigate the role of nerves in CRC and comprehend the mechanisms of PNI to impede tumor progression and improve patient survival.
Conclusion
This review elucidates the clinical significance of PNI, summarizes the underlying cellular and molecular mechanisms, introduces various experimental models suitable for studying PNI, and discusses the therapeutic potential of targeting this phenomenon. By delving into the intricate interactions between nerves and tumor cells, we hope this review can provide valuable insights for the future development of CRC treatments.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Colorectal cancer (CRC) consistently ranks as one of the most prevalent malignant gastrointestinal tumors and remains a leading cause of cancer-related deaths [1]. Despite advances in surgical techniques and adjuvant chemoradiotherapy, the long-term survival rates for CRC patients remain unsatisfactory [2, 81]. It is worth noting that the netrin-1 receptors, deleted in colorectal cancer (DCC) and uncoordinated 5 homolog (UNC5H), are often silenced in CRC through mechanisms such as loss of heterozygosity or epigenetic modifications. The methylation-mediated repression of several UNC5H genes holds promise as a diagnostic and prognostic marker in CRC [82, 83].
Ephrin/Eph signaling plays a crucial role in the regulation of cell adhesion, migration, and sorting. Ephrin-A proteins are anchored to the cell surface by a glycosylphosphatidylinositol anchor and interact with class A receptors (EphA), while ephrin-B proteins are transmembrane proteins that bind to class B receptors (EphB) [84]. Specifically, ephrin-A1 expression serves as a valuable marker for predicting a higher risk of recurrence and cancer-related mortality in patients who have undergone curative resection for CRC [85]. In vitro assays have demonstrated that decreased ephrin-A1 expression is associated with reduced proliferative activity, as well as decreased invasion and migration of CRC cell lines [85]. Furthermore, research has shown higher expression of ephrin-B2 in colon carcinoma compared to adjacent normal tissues [86]. Additionally, ephrin-B1 and ephrin-B2 are preferentially incorporated into exosomes derived from CRC [87]. These findings suggest that ephrin-B1 and ephrin-B2 could potentially serve as diagnostic biomarkers. Interestingly, the overexpression of ephrin-B2 in CRC has been found to be associated with increased tumor angiogenesis, unexpectedly resulting in reduced tumor growth due to the structural abnormalities of the new vessels [88].
SEMA proteins serve as cues for axons to navigate through their environment, with some acting as attractants and others as repellants [89]. SEMA3C is implicated in the development of the ENS. A loss-of-function mutation in SEMA3C leads to Hirschsprung’s disease, a congenital disease in which the ENS fails to form in parts of the intestine [90]. In Crohn’s disease, the increased SEMA3C expression in intestinal crypts is associated with a reduction in mucosal sympathetic nerve fibers [91]. SEMA3C is also involved in various oncogenic processes in colon, gastric, lung, liver, breast, and pancreatic cancers [92]. SEMA3D also plays a role in the formation of neuronal networks. Reducing SEMA3D or its receptor plexin D1 expression inhibits the invasion of tumor cells towards the nerves and decreases the nerve density in tumor tissues [93]. Additionally, SEMA4D and its receptor plexin B1 are highly expressed in tumor cells and nerves, respectively, promoting PNI in colon cancer [94].
Slits (Slit1, Slit2, Slit3) are secreted glycoproteins that bind to the membrane-bound neuronal guidance receptors Roundabout (Robo) family. The Slit-Robo pathway plays important roles in neuronal guidance and tumorigenesis [95]. Tomasini et al. discovered that Slit2 secreted by cancer-associated fibroblasts could increase dorsal root ganglion (DRG) neuron neurite outgrowth and Schwann cell proliferation/migration by regulating N-cadherin/β-catenin signaling [96]. Several studies have shown that Slit2 is downregulated in CRC tissues compared to adjacent tissues and can inhibit CRC cell migration in Robo-dependent manners [97, 98]. However, Yao et al. found that the serum levels of Slit2 were significantly increased in CRC patients compared to healthy controls. Blocking the binding of Slit2 to Robo1 could inhibit CRC cell migration and metastasis through the TGF-β/Smads signaling pathway [99]. Therefore, further exploration is needed to understand the functions of Slits in CRC.
4.3 Adhesion proteins
Cell adhesion molecules are a group of transmembrane proteins that mediate cell-cell and cell-extracellular matrix interactions. The neural cell adhesion molecule (NCAM), belonging to the immunoglobulin superfamily of adhesion molecules, is expressed on the surface of neurons and Schwann cells [100]. During nervous system maturation, NCAM plays an important role in facilitating neuronal migration, axon/dendrite outgrowth, and synaptogenesis [100]. NCAM is also implicated in tumor growth and metastasis and is associated with PNI in various types of cancers [101,102,103]. A recent study provided evidence that NCAM expression by Schwann cells serves as a guiding mechanism for cancer cells to migrate towards nerves, thereby promoting cancer cell invasion and dispersion, ultimately facilitating PNI [104]. Previously, NCAM was believed to act as a tumor suppressor in CRC; tumors lacking NCAM expression were associated with aggressive clinical behaviors [105]. However, subsequent studies have demonstrated that NCAM expression is elevated in human CRC tissues and cell lines, and its expression is positively correlated with the presence and number of lymph node metastases [106, 107]. Moreover, the NCAM gene has been identified as a target of β-catenin, and the induction of NCAM transcription is thought to play a role in colon cancer tumorigenesis, probably by promoting cell growth and motility [107].
The L1 family of cell adhesion molecule (L1CAM) shares important structural and functional features with NCAM and is also involved in signaling transductions related to neuron migration and neurite outgrowth [108]. L1CAM upregulates cell-matrix adhesion by initiating the MAPK ERK1/2 signaling downstream cascade, which triggers the transcription of integrins, thereby enhancing cell motility [109]. In CRC, L1CAM has been proven to be a potential marker for tumor invasiveness, metastasis, lymph node metastasis, and worse patient outcomes [110,111,112]. Moreover, L1CAM mRNA expression is significantly higher at the invasive front compared to the center of the tumor, indicating a supportive role of L1CAM in CRC dissemination [113]. L1CAM can also mediate tumor-nerve adhesion, allowing CRC cells to migrate along enteric neurons in vitro and potentially contributing to PNI [114].
4.4 Neurotransmitters
During tumor development, cancer cells and peripheral nerves release numerous neurotransmitters directly into the TME, which can activate corresponding receptors and influence various cellular signaling pathways, promoting cancer cell proliferation and progression. Neurotransmitters are mainly classified into four types based on their chemical composition: choline (Ach), catecholamines (adrenaline, NA, and dopamine), amino acids (excitatory transmitter such as glutamic acid and aspartic acid; inhibitory transmitters such as γ-aminobutyric acid and glycine), and neuropeptides (vasoactive intestinal peptide) [115]. These neurotransmitters play critical roles in maintaining various physiological functions and stress responses.
Adrenergic signaling is reported to be upregulated in CRC. Pathology reports have shown that β adrenergic receptors (ADRβ) are more abundant in the tumor site, which is usually correlated with worse prognosis [116]. Adrenergic transmitters can promote the proliferation of human colon cancer cell line HT-29 by inducing the expression of cyclooxygenase 2 (COX-2), VEGF, prostaglandin E2, and MMP-9. This effect can be rescued by ADRβ antagonists or COX-2 inhibitors [117]. Furthermore, NA has been shown to stimulate the proliferation and dissemination of colon cancer cells by inducing phosphorylation of cAMP response element-binding protein 1 (CREB1), thereby activating the CREB1/miRNA-373 axis [118]. Additionally, adrenaline (AD) can increase the expression of COX-2 and interleukin (IL)-10 in macrophages, suppressing CD8 + T lymphocytes proliferation and interferon-γ production, thus facilitating immune escape in colon cancer [119].
γ-aminobutyric acid (GABA) is a neuromodulator that helps restore homeostasis. It inhibits pro-inflammatory cytokines, stimulates anti-inflammatory cytokines, decreases intestinal permeability, and promotes neuronal survival [120]. The GABA shunt involves the enzymatic conversion of glutamate to GABA by glutamate decarboxylase 1 (GAD1) and GAD2, followed by the catabolism of GABA by GABA-transaminase (ABAT). Notably, GAD1 expression is upregulated in CRC cell lines and tumor tissues, while ABAT expression is downregulated, suggesting the accumulation of GABA in CRC may be attributed to elevated GAD1 and a lack of ABAT [121]. In a mouse model of colon cancer, GABA can directly bind to the GABA type A receptor (GABAAR) on CD8 + T lymphocytes, reducing anti-tumor immunity and facilitating tumor growth [122]. Additionally, Huang et al. discovered that GABA activates the GABA type B receptor (GABABR), leading to the enhancement of β-catenin signaling, promoting CRC cell proliferation, and inhibiting the intratumoral infiltration of CD8 + T lymphocytes, resulting in immunosuppression [121]. Furthermore, lower GABABR1 expression in tumor tissues impairs CRC cell migration and invasion by regulating epithelial-mesenchymal transition, while higher GABABR1 expression is associated with longer survival in CRC patients [123]. However, other studies show that GABA acting at the GABABR can inhibit colon cancer cell migration in vitro, enhance the anti-tumor efficacy of oxaliplatin, and reduce metastasis of CRC in mice [124, 125].
