
Abstract
The neuraminidase inhibitor oseltamivir (Tamiflu®) is currently the first-line therapy for patients with influenza virus infection. Common analysis of the prodrug and its active metabolite oseltamivircarboxylate is determined via extraction from plasma. Compared with these assays, dried blood spot (DBS) analysis provides several advantages, including a minimum sample volume required for the measurement of drugs in whole blood. Samples can easily be obtained via a simple, non-invasive finger or heel prick. Mainly, these characteristics make DBS an ideal tool for pediatrics and to measure multiple time points such as those needed in therapeutic drug monitoring or pharmacokinetic studies. Additionally, DBS sample preparation, stability, and storage are usually most convenient. In the present work, we developed and fully validated a DBS assay for the simultaneous determination of oseltamivir and oseltamivircarboxylate concentrations in human whole blood. We demonstrate the simplicity of DBS sample preparation, and a fast, accurate and reproducible analysis using ultra high-performance liquid chromatography coupled to a triple quadrupole mass spectrometer. A thorough validation on the basis of the most recent FDA guidelines for bioanalytical method validation showed that the method is selective, precise, and accurate (≤15% RSD), and sensitive over the relevant clinical range of 5–1,500 ng/mL for oseltamivir and 20–1,500 ng/mL for the oseltamivircarboxylate metabolite. As a proof of concept, oseltamivir and oseltamivircarboxylate levels were determined in DBS obtained from healthy volunteers who received a single oral dose of Tamiflu®.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The neuraminidase (NA) are a family of enzymes, which cleave the sialic acid (derivative of neuraminic acids) residue from cellular glycoproteins and lipids [1]. These enzymes are essential for influenza viruses in order to spread within the host. The process allows the virus to detach from the host cell after replication and consequently infects further host cells. Oseltamivircarboxylate (OSC), the active metabolite of oseltamivir (OS) (Tamiflu®, Hoffmann-La Roche, Nutley, NJ) provides a pharmacophore, which resembles sialic acid and selectively inhibits the activity of the enzyme, thus preventing the virus from spreading in the host.
Treatment of influenza virus infection is particularly important for patients who are at risk for a complicated course of influenza. These patient groups include neonates, infants, young children, pregnant women, elderly, and immunocompromised patients [2–4]. Over the last couple of years, Tamiflu® resistance has emerged in seasonal influenza A (H1N1) viruses. However, it seems that this influenza strain has been replaced by the pandemic influenza A H1N1/09 virus, which is still sensitive to Tamiflu®. As with most of the drugs, a precise dosing regime for infants and young children is based on calculations according to their body weight and due to empiric data on efficacy [5]. Determinations of a therapeutic window in this patient subgroup are often hampered by an inadequate blood/plasma sampling and sensitive analytical methods. Dried blood spot (DBS) analysis provides an ideal tool to monitor circulating drug levels and is also feasible to obtain drug pharmacokinetics (PK studies) [6, 7]. From a finger or heel prick minimum amounts of blood, usually 10–30 μL are spotted directly on specifically designed filter cards, which are subsequently extracted in an on- or off-line procedure [8] to be further analyzed in a high-throughput manner by gas chromatography–mass spectrometry (GC-MS) [9] or ultra high-performance liquid chromatography (UHPLC) coupled to ultra violet (UV), fluorescence, or mass spectrometer (MS) detectors [10]. The use of MS detectors applying multiple reaction monitoring (MRM) mode has the advantage of being usually very sensitive and more selective than other common LC detectors. Moreover, they provide the possibility to monitor multiple analytes simultaneously and do not depend on fluorescence or UV active chemical structures.
The current work describes a novel DBS UHPLC-MS/MS method which allows to simultaneously monitor OS and OSC concentrations in human whole blood samples. This work expands on a just recently published DBS method for measurement of OS and OSC in rodent blood [8]. Here, we describe for the first time a method to measure OS and OSC concentrations in human whole blood via DBS. The assay was validated on the basis of the latest Food and Drug Administration (FDA) guideline for bioanalytical method validation [11], and was applied to a small set of samples obtained from healthy subjects, as a proof of concept. The assay requires minimum blood volumes, which can easily be obtained by finger or heel prick, and it obviates plasma centrifugation to simplify sample preparation. Previously reported ester hydrolysis [12] was not observed on DBS during method development and validation, also shown by the various stability tests of OS and OSC on DBS cards.
