
Abstract
Background
t(8;21)(q22;q22) is one of the most frequent chromosomal abnormalities in acute myeloid leukemia (AML), leading to the generation of the fusion protein AML1-ETO. Despite t(8;21) AML being considered as a subtype with a favorable prognosis, approximately 30–50% of patients experience drug resistance and subsequent relapse. N6-methyladenosine (m6A) is demonstrated to be involved in the development of AML. However, the regulatory mechanisms between AML1-ETO and m6A-related enzymes and the roles of dysregulated m6A modifications in the t(8;21)-leukemogenesis and chemoresistance remain elusive.
Methods
Chromatin immunoprecipitation, dual-luciferase reporter assay, m6A-qPCR, RNA immunoprecipitation, and RNA stability assay were used to investigate a regulatory loop between AML1-ETO and FTO, an m6A demethylase. Gain- and loss-of-function experiments both in vitro and in vivo were further performed. Transcriptome-wide RNA sequencing and m6A sequencing were conducted to identify the potential targets of FTO.
Results
Here we show that FTO is highly expressed in t(8;21) AML, especially in patients with primary refractory disease. The expression of FTO is positively correlated with AML1-ETO, which is attributed to a positive regulatory loop between the AML1-ETO and FTO. Mechanistically, AML1-ETO upregulates FTO expression through inhibiting the transcriptional repression of FTO mediated by PU.1. Meanwhile, FTO promotes the expression of AML1-ETO by inhibiting YTHDF2-mediated AML1-ETO mRNA decay. Inactivation of FTO significantly suppresses cell proliferation, promotes cell differentiation and renders resistant t(8;21) AML cells sensitive to Ara-C. FTO exerts functions by regulating its mRNA targets, especially IGFBP2, in an m6A-dependent manner. Regain of Ara-C tolerance is observed when IGFBP2 is overexpressed in FTO-knockdown t(8;21) AML cells.
Conclusion
Our work reveals a therapeutic potential of targeting AML1-ETO/FTO/IGFBP2 minicircuitry in the treatment for t(8;21) patients with resistance to Ara-C.
Similar content being viewed by others

Introduction
Acute myeloid leukemia (AML) is a highly heterogeneous disease characterized by complex cytogenetic and molecular landscape, leading to diverse prognostic outcomes [1, 2]. The t(8;21)(q22;q22) chromosomal translocation is one of the most common cytogenetic abnormalities in AML [3]. Clinically, t(8;21) AML has been categorized as a subtype with favorable prognosis; nonetheless, the outcomes of patients with this subtype still exhibit significant heterogeneity. Approximately 30–50% of t(8;21) AML patients experience relapse, resulting in poor clinical outcomes [4], suggesting the presence of genetic and clonal heterogeneity within this specific subtype of AML. Therefore, an in-depth understanding of the pathogenesis of t(8;21) AML will aid in improving personalized clinical interventions.
The AML1-ETO fusion protein, generated by the t(8;21)(q22;q22) chromosomal translocation, plays the pivotal role in the initiation of t(8;21) AML. AML1-ETO disturbs the normal biofunctions of core binding factors, blocks differentiation and induces aberrant self-renewal of hematopoietic stem cells, transforms them into preleukemic cells [5]. Cooperating gene mutations (including c-KIT, FLT3 and N/KRAS, etc.) enhance the proliferative potential of the preleukemic cells as ‘second hit’ and eventually causes full-blown leukemia [3]. Additional pathological mechanisms, such as epigenetic regulation, work in collaboration to facilitate the development of t(8;21) AML [6, 7].