A vast number of enteric neurons are cholinergic and have excitatory or inhibitory control over gastrointestinal motility by releasing Ach [24]. Parasympathetic innervation via Ach and its receptors is observed in later stages of CRC and is associated with a poor prognosis, suggesting that cholinergic parasympathetic nerves could signal to CRC tissue and exacerbate the disease [126]. Muscarinic Ach receptors are the predominant Ach receptors distributed in the gut and can induce the growth of colon cancer by activating the EGFR/ERK signaling pathway, with muscarinic Ach receptors 3 (M3R) significantly overexpressed in CRC lesions [127]. The activation of nicotinic Ach receptors leads to increased cell proliferation and decreased apoptosis in human colon cell lines [128, 129].In gastric cancer, Ach released by cholinergic nerves and Tuft cells contributes to tumorigenesis, stimulating the production of NGF from epithelial cells, which further increases nerve density and Ach release in turn [63]. Notably, Ach can also be released by colon cancer cells in an autocrine manner [130], making it more complicated to observe neuron-cancer crosstalk.
The first neurotransmitter associated with CRC is vasoactive intestinal peptide (VIP) [131]. VIP is widely expressed in the central and peripheral nervous system, as well as in the gastrointestinal tract. In peripheral nerve crush or transection models, an increase in VIP immunoreactivity and mRNA expression has been observed, indicating the involvement of VIP in the process of nerve repair [132, 133]. In the gastrointestinal tract, VIP primarily acts as an intestinal relaxant and reduces intestinal permeability to counterbalance the effects of permeability-increasing factors such as Ach [134]. The role of VIP in the intestinal epithelium is controversial. On the one hand, the addition of VIP to cultures of the CRC cell lines stimulates cell proliferation via the activation of the MAPK pathway in a time- and concentration-dependent manner [135]. In an azoxymethane-induced mice CRC model, the administration of VIP before and during azoxymethane treatment leads to a significant increase in the incidence of colonic tumors [136]. On the other hand, contradictory evidence indicates that VIP attenuates the motility and invasiveness potential of colon cancer cells [137], and inhibits liver metastasis partly due to the prevention of tumor angiogenesis [138]. The mechanisms underlying these different effects of VIP are yet to be determined.
5 Cells in TME related to PNI
Emerging evidence highlights the critical role of TME in tumor progression [139]. The involvement of the nervous system within the TME has also gained attention [140,141,142,143,144]. Most cells in the TME express receptors for neurotransmitters such as NA and Ach, indicating that the crosstalk between different cells and neurons can potentially affect the pathogenesis of tumor. In this review, we will focus on the major cell types that support nerve-cancer interactions within and adjacent to the peripheral nerve sheath (Fig. 4).

Cellular crosstalk in the TME. Schwann cells regulate tumor cell proliferation, invasion, and PNI through membrane protein (NCAM) and secretory proteins (TGF-β, LICAM, IL-8, NGF). Tumor cells and Schwann cells secrete CSF-1 or CCL2 to recruit macrophages to the site of PNI, which aggravates nerve injury and tumor invasion. In turn, macrophages release GDNF, promoting cancer migration. Macrophages also secret Slit3, which guides Schwann cells and fibroblasts in forming a peripheral nerve bridge during nerve injury. Both macrophages and fibroblasts can release LIF, which promotes Schwann cell migration and neural plasticity. Fibroblasts also stimulate Schwann cell proliferation and neural remodeling via secreting Slit2. Additionally, the combination of CXCL12 and CXCR4 induces Schwann cell migration and infiltration
5.1 Schwann cells
Schwann cells, originating from neural crest cells, are the neurogliocytes of the periphery nervous system [145]. They are essential components of the nerve microenvironment, playing crucial roles in nurturing neurons during development, supporting neuronal repair, and regeneration processes [60, 146]. There is evidence that Schwann cells can migrate towards colon cancer even before any neural invasion, suggesting their involvement in initiating PNI [147]. During PNI, Schwann cells interact with tumor cells directly through plasma membrane proteins such as NCAM and indirectly via secretory proteins such as L1CAM and transforming growth factor-beta (TGF-β), thereby augmenting the aggressive capacity of tumor cells and promoting PNI in vitro and in vivo [104, 148, 149]. After contacting cancer cells, Schwann cells can intercalate between cancer cells, stimulating tumor protrusion and dispersion. These processes promote cancer cells migration and invasion along nerves [104]. Schwann cells can also assemble into “tumor-activated Schwann cell tracks”, envelop cancer cells and enhance their motility along the pathways of tissue innervation [150]. Additionally, Schwann cells secrete C-C motif chemokine ligand (CCL) 2, which recruits macrophages to the site of PNI, degrades collagen IV of the nerve perineurium, and ultimately aggravates nerve injury and tumor invasion [151]. Notably, recent studies have demonstrated reciprocal communication between Schwann cells and CRC cells. IL-8 and NGF from Schwann cell-conditioned medium could promote CRC proliferation, invasion and metastasis, and may serve as potential therapeutic targets for the treatment of CRC [152, 153].
5.2 Immune cells
The interaction among cancer cells, nerves, and immune cells is essential for tumor progression. During tumorigenesis, cancer cells secrete colony-stimulating factor 1 (CSF-1) to recruit macrophages, which, in turn, release GDNF, promoting cancer migration as well as nerve invasion [154]. It has been reported that tumor-associated macrophages (TAMs), a pro-tumor M2 subtype of macrophage, express more GDNF than resting macrophages [154]. Muscularis macrophages have been shown to interact directly with enteric neurons and enteric glial cells, regulating inflammation and peristaltic activity of the colon [155, 156]. Macrophages can secret Slit3, which guides Schwann cells and fibroblasts in forming an appropriate peripheral nerve bridge, ensuring axon targeting to the distal nerve stump following injury [157]. Besides, both macrophages and fibroblasts secrete the leukemia inhibitory factor (LIF), which promotes Schwann cell migration and neural plasticity, leading to enhanced neurite outgrowth [158].
Lymphocytes exert both pro- and anti-tumor effects during the dynamic modulation of the TME. At the intestinal mucosal barrier, lymphocytes have been observed to co-localize with neurons, glial cells, and neuroendocrine cells, and the crosstalk between lymphocytes and nerves maintains mucosal homeostasis [159]. Famulski et al. demonstrated a negative correlation between PNI and the infiltration of stromal tumor-infiltrating lymphocytes (TILs) in CRC, while no correlation was found with intraepithelial TIL infiltration [160]. Similarly, Huh et al. observed a correlation between a low proportion of TILs and the occurrence of PNI in CRC [161]. Moreover, CRC tumors without early metastatic invasion exhibit increased numbers of CD8 + T lymphocytes, along with elevated expression of markers associated with T lymphocyte migration, activation, and differentiation [20]. Mechanistically, the input of sympathetic nervous system into the TME inhibits cancer cell production of type I interferon and the recruitment of M1 macrophages, natural killer cells, and CD8 + T lymphocytes, thereby compromising tumor immune surveillance and restraining anti-tumor inflammation [162]. Conversely, the parasympathetic nervous system, specifically the vagus nerve, can modulate the immune microenvironment through cholinergic signaling, promoting tumor growth by impairing CD8 + T lymphocytes infiltration and Th1 differentiation, partly through HDAC-mediated inhibition of CCL5 expression [163].