Materials and methods
Chemicals and reagents
All solvents were of ULC/MS grade and were purchased from Biosolve (Valkenswaard, the Netherlands). The standards for OS and OSC (see Fig. 1) and their respective stable isotope-labeled internal standards d3-OS and d3-OSC were kindly provided by the Hoffmann-La Roche Ltd. (Basel, Switzerland). Filter paper from Schleicher & Schuell 2992 (Schleicher & Schuell BioScience GmbH, Dassel, Germany) was used for the validation and the patient samples.

Enzymatic activation of the prodrug oseltamivir (OS) to oseltamivircarboxylate (OSC). The dotted lines represent the fragmentation of the compounds during MRM measurements, including the m/z value of the selected fragment ions of the analytes and the respective stable isotope internal standards (values in brackets). For the stable isotope-labeled internal standards, the methyl group of the amid function is replaced by –CD3 group, respectively
Sample preparation
For the preparation of the DBS during the validation, blank whole blood (WB) was collected from two healthy donors (Sanquin, Rotterdam, the Netherlands). Blood was sampled in single-use evacuated fluoride/oxalate blood collection tubes containing 10 mg sodium fluoride and 8 mg potassium oxalate (BD VACUTAINER®) supplied by BD (Franklin Lakes, NJ, USA). Aliquots were kept in the freezer and were prepared freshly every day. Twenty microliters of blood were spotted with an Eppendorf pipette (Wesseling-Berzdorf, Germany) onto the DBS cards and dried for a minimum of 2 h at room temperature (RT).
For the measurement of the clinical samples, WB was collected by finger prick and spotted directly on DBS cards. The blood from the arm vein was collected in fluoride/oxalate tubes and spotted directly from the tubes onto the DBS paper as described above.
After drying, 4 μL of a mixture of the two internal standards (d-OS 50 ng/mL and d-OSC 250 ng/mL) were spotted onto the DBS and the cards were dried for another 30 min at RT.
The extraction of the DBS was conducted by manually punching out a disk (∼5 mm Ø) into an Eppendorf tube. Extraction was achieved by the addition of 75 μL 5% methanol (MeOH) in water, followed by 10 min sonication. Forty-five microliters of the extract were transferred into a fresh Eppendorf tube and combined with 30 μL of acetonitrile (kept at −20 °C) for protein precipitation. The mixture was centrifuged for 10 min (14.000 rpm, 4 °C), and the supernatant was diluted 1:4 with water prior to analysis.
Chromatographic conditions
An Ascentis® Express C18 reversed-phase column (100 × 2.1 mm, 2.7 μm) from Supelco (Munich, Germany) and an Ascentis® Express guard column (C18, 2.1 mm I.D.) from Supelco (Munich, Germany) were used for LC separations.
The chromatographic separation was carried out on an Ultimate 3000 Rapid Separation LC system from Dionex Benelux B.V. (Amsterdam, the Netherlands). Chromatographic conditions were as follows: gradient elution was performed with two solvents: solvent A, 0.1% formic acid (FA) in water and solvent B, 0.1% FA in MeOH. The program was initiated with a linear gradient from 25% to 100% B at 0.25 min and kept constant for 2.75 min. At 3 min, the column was equilibrated again with 25% solvent B for 3 min. Total run time was 6 min with a constant flow rate of 350 μL/min and a constant column oven temperature of 30 °C. The retention time of OSC was 1.88 min and for OS 3.20 min.