As the most abundant chemical modification in eukaryotic mRNAs, N6-methyladenosine (m6A) plays a crucial role in the initiation, progression, and drug resistance of human cancer [8,9,10,11,12]. Several studies have indicated that alterations in m6A modifications contribute to leukemia development: m6A writers (i.e., methyltransferase-like 3 (METTL3) [13, 14], methyltransferase-like 14 (METTL14) [15, 16] and Wilms’ tumor 1-associating protein (WTAP) [17]), erasers (i.e., fat mass and obesity-associated protein (FTO) [18, 19] and α-ketoglutarate-dependent dioxygenase AlkB homolog 5 (ALKBH5) [20, 21]), and readers (i.e., YTH domain family 2 (YTHDF2) [22, 23]) are found to act as oncogenes in AML. In particular, the m6A demethylase FTO is reported to be highly expressed in certain AML subtypes, including those carrying t(15;17)/PML-RARA, t(11q23)/MLL-rearrangements, NPM1, or FLT3-ITD mutations, and promote leukemogenesis by regulating its mRNA targets, especially RARA and ASB2, through m6A demethylation [18]. A recent study identifies that the high expression of FTO in NPM1 mutant AML patients may be attributed to an intragenic long noncoding RNA within FTO. It is a result of intragenic transcription activation occurring in the final intron of FTO, induced by a non-random epigenetic event [24]. R-2-hydroxyglutarate, produced by mutant isocitrate dehydrogenase 1/2 (IDH1/2), inhibits the proliferation of leukemic cells by suppressing the demethylase activity of FTO and its downstream CEBPA/MYC-relevant pathway [19]. Moreover, elevated expression of FTO contributes to resistance to tyrosine kinase inhibitor in AML [25]. However, the definite regulatory relationship between FTO and leukemia-initiating fusion protein AML1-ETO and the role of FTO in progression and chemoresistance in t(8;21) AML remain largely unknown.
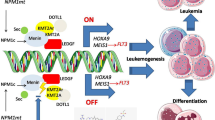
Here, we report a positive feedback regulatory loop between AML1-ETO and FTO: AML1-ETO promotes FTO expression via transcription factor PU.1; meanwhile, FTO stabilizes AML1-ETO mRNA via m6A reader YTHDF2, thus upregulating its expression. Suppression of FTO expression genetically or pharmacologically inhibits cell proliferation, promotes cell differentiation and resensitizes resistant t(8;21) AML cells to Cytosine arabinoside (Ara-C), via downregulating its functional downstream target IGFBP2. Our findings suggest a therapeutic potential of targeting AML1-ETO/FTO/IGFBP2 minicircuitry in the treatment of t(8;21) AML.
Methods
Samples from patients with leukemia and healthy donors
The collection of patient samples was approved by the Medical Ethics Committee of Shenzhen University General Hospitals. Mononuclear cell (MNC) isolated from bone marrow (BM) samples from patients with t(8;21) AML (Additional file 2: Table S1) and healthy donors were obtained with informed consent and stored in TRIzol reagent (Invitrogen) until use.
Cell culture
The t(8;21) AML cell lines SKNO-1 and Kasumi-1 were maintained in RPMI-1640 medium (Gibco) supplemented with 10% fetal bovine serum (FBS, Gibco) and 1% penicillin–streptomycin (Solarbio). Human embryonic kidney (HEK)-293T cells were maintained in DMEM (Gibco) supplemented with 10% FBS and 1% penicillin–streptomycin.
DNA site-directed mutagenesis
The pCMV6-wild type FTO-CDS (coding region sequence) plasmid was used to construct mutant FTO-CDS carrying H231A and D233A single-point mutations. The plasmid and mutation sites of FTO-CDS were kindly provided by Dr. Chuan He and Dr. Guifang Jia. DNA site-directed mutagenesis was performed using the Mut Express II Fast Mutagenesis Kit V2 (Vazyme), following the manufacturer’s instructions. Briefly, a pair of primers was designed based on the desired mutation sites. PCR amplification of target plasmids was performed using the designed primers. Amplification products were digested with DpnI, followed by homologous recombination reaction to cyclize the products. Cyclized products were used to transform competent bacterial cells, which were plated onto selective media overnight at 37 °C. Single bacterial colonies were identified by sequencing.