5.3 Fibroblasts
Cancer-associated fibroblasts (CAFs), the predominant stromal cells in CRC, can release growth factors, cytokines, pro-angiogenic factors, and extracellular matrix proteins, which contribute to carcinogenesis, angiogenesis, and PNI [164,165,166]. Importantly, fibroblasts are the main cellular constituents of the perineurium [167]. Alterations in the perineurium composition may increase its permeability and compromise the protective barrier against tumor cell invasion. In CRC, CAFs release stromal cell-derived factor 1, also known as C-X-C motif chemokine (CXCL) 12, which has been shown to accelerate CRC metastasis and cisplatin resistance [168, 169]. The CXCL12 and its receptor C-X-C motif chemokine receptor (CXCR) 4 axis can induce tumor infiltration by Schwann cells during early carcinogenesis and enhance Schwann cells migration via the p38/MAPK signaling pathway [170, 171]. Activation of CXCL12/CXCR4 axis significantly promotes cancer cell invasion and facilitates the outgrowth of DRG, eventually leading to PNI [172]. CAFs also secrete Slit2, which stimulates Schwann cell proliferation and neural remodeling, resulting in increased nerve density [96]. The expression of fibroblast specific protein-1 (FSP-1) in CAFs within the intra-tumoral stroma is associated with PNI, lymphatic invasion, and tumor stage of CRC [173]. Another widely used marker for CAFs in various tumor types is fibroblast activation protein alpha (FAPα), and its expression is correlated with PNI and poor prognosis [174]. In a study by Tassone et al., fibroblasts within the PNI microenvironment were found to express MMP-2, whereas fibroblasts in nerves without PNI did not, suggesting that MMP-2 expression by fibroblasts may be a potential mechanism promoting PNI [175].
6 Models to study PNI
Our current understanding of PNI pathogenesis has been constrained by the absence of effective models capable of capturing the intricate interactions between nerves, tumor cells, and the stroma. In our previous review, we provided an overview of commonly used in vitro and in vivo models employed in PNI research [176]. In this review, we will specifically highlight models utilized to investigate PNI in CRC and explore potential novel models for studying enteric nerves.
6.1 In Vitro models
Duchalais et al. initially cocultured tumor epithelial cells from human primary colon adenocarcinomas with human ENS plexus explants in a Transwell chamber coated with Matrigel to mimic tumor cells invasion into the neural sheath [114]. In this study, confocal and atomic force microscopy, as well as video microscopy, were employed to assess colon cancer cell adhesion and migration on the ENS. More recently, a colon-nerve organoid model was utilized to investigate the reciprocal signaling between neurons and cancer cells [177]. This model involved using L6 dorsal and ventral roots with the spinal cord removed from the spinal column. Through this model, the authors demonstrated that extrinsic parasympathetic pathways influence myenteric neuron activity and mediate smooth muscle contractions in the colon. This method can be adapted for studying sensory neuron hypersensitivity by recording neuronal activity electrophysiologically in the presence of cancer cells.
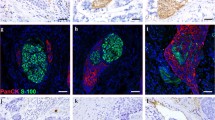
Tissue clearing followed by three-dimensional (3D) imaging is a technique that enables the simultaneous analysis of multiple cells within an organ. Masaki Mori et al. were the first to characterize the 3D structure of tumor invasion around nerve tissue in CRC using this method [178]. By staining CRC tissue with cytokeratin AE1/AE3 and anti-S100 protein antibody, virtual slides were scanned to reconstruct the tissue, effectively demonstrating the morphological features of intramural PNI in CRC [178].
In vitro experimental models allow investigators to control the experimental components and conditions of the cellular microenvironment, and capture specific aspects of the disease process. However, these models have limitations, particularly in replicating the neural microenvironment and culturing peripheral nerves.
6.2 In vivo models
The ideal in vivo models should recapitulate the neuropathic changes that occur in human tumors, such as neural hypertrophy and neural remodeling. Genetically engineered mouse models (GEMMs) are valuable tools for elucidating the molecular mechanisms underlying tumor initiation and development and have shown great promise in investigating PNI [179, 180]. GEMMs allow researchers to observe the neurogenesis, neural remodeling, and tumor-neural interactions at different stages of tumor progression. This can be achieved through the following methods: (1) Immunohistochemistry and immunofluorescence staining can be performed on tissues to label nerves and tumor cells. Additionally, tissue clearing followed by 3D imaging can be employed for neural reconstruction. (2) The expression levels of neurotrophic factors, neurotransmitters, chemokines, and other molecules related to neural function can be measured in tumor tissues and/or blood samples. Correlation analysis can be performed to examine their association with tumors and neural features. (3) The expression levels of RNAs and proteins in tumors can be examined to assess their correlation with PNI and other neural characteristics. (4) Researchers can test the effectiveness of drugs targeting specific molecular or pathways involved in PNI.
The most widely utilized GEMM of intestinal cancer is the ApcMin/+ model, which harbors a dominant nonsense mutation in one Apc allele, resulting in the development of multiple intestinal adenomas, primarily in the small intestine, with few occurring in the colorectum. Vasiliou et al. crossed Apcflox mice with Cdx2ERT2 − Cre mice to generate deficient Apc specifically in colon epithelial cells following tamoxifen treatment [181]. Kevin G et al. generated ApcMin/+; KrasLSL−G12D/+; VillinERT2−Cre compound mutant mice, carrying a Cre-dependent activated KrasLSL−G12D on the ApcMin/+ background, crossed with mice carrying the VillinERT2−Cre transgene, which expresses Cre recombinase throughout the intestine, and confirmed an enhancement of tumor development in the colon [182]. Similarly, an Apcflox; KrasLSL−G12D/+; TP53KO/KO; VillinERT2−Cre compound mutant mice model was used to study cancer stem cells in CRC [183]. Although GEMMs allow us analyze the complex interactions between nerves, tumor cells, and other cells in the TME, the incidence of PNI in them has not been reported.
Other in vivo models include the orthotopic model and heterotopic model. The orthotopic model has been used in the study of pancreatic and head and neck cancer, and the incidences of PNI have been recorded [184, 185]. The widely used heterotopic model is mouse sciatic nerve model, which can recapitulate cancer cell migration through invaded nerves [151, 186]. However, neither of these models is suitable for studying the interactions between cancer cells and nerves at pre-neoplastic stages. Furthermore, the sciatic nerve is a somatic nerve, which is different from the natural innervation of the actual end-organ. Thus, it is urgent to develop other models that can recapitulate the development of PNI.
7 Therapeutic potential
The strong clinical impact of PNI makes it a promising therapeutic target. While there have been limited active clinical trials on CRC patients so far, animal models have been used to investigate the main molecules involved in PNI. Under stress conditions, human CRC cells synthesize BDNF, which stimulates cell proliferation and exerts an anti-apoptotic effect. This effect can be suppressed by K252a, a pharmacologic inhibitor of Trk receptors [187]. In murine xenograft models, targeting BDNF/TrkB signaling with K252a results in reduced metabolic activity and enhanced apoptosis of CRC cells [188]. In the TrkA-expressing CRC cell line KM12, the selective TrkA inhibitor NMS-P626 effectively inhibits TrkA phosphorylation and downstream signaling, demonstrating significant antitumor activity in mice with xenograft tumors [189]. Additionally, inhibition of GDNF expression by miR-196a-5p mimics has been shown to reduce the migration of CRC cells in vitro [190]. In a study by Duchalais et al., blocking L1CAM and N-cadherin with antibodies resulted in decreased migration of human primary colon adenocarcinomas epithelial cells along ENS structures [114]. The CCL2 and C-C motif chemokine receptor (CCR) 2 axis has also been implicated in PNI through Akt and MAPK signaling pathways. Treatment with anti-CCL2 antibodies has been found to inhibit CRC angiogenesis and growth by blocking p38/MAPK signaling [191]. Although MMPs are considered potential therapeutic targets in PNI, several clinical trials have failed to demonstrate significant effects on tumor development using broad-spectrum MMP inhibitors [192].
Targeting tumor innervation has also been proposed as a therapeutic option for several cancers, including breast cancer [142], prostate cancer [8], and gastric cancer [143]. In CRC, denervation of the myenteric plexus through benzalkonium chloride (BAC) treatment reduces the number of both preneoplastic and neoplastic lesions, suggesting that colonic denervation could attenuate carcinogenesis in the early stages [193, 194]. Targeting M3R via selective or non-selective antagonists could repress colon cancer cell proliferation, whereas acetylcholinesterase inhibitors are capable of stimulating tumor growth [130]. Similarly, deficiency of CHRM3, the coding gene of M3R, reduces the epithelial proliferation and tumor size in murine colon cancer models [195]. ADRβ is described as a potential therapeutic target in multiple cancers as it can promote tumor progression when activated by sympathetic signaling [196]. Selective ADRβ antagonists suppress CRC cell proliferation and viability both in vivo and in vitro, probably through the EGFR-Akt/ERK1/2 signaling pathway [197]. Wang et al. found that trefoil factor 2 (TFF2) is essential for the anti-inflammatory neural arc involving the vagus nerve and memory T cells. Deletion of Tff2 or splenic denervation disrupts this arc, leading to pro-carcinogenic inflammation in CRC [198]. These results indicate the participation of cholinergic and adrenergic signaling in CRC development, suggesting the therapeutic potential of denervation in cancer treatment.