The system was coupled to an API 4000 QTRAP linear ion trap mass spectrometer (AB Sciex, Concord, ON, Canada). The mass spectrometer was run in positive MRM mode with nitrogen as nebulizer, drying and collision gas. Following parameters were used: curtain gas, 25 (arbitrary units); collision gas pressure, medium; ion-spray voltage (IS), 5,000 V; temperature, 650 °C; nebulizer gas, 20 (arbitrary units); declustering potential, 20 V; entrance potential, 10 V; collision energy, 15 eV for d-OS, OS, and OSC and 30 eV for d-OSC; collision cell exit potential, 10 V for all analytes, except OSC, for which it was set at 20 eV. The MRM transitions were (OS) m/z 313+ → 225, (d-OS) m/z 316+ → 228, (OSC) m/z 285+ → 138, and (d-OSC) m/z 288+ → 200, respectively (see also Fig. 1). Selection of each MRM transition was based on optimal intensities obtained for the fragment ion during method development (data not shown).
Post-column infusion experiments
A mixture of the two analytes OS (50 ng/mL) and OSC (200 ng/mL) was prepared in water and the solution was directly infused from a syringe pump (Harvard Instruments, Holliston, MA) into the ESI source via a T-piece, where the mobile phase was introduced from the UHPLC system at assay flow rate, column and gradient conditions. Once a steady selected ion current (XIC) for the MRM transitions of OS and OSC was achieved, 60 μL extract from blank blood spot sample were injected onto the column (Fig. 2).

Post-column infusion chromatograms. Indicated by the arrows are the retention times of OSC t R1 = 1.88 min and OS t R2 = 3.20 min. The major drops in the XIC signal (for OS m/z 313+ → 225 and OSC m/z 285+ → 138) are due to ion suppression of eluting matrix compounds at the beginning of the gradient and due to a sudden change in solvent composition after ∼3.4 min
Method validation
Validation samples
For the validation samples, fresh WB was collected in BD Vacutainer® fluoride/oxalate tubes from two healthy donors and aliquots were stored at −20 °C. Blank blood samples (spiked with 3 μL of water instead of analyte solution) did not show any interfering signals under assay conditions. Furthermore, quality control (QC) samples prepared (see text below) in blood of donor 1 were calculated with a calibration curve of standards prepared in blood from donor 2, and vice versa to demonstrate the independency of the assay results from sample matrix.
Stock solutions and dilutions thereof were prepared in water for all analytes. Calibration standards were prepared daily by adding 3 μL of the respective OS–OSC mixture to 27 μL of blood to obtain one sample. Calibration standards were chosen at the following concentrations for OS: 5, 10, 20, 30, 50, 100, 250, 500, 1,000, and 1,500 ng/mL and for OSC: 20, 30, 50, 100, 250, 500, 1,000, and 1,500 ng/mL. Calibration curves included a blank sample and two zero samples, spiked with each internal standard separately. QC samples were prepared in the following concentrations (OS/OSC): 0/750, 75/200, and 150/75 ng/mL and measured with n = 4 for each QC sample.
Validation protocol
The method validation was performed in respect to the latest FDA guideline for method validation of bioanalytical assays [11].
The selectivity of the method was determined by the comparison of blood blank and two zero samples from two donors, respectively. Linearity of the DBS assay was verified by five calibration curves measured on five separate days. Each one consisted of ten nonzero calibration standards for OS and eight for OSC, covering the concentration range from 5–1,500 ng/mL for OS and 20–1,500 ng/mL for OSC measured in the identical calibration standards. The calibration curves were calculated by linear regression using weighting for both analytes. The back-calculated values are required to be within 15% relative standard deviation (RSD) and 20% RSD at the lower limit of quantification (LLOQ), from the nominal concentration. The acceptance criteria for the correlation coefficients (r 2) of the calculated regression curves were 0.99 or higher.
The determination of the LLOQ was conducted by spiking six WB samples with OS and OSC at the lower end of the calibration curves (5 and 20 ng/mL, respectively). Results were recalculated with a freshly prepared calibration curve.
Intra- and interday accuracy and precision were determined by analyzing three times four replicates of each QC sample together with a freshly prepared calibration curve in one analytical run within 1 day (intraday) and by analyzing three times four replicates of each QC sample together with a freshly prepared calibration curve in one analytical run on three consecutive days (interday). The mean, the standard deviation, the RSD, and bias were calculated.