Plasmid construction
Wild-type and mutant FTO-CDS were PCR-amplified from pCMV6-wtFTO and pCMV6-mutFTO, respectively, using the following primers: forward 5′-CTAGACTAGTATGAAGCGCACCCCGACTGCCGAGGAACGA-3′ and reverse 5′-ATTTGCGGCCGCCTAGGGTTTTGCTTCCAGAAGCTGA-3′, and then inserted into the pLVX lentiviral plasmid between SpeI and NotI. The DNA sequences encoding shRNAs targeting human FTO (shFTO#1 TRCN0000255405 and shFTO#2 TRCN0000255403), mouse Fto (TRCN0000277195, TRCN0000216341, and TRCN0000277143 mixed), human YTHDF2 (shYTHDF2#1 TRCN0000254336, shYTHDF2#2 TRCN0000254410), human IGFBP2 (shIGFBP2#1 TRCN0000006574 and shIGFBP2#2 TRCN0000006577), and mouse Igfbp2 (TRCN0000420942, TRCN0000422736, and TRCN0000012860 mixed) were commercially synthesized by TranSheep Bio Co. Ltd., and cloned into the pLKO.1 plasmid. Likewise, human PU.1-CDS and IGFBP2-CDS were commercially synthesized and cloned into the pTSB vector. For the experiments of silencing the three YTHDF proteins, the shRNA sequences targeting the three proteins were collectively inserted into a single pLKO.1 plasmid (DF1: TRCN0000294275; DF2: TRCN000254410; DF3: TRCN000365173 as #1 and DF1: TRCN0000286871; DF2: TRCN0000254336; DF3: TRCN0000167772 as #2).
Lentiviral infection and siRNA transfection
Lentiviruses were generated from HEK-293T cells via transfection of lentiviral expression vectors together with plp-VSV-G:psPAX2 packaging mix (Addgene) using 15 μM PEI. Supernatants containing the virus were harvested 48 and 72 h after cotransfection and were added to target cells along with polybrene (8 μg/mL, Solarbio). Selection of infected cells was performed using puromycin (Solarbio, 1.5 μg/mL) 96 h after infection.
The siRNA targeting the fusion site of the AML1-ETO mRNA (siAE#1, sense, 5′-CCUCGAAAUCGUACUGAGAAG-3′, antisense, 5′-UCUCAGUACGAUUUCGAGGUU-3′; siAE#2, sense, 5′-CCUCGAAAUCGUACUGAGATT-3′, antisense, 5′-UCUCAGUACGAUUUCGAGGTT-3′) was designed according to previous studies [26] and was commercially synthesized by Sangon Biotech. siRNAs against human FTO (sc-75002), IGFBP2 (sc-37195), YTHDF2 (sc-78661) and YTHDF3 (sc-77724) were purchased from Santa Cruz Biotechnology. siRNAs were transfected into cells using the 4D-Nucleofector System (Lonza, Cologne, Germany) with the SF Cell Line 4D-Nucleofector X Kit by DS-138 for SKNO-1 and CM137 for Kasumi-1. Cells were harvested at 48 or 72 h after transfection.
The SKNO-1-siAE cell line was kindly provided by Dr. Clara Nervi which was generated by transfected with the lentiviral vector pRRLcPPT.hPGK encoding siAE oligonucleotides to silence the expression of AML1-ETO in SKNO-1 cells [6]. Kasumi-1-siAE cells were generated by siRNA transfection using the 4D-Nucleofector system.