8 Discussion and perspectives
Despite the dense innervation of the colon and rectum, the prevalence of PNI in CRC remains low, and the reasons for this phenomenon are not yet fully understood. One possible explanation is that the natural organ-innervation of the tumor origin may lack significant biologic affinity between cancer cells and neurons. The expression of neurotrophins, chemokines, and their receptors in colonic nerves and tumor cells is much lower in CRC compared to PDAC [199]. Additionally, the absence of remarkable neuroplasticity in CRC may contribute to the low occurrence of PNI. Furthermore, PNI is often underreported due to the lack of standardized reporting standards. The incidence of PNI in rectal cancer is higher than in colon cancer, which can be attributed to the presence of numerous autonomic nerve plexuses surrounding the rectum [18]. The rectum is localized extra-peritoneally and possesses its own rectal plexus, while several parts of the colon are intraperitoneal and lack an external plexus, resulting in differences in innervation density [200]. Moreover, more extensive examination of the mesorectal fat in rectal cancer to investigate circumferential resection margin involvement may increase the detection rate of PNI.
Despite the relatively low prevalence of PNI in CRC, its presence exerts a significant influence on the prognosis of the disease, comparable to well-established prognostic factors such as tumor invasion depth and lymphatic invasion. The significance of PNI in CRC lies in its capacity to facilitate tumor invasion along nerves, leading to the dissemination of cancer cells to nearby tissues and distant organs. By allowing tumor cells to interact directly with nerves, PNI creates a favorable microenvironment that supports tumor growth and survival. Furthermore, PNI can contribute to cancer-related pain and serve as a valuable pathological marker for early metastasis, offering crucial prognostic information.
Overall, PNI involves complex communication between nerves, cancer cells and other components of the neoplastic microenvironment. These reciprocal interactions contribute to tumor growth, migration, dissemination, and metastasis. Understanding the underlying pathological mechanisms of PNI and identifying specific molecular targets may have a potential impact on CRC treatment.
Data Availability
Not applicable.
Abbreviations
- Ach:
-
Acetylcholine
- AD:
-
Adrenaline
- ADRβ:
-
β adrenergic receptors
- BDNF:
-
Brain-derived neurotropic factor
- CCL:
-
C-C motif chemokine ligand
- CM:
-
Circular muscle
- CNS:
-
Central nervous system
- COX-2:
-
Cyclooxygenase 2
- CRC:
-
Colorectal cancer
- CXCL:
-
C-X-C motif chemokine
- CXCR:
-
C-X-C motif chemokine receptor
- DFS:
-
Disease free survival
- DRG:
-
Dorsal root ganglion
- ENS:
-
Enteric nervous system
- GABA:
-
γ-aminobutyric acid
- GABAAR:
-
GABA type A receptor
- GABABR:
-
GABA type B receptor
- GDNF:
-
Glial cell line-derived neurotrophic factor
- GEMM:
-
Genetically engineered mouse models
- HNSCC:
-
Head and neck squamous cell carcinoma
- L1CAM:
-
L1 family of cell adhesion molecule
- LM:
-
Longitudinal muscle
- LNI:
-
Lymph node involvement
- M3R:
-
Muscarinic Ach receptor 3
- MMP:
-
Matrix metalloproteinase
- MP:
-
Myenteric plexus
- NA:
-
Noradrenaline
- NCAM:
-
Neural cell adhesion molecule
- NGF:
-
Nerve growth factor
- OS:
-
Overall survival
- PDAC:
-
Pancreatic ductal adenocarcinoma
- PNI:
-
Perineural invasion
- SEMA:
-
Semaphorin
- SMP:
-
Submucosal plexus
- TIL:
-
Tumor-infiltrating lymphocyte
- TME:
-
Tumor microenvironment
- TrkA:
-
Tropomyosin-related kinase A
- TrkB:
-
Tropomyosin-related kinase B
- VEGF:
-
Vascular endothelial growth factor
- VIP:
-
Vasoactive intestinal peptide
References
R.L. Siegel et al., Colorectal cancer statistics, 2020. CA-Cancer J. Clin. 70(3), 145–164 (2020)
T. Olenius et al., Long-term survival among colorectal cancer patients in Finland, 1991–2015: a nationwide population-based registry study. BMC Cancer. 22(1), 356 (2022)
Y.-H. **e, Y.-X. Chen, J.-Y. Fang, Comprehensive review of targeted therapy for colorectal cancer. Signal Transduct. Target. Therapy. 5(1), 22 (2020)
L.H. Biller, D. Schrag, Diagnosis and treatment of metastatic colorectal Cancer: a review. JAMA 325(7), 669–685 (2021)
P. Hernández-Camarero et al., Cancer: a mirrored room between tumor bulk and tumor microenvironment. J. Experimental Clin. Cancer Res. 40(1), 217 (2021)
M. Dinevska et al., Cell Signaling Activation and Extracellular Matrix Remodeling Underpin Glioma Tumor Microenvironment Heterogeneity and Organization. Cell. Oncol. 46(3), 589-602
C. Hutchings, J.A. Phillips, M.B.A. Djamgoz, Nerve input to tumours: pathophysiological consequences of a dynamic relationship. Biochim. Biophys. Acta Rev. Cancer. 1874(2), 188411 (2020)
C. Magnon et al., Autonomic nerve development contributes to prostate cancer progression. Science. 341(6142), 1236361 (2013)
J.H. Baraldi et al., Tumor innervation: history, methodologies, and significance. Cancers (Basel), 14(8) (2022)
H.D. Reavis, H.I. Chen, R. Drapkin, Tumor Innervation: Cancer Has Some Nerve. Trends Cancer. 6(12), 1059–1067 (2020)
J.G. Batsakis, Nerves and neurotropic carcinomas. Ann. Otol Rhinol Laryngol. 94(4 Pt 1), 426–427 (1985)
C. Liebig et al., Perineural invasion in cancer: a review of the literature. Cancer. 115(15), 3379–3391 (2009)
D.E. Bockman, M. Büchler, H.G. Beger, Interaction of pancreatic ductal carcinoma with nerves leads to nerve damage. Gastroenterology. 107(1), 219–230 (1994)
S.H. Jiang et al., The genomic, transcriptomic, and immunological profiles of perineural invasion in pancreatic ductal adenocarcinoma. Sci. China Life Sci. 66(1), 183–186 (2023)
G. Hu, L. Li, K. Hu, Clinical implications of perineural invasion in patients with colorectal cancer. Med. (Baltim). 99(17), e19860 (2020)
T. Matsushima et al., Preoperative estimation of neural invasion in rectal carcinoma. Oncol. Rep. 5(1), 73–76 (1998)
G. Burdy et al., Identifying patients with T3-T4 node-negative colon cancer at high risk of recurrence. Dis. Colon Rectum. 44(11), 1682–1688 (2001)
Knijn N, Mogk SC, Teerenstra S, Simmer F, Nagtegaal ID. Perineural invasion is a strong prognostic factor in colorectal cancer: a systematic review. Am. J. Surg. Pathol. 40(1):103–12 (2016). https://doi.org/10.1097/PAS.0000000000000518. PMID: 26426380
L. Zhang et al., Nerve dependence in Colorectal Cancer. Front. Cell. Dev. Biol. 10, 766653 (2022)
F. Pagès et al., Effector memory T cells, early metastasis, and Survival in Colorectal Cancer. N. Engl. J. Med. 353(25), 2654–2666 (2005)
H.S.V. Frank Winkler, M. Amit, T. Batchelor, I.E. Demir, B. Deneen, D.H. Gutmann, S. Hervey-Jumper, T. Kuner, D. Mabbott, M. Platten, A. Rolls,Erica, K. Sloan, T.C. Wang, Wolfgang Wick,Varun Venkataramani, and Michelle Monje, Cancer neuroscience: state of the field, emerging directions. Cell 186, 1689–1707 (2023)
D. Hanahan, M. Monje, Cancer hallmarks intersect with neuroscience in the tumor microenvironment. Cancer Cell. 41(3), 573–580 (2023)
A. Kamiya et al., Sympathetic and parasympathetic innervation in cancer: therapeutic implications. Clin. Auton. Res. 31(2), 165–178 (2021)
K.N. Browning, R.A. Travagli, Central nervous system control of gastrointestinal motility and secretion and modulation of gastrointestinal functions. Compr. Physiol. 4(4), 1339–1368 (2014)
S.L. Schonkeren et al., The Emerging Role of Nerves and Glia in Colorectal Cancer. Cancers (Basel) 13(1) (2021)
A.E. Lomax, K.A. Sharkey, J.B. Furness, The participation of the sympathetic innervation of the gastrointestinal tract in disease states. Neurogastroenterol Motil. 22(1), 7–18 (2010)
M.D. Gershon, The enteric nervous system: a second brain. Hosp. Pract. 34(7), 31–52 (1999)
F. Obermayr et al., Development and developmental disorders of the enteric nervous system. Nat. Rev. Gastroenterol. Hepatol. 10(1), 43–57 (2013)
M.B. Hansen, The enteric nervous system II: gastrointestinal functions. Pharmacol. Toxicol. 92(6), 249–257 (2003)