The recovery of the method was determined by spiking three WB samples with OS and OSC (75 and 200 ng/mL, respectively), spotting and cutting out the whole blood spot. As a comparison, three blank blood spots, completely extracted and spiked in the extraction solvent with the same concentration were used. Samples were measured and results calculated by setting the samples spiked after extraction as 100%.
Stability of OS and OSC was determined for all QC samples with n = 4, for 0, 1, 3, and 7 days at RT and in the fridge at 4 °C; at −20 and +40 °C for 24 h. All samples were kept in air-tight plastic containers for time of storage. Additionally, 12 h autosampler stability was tested for the extracted solution.
Clinical samples
As a proof of concept study, three healthy volunteers (three male adults) took a single Tamiflu® capsule (75 mg), after signing written informed consent. Blood obtained via finger prick and from the median cubital vein was collected 2.5 (±15 min) and 4.25 h (±15 min) after administration of the capsule by trained personnel.
Results and discussion
The method described in the current work shows the development of a Tamiflu® DBS assay in combination with a fast, accurate, precise, and reproducible UHPLC-MS/MS application. The assay was validated based on criteria described by the most recent FDA guideline for bioanalytical method validation [11]. The feasibility of the analytical procedure was demonstrated in a clinical proof of concept study. To the best of the author’s knowledge, this study is the first report in the field of DBS analysis which assesses the effect of the type of whole blood, i.e., venous WB in blood collection tubes containing additives or capillary WB obtained by finger prick, on the measured concentrations of OS and OSC. This comparison demonstrated that results obtained with the newly developed assay were not influenced by additives (fluoride/oxalate) in the blood collection tubes. Fluoride/oxalate tubes are the tubes of choice for OS blood sampling [13]. Further proof of the accurate and reproducible analytical method was obtained via post-column infusion chromatograms, demonstrating no interference (ion suppression or enhancement) at the respective retention times of the analytes (see Fig. 2), despite an extraction protocol for the DBS using 5% methanol in water. Watery extraction solvents are technically more prone to show matrix effects, as endogenous, hydrophilic compounds get easily extracted together with the drug of interest [14]. This interference can mainly be observed in the infusion chromatogram (see Fig. 2) during the first minute.
Method validation
Selectivity
No interfering signals were observed at the respective retention times of t = 1.88 min (OSC) and t = 3.20 min (OS) when blank or zero sample (spiked with IS) blood spots were analyzed.
Linearity
Five measurements of the calibration standards, freshly prepared on five separate days, showed good linearity over the entire calibration range from 5–1,500 ng/mL for OS and 20–1,500 ng/mL for OSC. To obtain the best fit of the calibration standards a weighting factor of 1/x 2 was used for each analyte. This factor was determined through a comparison of the calculated correlation coefficients (r 2 values) and the sums of the absolute values of the relative errors for the calibration curves when different weighting factors were applied in the calculation (data not shown). The r 2 values were consistently greater than 0.991, with a mean of 0.9938 for OS and 0.9987 for OSC, respectively. The RSD values of the calculated standard concentrations from their nominal values were constantly ≤15% for all values, including the respective LLOQs. These results were well in line with the FDA specifications [11].
Lower limit of quantification
The lower limit of quantification, determined in six spiked DBS samples was 5 ng/mL for OS and 20 ng/mL for OSC. Measurements for OS showed an average of 5.7 ng/mL ± 3.3% (bias = 5.1%) and for OSC an average of 21.3 ng/mL ± 8.5% (bias = 6.7%). These values are well below the general FDA acceptance criterion of 20% RSD for the LLOQ [11]. A representative chromatogram at the LLOQ is shown in Fig. 3.