AML1-ETO9a-driven AML mice model
The animal experiments in our study were approved by the Ethics Committee of Shenzhen University General Hospital. BM and spleen cells isolated from AML1-ETO9a-driven AML mice were kindly provided by Dr. Lan Wang [27] and were further amplified by retransplantation into sublethally irradiated (450 cGy γ-rays) C57BL/6 recipient mice via tail vein injection (2 × 104 cells/mouse). Freshly harvested BM and spleen cells were infected with pLKO.1-based lentiviral shRNA targeting mouse Fto or mouse Igfbp2, or pLKO.1-scrambled shRNA, and injected into sublethally irradiated (450 cGy) C57BL/6 recipient mice via tail vein injection (2 × 104 cells/mouse). For FB23-2 treatment, FB23-2 was administered at 6 mg/kg daily for 30 days from the third day after transplantation.
Xenograft model
Four- to six-week-old female BALB/c nude mice were used and maintained in a pathogen-free environment. After 3–5 days of adaptive feeding, each mouse received subcutaneous inoculation into the right flank with 2 × 107 SKNO-1 cells in 200 µL of PBS. When the tumors reached a volume of approximately 150 mm3, the mice were randomly divided into 4 groups (n = 6 in each group). Then the mice in each group received intraperitoneal injection once a day of DMSO, Ara-C (75 mg/kg/day), FB23-2 (2 mg/kg/day), or a combination of both. The mice were sacrificed by cervical dislocation under anesthesia after two-week drug injection and the harvested xenograft tumors were analyzed further.
NOD/SCID/IL2rγnull immunodeficient mice model
The NOD/SCID/IL2rγnull immunodeficient NSG mice (6–8 weeks old, female) were irradiated at 200 cGy and injected with GFP+ SKNO-1 cells (5 × 106) via tail vein. The mice were randomly divided into 4 groups (n = 6 in each group), receiving intraperitoneal injection every other day of DMSO, Ara-C (75 mg/kg/day), FB23-2 (4 mg/kg/day), or a combination of both, starting one week after injection. The mice were sacrificed by cervical dislocation under anesthesia 6 weeks after AML cell injection.
Cell proliferation and colony forming unit assays
The Cell Counting Kit-8 (Do**do) was used to conduct cell proliferation assays. Cells were seeded at 30,000 cells/100 μL per well into 96-well plates in triplicate and incubated with 10 μL Cell Counting Kit-8 solution at 37 °C for 4 h. Absorbance was read using a microplate reader (Multiskan FC, Thermo Fisher) at 450 nm.
The MethoCult™ H4434 Classic (StemCell Technologies) culture system was used to perform colony-forming unit assays. Briefly, 1000 cells were mixed thoroughly with 4 mL MethoCult™ medium, seeded into 35 mm dishes in triplicate, and incubated for 14 days at 37 °C and 5% CO2 with humidification. Clusters that showed morphological hematopoietic characteristics containing more than 50 cells were counted as colonies.
Flow cytometric analysis of apoptosis, cell cycle, and differentiation
The Annexin V-FITC Apoptosis Detection kit (Do**do) was used for apoptosis assays. Cells were harvested and washed twice with cold PBS. After being suspended in 1 × Annexin V binding buffer, cells were stained with Annexin V-FITC and propidium iodide (PI) for 15 min on ice. For cell cycle assays, a DNA labeling solution (Cytognos) was used. Briefly, after collection and washing with cold PBS, cells were stained with DNA labeling solution for 15 min at 25 °C. For cell differentiation assays, human AML cells were harvested and washed with cold PBS; peripheral blood (PB), BM, and spleen cells were harvested from AML1-ETO9a AML mice, and erythrocytes were lysed using RBC Lysis Buffer (00-4333-57, Invitrogen). Human AML cells were stained with anti-human CD11b (301310, Biolegend) and isolated mice cells were stained with anti-CD11b-BV421 (101251, Biolegend) for 30 min on ice. Stained cells were analyzed using a CytoFLEX S Flow Cytometer (Beckman Coulter).