T. Wedel et al., Organization of the enteric nervous system in the human colon demonstrated by wholemount immunohistochemistry with special reference to the submucous plexus. Annals of Anatomy - Anatomischer Anzeiger. 181(4), 327–337 (1999)
J.B. Furness et al., The enteric nervous system and gastrointestinal innervation: integrated local and central control. Adv. Exp. Med. Biol. 817, 39–71 (2014)
A Dissertation on the Sensible and Irritable Parts of Animals. JAMA 108(4), 328–328 (1937)
L.J. Burgart, S. Kakar, C. Shi, M.E. Berho, D.K. Driman, P. Fitzgibbons, W.L. Frankel, K.A. Hill, J. Jessup, A.M. Krasinskas, M.K. Washington, Protocol for the Examination of Resection Specimens from Patients with Primary Carcinoma of the Colon and Rectum (College of American Pathologists, 2020)
Di F. Fabio et al., Prognostic variables for cancer-related survival in node-negative colorectal carcinomas. Dig. Surg. 21(2), 128–133 (2004)
E. Al-Sukhni et al., Lymphovascular and perineural invasion are associated with poor prognostic features and outcomes in colorectal cancer: a retrospective cohort study. Int. J. Surg. 37, 42–49 (2017)
Y.J. Lee et al., Risk factors for lymph node metastasis in early colon cancer. Int. J. Colorectal Dis. 35(8), 1607–1613 (2020)
J.W. Huh, H.R. Kim, Y.J. Kim, Lymphovascular or perineural invasion may predict lymph node metastasis in patients with T1 and T2 colorectal cancer. J. Gastrointest. Surg. 14(7), 1074–1080 (2010)
S. Kim et al., Lymphovascular invasion, perineural invasion, and tumor budding are prognostic factors for stage I colon cancer recurrence. Int. J. Colorectal Dis. 35(5), 881–885 (2020)
M. Skancke et al., Lymphovascular Invasion and Perineural Invasion negatively Impact overall survival for stage II adenocarcinoma of the Colon. Dis. Colon Rectum. 62(2), 181–188 (2019)
A.B. Benson et al., NCCN Guidelines Insights: Colon cancer, Version 2.2018. J. Natl. Compr. Canc Netw. 16(4), 359–369 (2018)
T. Suzuki et al., Adjuvant chemotherapy for the perineural invasion of colorectal cancer. J. Surg. Res. 199(1), 84–89 (2015)
J.H. Song et al., Significance of perineural and lymphovascular invasion in locally advanced rectal cancer treated by preoperative chemoradiotherapy and radical surgery: can perineural invasion be an indication of adjuvant chemotherapy? Radiother Oncol. 133, 125–131 (2019)
J.W. Huh, H.R. Kim, Y.J. Kim, Prognostic value of perineural invasion in patients with stage II colorectal cancer. Ann. Surg. Oncol. 17(8), 2066–2072 (2010)
M.J. Krasna et al., Vascular and neural invasion in colorectal carcinoma. Incidence and prognostic significance. Cancer. 61(5), 1018–1023 (1988)
L.F. Oñate-Ocaña et al., Identification of patients with high-risk lymph node-negative colorectal cancer and potential benefit from adjuvant chemotherapy. Jpn J. Clin. Oncol. 34(6), 323–328 (2004)
Y. Yang et al., Prognostic value of perineural invasion in colorectal cancer: a meta-analysis. J. Gastrointest. Surg. 19(6), 1113–1122 (2015)
S. Mo et al., Development and external validation of a predictive scoring system associated with metastasis of T1-2 colorectal tumors to lymph nodes. Clin. Transl Med. 10(1), 275–287 (2020)
A. Horn, O. Dahl, I. Morild, The role of venous and neural invasion on survival in rectal adenocarcinoma. Dis. Colon Rectum. 33(7), 598–601 (1990)
K. Shirouzu, H. Isomoto, T. Kakegawa, Prognostic evaluation of perineural invasion in rectal cancer. Am. J. Surg. 165(2), 233–237 (1993)
A. Ross et al., Recurrence and survival after surgical management of rectal cancer. Am. J. Surg. 177(5), 392–395 (1999)
W.L. Law, K.W. Chu, Anterior resection for rectal cancer with mesorectal excision: a prospective evaluation of 622 patients. Ann. Surg. 240(2), 260–268 (2004)
Q. Sun et al., Perineural and lymphovascular invasion predicts for poor prognosis in locally advanced rectal cancer after neoadjuvant chemoradiotherapy and surgery. J. Cancer. 10(10), 2243–2249 (2019)
M. Stojkovic Lalosevic et al., Perineural invasion as a prognostic factor in patients with stage I-III rectal cancer – 5-year follow up. World J. Gastrointest. Oncol. 12(5), 592–600 (2020)
Y.I. Kim et al., Clinical Implication of Perineural and Lymphovascular Invasion in Rectal Cancer Patients Who Underwent Surgery After Preoperative Chemoradiotherapy. Dis. Colon. Rectum. 65(11), 1325-1334 (2022)
J.A. Yun et al., Prognostic significance of perineural invasion in stage IIA colon cancer. ANZ J. Surg. 86(12), 1007–1013 (2016)
L.G.J. Leijssen et al., Perineural Invasion is a prognostic but not a predictive factor in nonmetastatic Colon cancer. Dis. Colon Rectum. 62(10), 1212–1221 (2019)
H. Nozawa et al., Obstruction is associated with perineural invasion in T3/T4 colon cancer. Colorectal Dis. 21(8), 917–924 (2019)
S.Y. Lee et al., Perineural invasion and number of retrieved lymph nodes are prognostic factors for T2N0 colon cancer. Langenbecks Arch. Surg. 406(6), 1979–1985 (2021)
R. Klein, Role of neurotrophins in mouse neuronal development. Faseb j. 8(10), 738–744 (1994)
M. Amit, S. Na’ara, Z. Gil, Mechanisms of cancer dissemination along nerves. Nat. Rev. Cancer. 16(6), 399–408 (2016)
A. Kolokythas et al., Nerve growth factor and tyrosine kinase A receptor in oral squamous cell carcinoma: is there an association with perineural invasion? J. Oral Maxillofac. Surg. 68(6), 1290–1295 (2010)
Y. Lei et al., Nerve growth factor orchestrates NGAL and matrix metalloproteinases activity to promote colorectal cancer metastasis. Clin. Transl Oncol. 24(1), 34–47 (2022)
Y. Hayakawa et al., Nerve growth factor promotes gastric tumorigenesis through aberrant Cholinergic Signaling. Cancer cell. 31(1), 21–34 (2017)
W. Wang, J. Chen, X. Guo, The role of nerve growth factor and its receptors in tumorigenesis and cancer pain. Biosci. Trends. 8(2), 68–74 (2014)
V.L. Chamary, T.R.M. Loizidou, P.B. Boulos, G. Burnstock†, Progressive loss of perivascular nerves adjacent to colorectal cancer. Eur. J. Surg. Oncol. 26, 588–593 (2000)
A. Nakagawara, Trk receptor tyrosine kinases: a bridge between cancer and neural development. Cancer Lett. 169(2), 107–114 (2001)
C. de Brunetto et al., BDNF/TrkB content and interaction with gastrin-releasing peptide receptor blockade in colorectal cancer. Oncology. 79(5–6), 430–439 (2010)
X. Yang, T.A. Martin, W.G. Jiang, Biological influence of brain-derived neurotrophic factor (BDNF) on colon cancer cells. Exp. Ther. Med. 6(6), 1475–1481 (2013)
Y. Yu et al., Overexpression of TrkB promotes the progression of colon cancer. Apmis. 118(3), 188–195 (2010)
S.M. Huang et al., Brain-derived neurotrophic factor regulates cell motility in human colon cancer. Endocr. Relat. Cancer. 22(3), 455–464 (2015)
D.-Y. Lu et al., Glial cell line-derived neurotrophic factor induces cell migration and matrix metalloproteinase-13 expression in glioma cells. Biochem. Pharmacol. 80(8), 1201–1209 (2010)
J.-Y. Chuang et al., Glial cell line-derived neurotrophic factor induces cell migration in human oral squamous cell carcinoma. Oral Oncol. 49(12), 1103–1112 (2013)
S. Qiao et al., Increased expression of glial cell line-derived neurotrophic factor and neurturin in a case of colon adenocarcinoma associated with diffuse ganglioneuromatosis. Clin. Neuropathol. 28(2), 105–112 (2009)
A. Furuta et al., The relationship between GDNF and integrins in human colorectal cancer cell activity. Hepatogastroenterology. 54(77), 1398–1402 (2007)
S.M. Huang et al., GDNF increases cell motility in human colon cancer through VEGF-VEGFR1 interaction. Endocr. Relat. Cancer. 21(1), 73–84 (2014)
S. Wang et al., Neurturin promotes tumor cell motility and angiogenesis in colorectal cancer. Exp. Cell. Res. 413(1), 113049 (2022)
C.S. Scanlon et al., Galanin modulates the neural niche to favour perineural invasion in head and neck cancer. Nat. Commun. 6, 6885 (2015)
Chen SH, Zhang BY, Zhou B, Zhu CZ, Sun LQ, Feng YJ. Perineural invasion of cancer: a complex crosstalk between cells and molecules in the perineural niche. Am. J. Cancer Res. 9(1):1–21 (2019). PMID: 30755808; PMCID: PMC6356921.