Exemplified chromatogram of OS (R t = 3.20 min) and OSC (R t = 1.88 min) at the LLOQ, monitored with scheduled MRMs for the following mass transitions OS m/z 313+ → 225 and OSC m/z 285+ → 138
Accuracy and precision
Accuracy and precision were determined for three independent measurements within 1 day and on three consecutive days. Results can be found in Table 1, which demonstrates that the acceptance criteria of a maximum RSD of 15% are fulfilled for all QC samples.
Recovery
The relative recoveries for OS (75 ng/mL) and OSC (200 ng/mL) were determined with 92.8% and 100.2%, respectively. These results demonstrate that there was almost no sample loss during sample preparation.
Stability
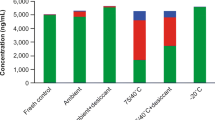
All stability tests showed sufficient stability of both analytes, OS and OSC under various test conditions. As shown in Table 2, on card stability of both analytes showed no significant sample loss over 7 days at RT and at 4 °C, simulating collection, shipment, and storage condition. Values for 1 and 3 days stability were well below 15% accuracy (bias) and precision (data not shown). Moreover, stability could be demonstrated for 24 h at −20 and +40 °C, simulating shipment conditions. Autosampler stability was shown for the extracted QC samples at 4 °C in the dark to guarantee stability during higher sample throughput.
No significant sample loss was shown under any of the test conditions.
Analysis of clinical samples
Three healthy volunteers were treated once daily with 75 mg Tamiflu® and blood samples were collected at two different time points, ∼2.5 and 4.25 h after oral administration. These time points were chosen in a time window in which the t max of the prodrug OS and the active metabolite OSC were expected, as previously described [15]. Concentrations were determined in DBS obtained from finger pricks (capillary WB) and in DBS from in BD Vacutainer® fluoride/oxalate tubes collected blood (venous WB). These tubes were also used for the collection of the blank blood used during the validation. Results are shown in Table 3. The values are well within the plasma concentrations described in literature [16]. The listed delta values describe the relative difference between the values obtained from the finger pricks and via the blood collected in fluoride/oxalate blood collection tubes from the cubital veins, prior spotting on DBS paper. The OS/OSC levels were comparable in DBS and venous WB, as judged by the delta values.
Conclusions
The current work describe for the first time a complete method validation for the simultaneous measurement of OS and OSC in WB using DBS. In the fast growing field of pharmaceutical DBS analysis, we demonstrated with the current work that DBS are feasible for Tamiflu® concentration measurements in humans. The importance of this novel approach lies in the greatly facilitated approach for blood collection, especially in neonates, infants, and small children (finger or heel prick), where collection of larger blood volumes may be problematic. The work described herein will also open possibilities for therapeutic drug monitoring (TDM), as again only small sample volumes (20 μL/spot) are needed and a more invasive venous puncture can be avoided.
Furthermore, OS potentially undergoes further ester hydrolysis (esterase activity) in collected WB samples [12] and could therefore influence accurate determinations of plasma OS and OSC (OSC is the hydrolysis product of OS) concentrations. A recently published, elaborate study by Heinig et al. of Tamiflu DBS analysis in rat blood specifically investigated stability of OS during drying time of different DBS cards and found treated (DMPK-A and DMPK-B) in contrast to chemically untreated cards to stabilize the drug [8]. This stands in contrast to a report by D’Arienzo et al. who could not observe ester hydrolysis on untreated cards (Whatman 903 Protein Saver) [17]. In order to investigate this stability issue, we compared DBS spotted from fluoride/oxalate tubes to directly spotted blood from the finger prick. The fluoride/oxalate blood collection tubes were shown to be necessary to prevent ester hydrolysis of OS after blood sampling [13, 18]. The small pilot study presented in the current work did not show any significant differences in OS or OSC concentrations in DBS collected via these two ways. Furthermore, we did not observe any significant degradation during method development or in a separate calibration curve for OS. However, since these calibration samples were prepared in previously frozen blood a loss of esterase activity during as a consequence of freeze–thaw cycles cannot be ruled out. Further research comparing a validated plasma assay and the validated DBS assay, described in the current work will be necessary to specifically address this issue in a larger patient collective.