K.L.W. Sun, J.P. Correia, T.E. Kennedy, Netrins: versatile extracellular cues with diverse functions. Development. 138(11), 2153–2169 (2011)
B. Li et al., Serum netrin-1 as a biomarker for colorectal cancer detection. Cancer Biomark. 28(3), 391–396 (2020)
H. Nakayama et al., Inactivation of axon guidance molecule netrin-1 in human colorectal cancer by an epigenetic mechanism. Biochem. Biophys. Res. Commun. 611, 146–150 (2022)
S.K. Shin et al., Epigenetic and genetic alterations in Netrin-1 receptors UNC5C and DCC in human colon cancer. Gastroenterology. 133(6), 1849–1857 (2007)
D. Dong et al., Promoter methylation-mediated repression of UNC5 receptors and the associated clinical significance in human colorectal cancer. Clin. Epigenetics. 13(1), 225 (2021)
N.W. Gale et al., Eph receptors and ligands comprise two major specificity subclasses and are reciprocally compartmentalized during embryogenesis. Neuron. 17(1), 9–19 (1996)
H. Yamamoto et al., Ephrin-A1 mRNA is associated with poor prognosis of colorectal cancer. Int. J. Oncol. 42(2), 549–555 (2013)
W. Liu et al., Coexpression of ephrin-Bs and their receptors in colon carcinoma. Cancer. 94(4), 934–939 (2002)
H. Ji et al., Proteome profiling of exosomes derived from human primary and metastatic colorectal cancer cells reveal differential expression of key metastatic factors and signal transduction components. Proteomics. 13(10–11), 1672–1686 (2013)
W. Liu et al., Effects of overexpression of ephrin-B2 on tumour growth in human colorectal cancer. Br. J. Cancer. 90(8), 1620–1626 (2004)
J. Hao, J. Yu, Semaphorin 3 C and its receptors in Cancer and Cancer Stem-Like cells. Biomedicines. 6(2), 42 (2018)
Q. Jiang et al., Functional loss of semaphorin 3 C and/or semaphorin 3D and their epistatic interaction with ret are critical to Hirschsprung disease liability. Am. J. Hum. Genet. 96(4), 581–596 (2015)
R.H. Straub et al., Anti-inflammatory role of sympathetic nerves in chronic intestinal inflammation. Gut. 57(7), 911–921 (2008)
D. Hui et al., Semaphorin 3 C as a therapeutic target in prostate and other cancers. Int. J. Mol. Sci. 20(3), 774 (2019)
N.R. Jurcak et al., Axon Guidance Molecules promote Perineural Invasion and Metastasis of Orthotopic pancreatic tumors in mice. Gastroenterology. 157(3), 838–850e6 (2019)
N.O. Binmadi et al., Plexin-B1 and semaphorin 4D cooperate to promote perineural invasion in a RhoA/ROK-dependent manner. Am. J. Pathol. 180(3), 1232–1242 (2012)
R.E. Dickinson et al., Epigenetic inactivation of SLIT3 and SLIT1 genes in human cancers. Br. J. Cancer. 91(12), 2071–2078 (2004)
V. Secq et al., Stromal SLIT2 impacts on pancreatic cancer-associated neural remodeling. Cell. Death Dis. 6(1), e1592 (2015)
Z. Huang et al., USP33 mediates Slit-Robo signaling in inhibiting colorectal cancer cell migration. Int. J. Cancer. 136(8), 1792–1802 (2015)
Y. **a et al., Reduced USP33 expression in gastric cancer decreases inhibitory effects of Slit2-Robo1 signalling on cell migration and EMT. Cell Prolif. 52(3), e12606 (2019)
Y. Yao et al., Activation of Slit2/Robo1 signaling promotes Tumor Metastasis in Colorectal Carcinoma through activation of the TGF-β/Smads pathway. Cells, 2019. 8(6)
T.J. Neuberger, C.J. Cornbrooks, Transient modulation of Schwann cell antigens after peripheral nerve transection and subsequent regeneration. J. Neurocytol. 18(5), 695–710 (1989)
R. Li et al., Neural cell adhesion molecule is upregulated in nerves with prostate cancer invasion. Hum. Pathol. 34(5), 457–461 (2003)
K. Kameda et al., Expression of highly polysialylated neural cell adhesion molecule in pancreatic cancer neural invasive lesion. Cancer Lett. 137(2), 201–207 (1999)
J. Shang et al., Expression of neural cell adhesion molecule in salivary adenoid cystic carcinoma and its correlation with perineural invasion. Oncol. Rep. 18(6), 1413–1416 (2007)
S. Deborde et al., Schwann cells induce cancer cell dispersion and invasion. J. Clin. Invest. 126(4), 1538–1554 (2016)
J. Roesler et al., Tumor suppressor activity of neural cell adhesion molecule in colon carcinoma. Am. J. Surg. 174(3), 251–257 (1997)
A. Fernandez-Briera et al., Effect of human colorectal carcinogenesis on the neural cell adhesion molecule expression and polysialylation. Oncology. 78(3–4), 196–204 (2010)
M.E. Conacci-Sorrell et al., Nr-CAM is a target gene of the beta-catenin/LEF-1 pathway in melanoma and colon cancer and its expression enhances motility and confers tumorigenesis. Genes Dev. 16(16), 2058–2072 (2002)
V. Sytnyk, I. Leshchyns’ka, M. Schachner, Neural cell adhesion molecules of the Immunoglobulin Superfamily regulate synapse formation, maintenance, and function. Trends Neurosci. 40(5), 295–308 (2017)
R.S. Schmid, P.F. Maness, L1 and NCAM adhesion molecules as signaling coreceptors in neuronal migration and process outgrowth. Curr. Opin. Neurobiol. 18(3), 245–250 (2008)
A. Tampakis et al., L1CAM expression in colorectal cancer identifies a high-risk group of patients with dismal prognosis already in early-stage disease. Acta Oncol. 59(1), 55–59 (2020)
F.Y.F. Tieng et al., L1CAM, CA9, KLK6, HPN, and ALDH1A1 as potential serum markers in primary and metastatic colorectal Cancer screening. Diagnostics (Basel), 10(7) (2020)
Q.X. Fang, X.C. Zheng, H.J. Zhao, L1CAM is involved in lymph node metastasis via ERK1/2 signaling in colorectal cancer. Am. J. Transl Res. 12(3), 837–846 (2020)
Y. Kajiwara et al., Expression of l1 cell adhesion molecule and morphologic features at the invasive front of colorectal cancer. Am. J. Clin. Pathol. 136(1), 138–144 (2011)
E. Duchalais et al., Colorectal Cancer cells adhere to and migrate along the neurons of the enteric nervous system. Cell. Mol. Gastroenterol. Hepatol. 5(1), 31–49 (2018)
S.H. Jiang et al., Neurotransmitters: emerging targets in cancer. Oncogene. 39(3), 503–515 (2020)
R.N. Ciurea et al., B2 adrenergic receptors and morphological changes of the enteric nervous system in colorectal adenocarcinoma. World J. Gastroenterol. 23(7), 1250–1261 (2017)
H.P. Wong et al., Effects of adrenaline in human colon adenocarcinoma HT-29 cells. Life Sci. 88(25–26), 1108–1112 (2011)
J. Han et al., Norepinephrine-CREB1-miR-373 axis promotes progression of colon cancer. Mol. Oncol. 14(5), 1059–1073 (2020)
R. Muthuswamy et al., Epinephrine promotes COX-2-dependent immune suppression in myeloid cells and cancer tissues. Brain Behav. Immun. 62, 78–86 (2017)
J.T. Dudley et al., Computational repositioning of the anticonvulsant topiramate for inflammatory bowel disease. Sci. Transl Med. 3(96), 96ra76 (2011)
D. Huang et al., Cancer-cell-derived GABA promotes beta-catenin-mediated tumour growth and immunosuppression. Nat. Cell. Biol. 24(2), 230–241 (2022)
B. Zhang et al., B cell-derived GABA elicits IL-10(+) macrophages to limit anti-tumour immunity. Nature. 599(7885), 471–476 (2021)
H. Wang et al., GABAB receptor inhibits tumor progression and epithelial-mesenchymal transition via the regulation of Hippo/YAP1 pathway in colorectal cancer. Int. J. Biol. Sci. 17(8), 1953–1962 (2021)
P.H. Thaker et al., Inhibition of experimental colon cancer metastasis by the GABA-receptor agonist nembutal. Cancer Biol. Ther. 4(7), 753–758 (2005)
L. Song et al., γ-Aminobutyric acid inhibits the proliferation and increases oxaliplatin sensitivity in human colon cancer cells. Tumour Biol. 37(11), 14885–14894 (2016)
H. Zhou et al., Expression and significance of autonomic nerves and α9 nicotinic acetylcholine receptor in colorectal cancer. Mol. Med. Rep. 17(6), 8423–8431 (2018)
K. Cheng et al., Differential expression of M3 muscarinic receptors in progressive colon neoplasia and metastasis. Oncotarget. 8(13), 21106–21114 (2017)
H.P. Wong et al., Nicotine promotes cell proliferation via alpha7-nicotinic acetylcholine receptor and catecholamine-synthesizing enzymes-mediated pathway in human colon adenocarcinoma HT-29 cells. Toxicol. Appl. Pharmacol. 221(3), 261–267 (2007)
A. Cucina et al., Nicotine stimulates proliferation and inhibits apoptosis in colon cancer cell lines through activation of survival pathways. J. Surg. Res. 178(1), 233–241 (2012)
K. Cheng et al., Acetylcholine release by human colon cancer cells mediates autocrine stimulation of cell proliferation. Am. J. Physiol. Gastrointest. Liver Physiol. 295(3), G591–G597 (2008)
M. Laburthe et al., Vasoactive intestinal peptide: a potent stimulator of adenosine 3’:5’-cyclic monophosphate accumulation in gut carcinoma cell lines in culture. Proc. Natl. Acad. Sci. U S A 75(6), 2772–2775 (1978)
R.P. Mohney, R.E. Siegel, R.E. Zigmond, Galanin and vasoactive intestinal peptide messenger RNAs increase following axotomy of adult sympathetic neurons. J. Neurobiol. 25(2), 108–118 (1994)
H. Hyatt-Sachs et al., Phenotypic plasticity in adult sympathetic ganglia in vivo: effects of deafferentation and axotomy on the expression of vasoactive intestinal peptide. J. Neurosci. 13(4), 1642–1653 (1993)
M. Neunlist et al., Human ENS regulates the intestinal epithelial barrier permeability and a tight junction-associated protein ZO-1 via VIPergic pathways. Am. J. Physiol. Gastrointest. Liver Physiol. 285(5), G1028–G1036 (2003)
C. Alleaume et al., Vasoactive intestinal peptide stimulates proliferation in HT29 human colonic adenocarcinoma cells: concomitant activation of Ras/Rap1–B-Raf–ERK signalling pathway. Neuropeptides. 37(2), 98–104 (2003)
H. Iishi et al., Enhancement by vasoactive intestinal peptide of experimental carcinogenesis induced by azoxymethane in rat colon. Cancer Res. 47(18), 4890–4893 (1987)
M. Ogasawara et al., Differential effect of intestinal neuropeptides on invasion and migration of colon carcinoma cells in vitro. Cancer Lett. 116(1), 111–116 (1997)
M. Ogasawara et al., Inhibition by vasoactive intestinal polypeptide (VIP) of angiogenesis induced by murine Colon 26-L5 carcinoma cells metastasized in liver. Clin. Exp. Metastasis. 17(4), 283–291 (1999)
R. Mroue, M.J. Bissell, Three-dimensional cultures of mouse mammary epithelial cells, Epithelial Cell Culture Protocols: Second Edition, in S.H. Randell and M.L. Fulcher, Editors. 2013, Humana Press: Totowa, NJ. 221–250
J.L. Saloman et al., Ablation of sensory neurons in a genetic model of pancreatic ductal adenocarcinoma slows initiation and progression of cancer. Proc Natl Acad Sci U S A 113(11), 3078-3083 (2016).
A.H. Zahalka et al., Adrenergic nerves activate an angio-metabolic switch in prostate cancer. Science. 358(6361), 321–326 (2017)
E.A. Kappos et al., Denervation leads to volume regression in breast cancer. J. Plast. Reconstr. Aesthetic Surg. 71(6), 833–839 (2018)
C.M. Zhao et al., Denervation suppresses gastric tumorigenesis. Sci. Transl Med. 6(250), 250ra115 (2014)
D. Albo et al., Neurogenesis in colorectal cancer is a marker of aggressive tumor behavior and poor outcomes. Cancer. 117(21), 4834–4845 (2011)
K.R. Jessen, R. Mirsky, The origin and development of glial cells in peripheral nerves. Nat. Rev. Neurosci. 6(9), 671–682 (2005)
M. Pellegatta, C. Taveggia, The Complex Work of Proteases and Secretases in Wallerian Degeneration: Beyond Neuregulin-1. Front. Cell. Neurosci. 13, 93 (2019)
I.E. Demir et al., Investigation of Schwann cells at neoplastic cell sites before the onset of cancer invasion. J. Natl. Cancer Inst., 2014. 106(8)
S. Na’ara, M. Amit, Z. Gil, L1CAM induces perineural invasion of pancreas cancer cells by upregulation of metalloproteinase expression. Oncogene, 2019. 38(4): p. 596–608
E. Roger et al., Schwann cells support oncogenic potential of pancreatic cancer cells through TGFβ signaling. Cell. Death Dis. 10(12), 886 (2019)
S. Deborde et al., Reprogrammed Schwann cells organize into dynamic tracks that promote pancreatic Cancer Invasion. Cancer Discov. 12(10), 2454–2473 (2022)
R.L. Bakst et al., Inflammatory monocytes promote Perineural Invasion via CCL2-Mediated recruitment and cathepsin B expression. Cancer Res. 77(22), 6400–6414 (2017)
S. Chen, M. Chen, Schwann cells promote the migration and invasion of colorectal cancer cells via the activated NF-kappaB/IL-8 axis in the tumor microenvironment. Front. Oncol. 12, 1026670 (2022)
S. Han et al., A reciprocal feedback between colon cancer cells and Schwann cells promotes the proliferation and metastasis of colon cancer. J. Exp. Clin. Cancer Res. 41(1), 348 (2022)
O. Cavel et al., Endoneurial macrophages induce perineural invasion of pancreatic cancer cells by secretion of GDNF and activation of RET tyrosine kinase receptor. Cancer Res. 72(22), 5733–5743 (2012)
P.A. Muller et al., Crosstalk between muscularis macrophages and enteric neurons regulates gastrointestinal motility. Cell. 158(2), 300–313 (2014)
V. Grubišić et al., Enteric glia modulate macrophage phenotype and visceral sensitivity following inflammation. Cell. Rep. 32(10), 108100 (2020)
X.P. Dun et al., Macrophage-derived Slit3 controls Cell Migration and Axon Pathfinding in the peripheral nerve bridge. Cell. Rep. 26(6), 1458–1472e4 (2019)
J. Albrengues et al., LIF mediates proinvasive activation of stromal fibroblasts in cancer. Cell. Rep. 7(5), 1664–1678 (2014)
Y. Zhang, R. Grazda, Q. Yang, Interaction between Innate Lymphoid cells and the nervous system. Adv. Exp. Med. Biol. 1365, 135–148 (2022)
K. Jakubowska et al., Stromal and intraepithelial tumor-infiltrating lymphocytes in colorectal carcinoma. Oncol. Lett. 14(6), 6421–6432 (2017)
J.W. Huh, J.H. Lee, H.R. Kim, Prognostic significance of tumor-infiltrating lymphocytes for patients with colorectal cancer. Arch. Surg. 147(4), 366–372 (2012)
S.W. Cole et al., Sympathetic nervous system regulation of the tumour microenvironment. Nat. Rev. Cancer. 15(9), 563–572 (2015)
M.W. Yang et al., Perineural Invasion reprograms the Immune Microenvironment through Cholinergic Signaling in Pancreatic Ductal Adenocarcinoma. Cancer Res. 80(10), 1991–2003 (2020)
C. Heichler et al., STAT3 activation through IL-6/IL-11 in cancer-associated fibroblasts promotes colorectal tumour development and correlates with poor prognosis. Gut. 69(7), 1269–1282 (2020)
H. Kobayashi et al., Cancer-associated fibroblasts in gastrointestinal cancer. Nat. Reviews Gastroenterol. Hepatol. 16(5), 282–295 (2019)
D. Unterleuthner et al., Cancer-associated fibroblast-derived WNT2 increases tumor angiogenesis in colon cancer. Angiogenesis. 23(2), 159–177 (2020)