In the future, studies will also be performed to determine ideal dosing regimes in infants and small children [19], where dosing is currently calculated according to the bodyweight or according to empiric amounts, but not based on actual blood levels in influenza-infected children.
Besides pediatrics, this assay will also provide the possibility for future PK and TDM studies in clinical and nonclinical environments.
Taken together, further research in the context of DBS analysis of Tamiflu® will have to address more technical issues like, e.g., the influence of different hematocrit (Ht) values, paper quality, sampling instructions, etc. [14]. These investigations will also depend on the study of interest and the setting of the sample collection.
References
Gamblin SJ, Skehel JJ (2010) J Biol Chem 285:28403–28409
Reddy D (2010) J Antimicrob Chemother 65(Suppl 2):ii35–ii40
Satpathy HK, Lindsay M, Kawwass JF (2009) Postgrad Med 121:106–112
Smith SM, Gums JG (2010) Paediatr. Drugs 12:285–299
Gonzales K (2010) J Pediatr Nurs 25:555–565
Meesters RJ, van Kampen JJ, Reedijk ML, Scheuer RD, Dekker LJ, Burger DM, Hartwig NG, Osterhaus AD, Luider TM, Gruters RA (2010) Anal Bioanal Chem 398:319–328
Patel P, Mulla H, Tanna S, Pandya H (2010) Arch Dis Child 95:484–487
Heinig K, Wirz T, Bucheli F, Gajate-Perez A (2011) Bioanalysis 3:421–437
Ingels AS, Lambert WE, Stove CP (2010) Anal Bioanal Chem 398:2173–2182
Thomas A, Deglon J, Steimer T, Mangin P, Daali Y, Staub C (2010) J Sep Sci 33:873–879
U.S. Department of Health and Human Services, Food and Drug Administration, Center for Drug Evaluation and Research (CDER), Center for Veterinary Medicine (CVM) (2001) Guidance for industry—bioanalytical method validation. Center for Drug Evaluation and Research (CDER), Rockville. pp 1–22
Wiltshire H, Wiltshire B, Citron A, Clarke T, Serpe C, Gray D, Herron W (2000) J Chromatogr B Biomed Sci Appl 745:373–388
Lindegardh N, Davies GR, Hien TT, Farrar J, Singhasivanon P, Day NP, White NJ (2007) Antimicrob Agents Chemother 51:1835–1836
Edelbroek PM, van der Heijden J, Stolk LM (2009) Ther Drug Monit 31:327–336
Wildschut ED, de Hoog M, Ahsman MJ, Tibboel D, Osterhaus AD, Fraaij PL (2010) PLoS ONE 5:e10938
Massarella JW, He GZ, Dorr A, Nieforth K, Ward P, Brown A (2000) J Clin Pharmacol 40:836–843
D’Arienzo CJ, Ji QC, Discenza L, Cornelius G, Hynes J, Cornelius L, Santella JB, Olah T (2010) Bioanalysis 2:1415–1422
Lindegardh N, Davies GR, Tran TH, Farrar J, Singhasivanon P, Day NP, White NJ (2006) Antimicrob Agents Chemother 50:3197–3199
Pandya HC, Spooner N, Mulla H (2011) Bioanalysis 3:779–786
Acknowledgments
We would like to thank Rachel Scheuer and Marleen Reedijk for their help with the blood collection. We would also like to acknowledge Dr. Pieter Fraaij and especially Dr. James Smith at Hoffmann-La Roche (Basel, Switzerland) who kindly provided us with the reference standards. RAG is supported by the VIRGO Consortium, an Innovative Cluster approved by the Netherlands Genomics Initiative and partially funded by the Dutch Government (BSIK 03012), the Netherlands.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Hooff, G.P., Meesters, R.J.W., van Kampen, J.J.A. et al. Dried blood spot UHPLC-MS/MS analysis of oseltamivir and oseltamivircarboxylate—a validated assay for the clinic. Anal Bioanal Chem 400, 3473–3479 (2011). https://doi.org/10.1007/s00216-011-5050-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00216-011-5050-z