M.B. Bunge et al., Perineurium originates from fibroblasts: demonstration in vitro with a retroviral marker. Science. 243(4888), 229–231 (1989)
H. Jiang et al., CAFs secrete CXCL12 to accelerate the progression and cisplatin resistance of colorectal cancer through promoting M2 polarization of macrophages. Med. Oncol. 40(3), 90 (2023)
D. Wang et al., Exosomal miR-146a-5p and mir-155-5p promote CXCL12/CXCR7-induced metastasis of colorectal cancer by crosstalk with cancer-associated fibroblasts. Cell. Death Dis. 13(4), 380 (2022)
D. Gao et al., CXCL12 induces migration of Schwann cells via p38 MAPK and autocrine of CXCL12 by the CXCR4 receptor. Int. J. Clin. Exp. Pathol. 11(6), 3119–3125 (2018)
I.E. Demir et al., Early pancreatic cancer lesions suppress pain through CXCL12-mediated chemoattraction of Schwann cells. Proc. Natl. Acad. Sci. U S A 114(1), E85–e94 (2017)
Q. Xu et al., Stromal-derived factor-1α/CXCL12-CXCR4 chemotactic pathway promotes perineural invasion in pancreatic cancer. Oncotarget. 6(7), 4717–4732 (2015)
G.M. Son et al., Comparisons of cancer-associated fibroblasts in the intratumoral stroma and invasive front in colorectal cancer. Med. (Baltim). 98(18), e15164 (2019)
M. Shi et al., Expression of fibroblast activation protein in human pancreatic adenocarcinoma and its clinicopathological significance. World J. Gastroenterol. 18(8), 840–846 (2012)
P. Tassone et al., The role of matrixmetalloproteinase-2 expression by fibroblasts in perineural invasion by oral cavity squamous cell carcinoma. Oral Oncol. 132, 106002 (2022)
S.H. Jiang et al., Emerging experimental models for assessing perineural invasion in human cancers. Cancer Lett. 535, 215610 (2022)
K.M. Smith-Edwards et al., Extrinsic primary afferent neurons Link Visceral Pain to Colon motility through a spinal Reflex in mice. Gastroenterology. 157(2), 522–536e2 (2019)
Y. Miyashita et al., Three-dimensional imaging of intramural perineural invasion in colorectal cancer: three-dimensional reconstruction approach with multiple immunohistochemically stained sections. Pathol. Int. 72(5), 293–299 (2022)
I.E. Demir, H. Friess, G.O. Ceyhan, Neural plasticity in pancreatitis and pancreatic cancer. Nat. Rev. Gastroenterol. Hepatol. 12(11), 649–659 (2015)
Le C. Magnen, A. Dutta, C. Abate-Shen, Optimizing mouse models for precision cancer prevention. Nat. Rev. Cancer. 16(3), 187–196 (2016)
J.P. Golla et al., Interplay between APC and ALDH1B1 in a newly developed mouse model of colorectal cancer. Chem. Biol. Interact. 331, 109274 (2020)
I.B. Enquist et al., Lymph node-independent liver metastasis in a model of metastatic colorectal cancer. Nat. Commun. 5(1), 3530 (2014)
A. Fumagalli et al., Plasticity of Lgr5-Negative Cancer cells drives Metastasis in Colorectal Cancer. Cell. Stem Cell. 26(4), 569–578e7 (2020)
J. Yao et al., Pleiotrophin and N-syndecan promote perineural invasion and tumor progression in an orthotopic mouse model of pancreatic cancer. World J. Gastroenterol. 23(21), 3907–3914 (2017)
M. Amit et al., Loss of p53 drives neuron reprogramming in head and neck cancer. Nature. 578(7795), 449–454 (2020)
S. Deborde et al., An in vivo murine sciatic nerve model of Perineural Invasion. J. Vis. Exp., 2018(134).
H. Akil et al., Fine-tuning roles of endogenous brain-derived neurotrophic factor, TrkB and sortilin in colorectal cancer cell survival. PLoS One. 6(9), e25097 (2011)
C. Mazouffre et al., Dual inhibition of BDNF/TrkB and autophagy: a promising therapeutic approach for colorectal cancer. J. Cell. Mol. Med. 21(10), 2610–2622 (2017)
E. Ardini et al., The TPM3-NTRK1 rearrangement is a recurring event in colorectal carcinoma and is associated with tumor sensitivity to TRKA kinase inhibition. Mol. Oncol. 8(8), 1495–1507 (2014)
F. Zhong et al., LncRNA NEAT1 promotes colorectal cancer cell proliferation and migration via regulating glial cell-derived neurotrophic factor by sponging miR-196a-5p. Acta Biochim Biophys Sin (Shanghai), 2018. 50(12): p. 1190–1199
H. Feng et al., Targeting tumor cell-derived CCL2 as a strategy to overcome Bevacizumab resistance in ETV5(+) colorectal cancer. Cell. Death Dis. 11(10), 916 (2020)
C.M. Overall, O. Kleifeld, Tumour microenvironment - opinion: validating matrix metalloproteinases as drug targets and anti-targets for cancer therapy. Nat. Rev. Cancer. 6(3), 227–239 (2006)
M.V. Vespúcio et al., Intrinsic denervation of the colon is associated with a decrease of some colonic preneoplastic markers in rats treated with a chemical carcinogen. Braz J. Med. Biol. Res. 41(4), 311–317 (2008)
V. Kannen et al., Trypanosomiasis-induced megacolon illustrates how myenteric neurons modulate the risk for colon cancer in rats and humans. PLoS Negl. Trop. Dis. 9(4), e0003744 (2015)
J.P. Raufman et al., Genetic ablation of M3 muscarinic receptors attenuates murine colon epithelial cell proliferation and neoplasia. Cancer Res. 68(10), 3573–3578 (2008)
S.W. Cole, A.K. Sood, Molecular pathways: beta-adrenergic signaling in cancer. Clin. Cancer Res. 18(5), 1201–1206 (2012)
C.C. Chin et al., Selective β2-AR blockage suppresses colorectal Cancer growth through regulation of EGFR-Akt/ERK1/2 signaling, G1-Phase arrest, and apoptosis. J. Cell. Physiol. 231(2), 459–472 (2016)
Z. Dubeykovskaya et al., Neural innervation stimulates splenic TFF2 to arrest myeloid cell expansion and cancer. Nat. Commun. 7, 10517 (2016)
F. Liebl et al., The severity of neural invasion is associated with shortened survival in colon cancer. Clin. Cancer Res. 19(1), 50–61 (2013)
G.O. Ceyhan et al., The severity of neural invasion is a crucial prognostic factor in rectal cancer independent of neoadjuvant radiochemotherapy. Ann. Surg. 252(5), 797–804 (2010)
Acknowledgements
The Figs. 1, 3 and 4 are assembled by BioRender.com (https://app.biorender.com/). We appreciate the free use of BioRender.com.
Funding
This work is supported by the grant from the Natural Science Foundation of China (82260562), Academic Leaders Training Program of Pudong Health Bureau of Shanghai (PWRd2021-09), CSCO research funding (Y-xsk2021-0003), the Science and Technology Commission of Shanghai Municipality (19DZ1910502), CSCO-**nda research funding (Y-XD2019-248), and the Top-level Clinical Discipline Project of Shanghai Pudong (PWYgf2021-07).
Author information
Authors and Affiliations
Contributions
Hao Wang, Ruixue Huo, and Kexin He: designing the work, literature retrieval, drafting the work; Li Cheng, Shan Zhang, and Minhao Yu: literature arrangement, drawing the figures; Junli Xue, Hui Li, and Wei Zhao: reviewing and editing.
Corresponding authors
Ethics declarations
Ethical approval
Not applicable.
Competing interests
The authors declare that no personal relationships or conflict of interests influence the work reported in this paper.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wang, H., Huo, R., He, K. et al. Perineural invasion in colorectal cancer: mechanisms of action and clinical relevance. Cell Oncol. 47, 1–17 (2024). https://doi.org/10.1007/s13402-023-00857-y
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13402-023-00857-y