RNA extraction and quantitative RT-PCR
Total RNA was extracted using TRIzol reagent (Invitrogen), in accordance with the manufacturer’s instructions. Briefly, 1 µg of total RNA was reverse-transcribed into cDNA using the GoScript™ Reverse Transcription System (Promega). Quantitative real-time PCR (qPCR) analysis was performed using 1 µL diluted cDNA (fivefold dilution) with the SYBR Fast Universal (KAPA Biosystems) on the ABI Prism 7500 sequence detection system (Applied Biosystems). Samples were run in triplicate, with GAPDH as the endogenous control. Additional file 2: Table S3 lists the primers used.
Western blotting
Western blotting was performed with equal amounts of cell lysates (~ 20 μg), using the following antibodies: anti-FTO (#45980, 1:1000; Cell Signaling Technology), anti-AML1 (#4334, 1:1000; Cell Signaling Technology), anti-PU.1 (#2266, 1:1000; Cell Signaling Technology), anti-YTHDF2 (24744-1-AP, 1:1000; Proteintech), anti-YTHDF3 (sc-377119, 1:200; Santa Cruz Biotechnology), anti-IGFBP2 (#3922, 1:1000; Cell Signaling Technology), anti-Igfbp2 (sc-515134, 1:200; Santa Cruz Biotechnology), anti-β-actin hFAB rhodamine (12004163, 1:1000; Bio-Rad), and anti-β-actin (#4970, 1:1000; Cell Signaling Technology).
m6A dot blot
Polyadenylated (poly(A)) mRNA was isolated from total RNA using the GenElute™ mRNA Miniprep Kit (Sigma-Aldrich). Isolated mRNA was denatured at 95 °C for 3 min and then dotted onto an Amersham Hybond™-N+ membrane (RPN203B, GE Healthcare) using a Bio-Dot Apparatus (#170-6545, Bio-Rad). After UV cross-linking, the membrane was blocked for 1 h using 5% nonfat milk, incubated at 4 °C with m6A antibody (ab232905, 1:400; Abcam) overnight, and then incubated with HRP-linked anti-rabbit immunoglobulin G (IgG, #7074, Cell Signaling Technology) for 1 h. Finally, the membrane was imaged using a ChemiDoc Touch Imaging System (Bio-Rad). For loading control, the RNA dotted membrane was stained with 0.02% methylene blue (Solarbio) diluted with 0.5M sodium acetate (PH 5.0).
Chromatin immunoprecipitation (ChIP)
The SimpleChIP® Plus Enzymatic Chromatin IP Kit (#9005, Cell Signaling Technology) was used to conduct ChIP assays in accordance with the manufacturer’s instructions. Briefly, to crosslink proteins to DNA, cells were treated with formaldehyde (final concentration 1.5%) for 10 min. Micrococcal nuclease and sonication were used to digest chromatin to fragments of approximately 150–900 bp, which were then incubated with PU.1 antibody (#2266, Cell Signaling Technology), AML1 antibody (#sc-8563, Santa Cruz Biotechnology), ETO antibody (#sc-9737, Santa Cruz Biotechnology) or normal rabbit IgG antibody at 4 °C overnight with rotation. Finally, 1% of digested chromatin was preserved as an input sample and purified along with the immunoprecipitated complex. The primers using for ChIP-qPCR the listed in Additional file 2: Table S3.
Dual-luciferase reporter assays
The DNA fragments of different lengths of the FTO promoter were amplified from human genomic cDNA by PCR using the primers listed in Additional file 2: Table S3 and inserted into the pGL3-basic vector (Promega) between KpnI and HindIII enzyme sites. The pcDNA3.1 vector containing DNA fragments of human PU.1-CDS were synthesized by Vigene Biosciences, Inc. Total 400 ng of pcDNA3.1 vector (i.e., pcDNA3.1-mock or pcDNA3.1-PU.1-CDS) and pGL3-basic vector (i.e., PGL3-P1 to PGL3-P4), and 20 ng pRL-TK Renilla luciferase reporter vector were cotransfected into HEK293T cells in 24-well plates using the SuperFect® Transfection Reagent (Qiagen). Luciferase activity was assessed 48 h after transfection using a Dual-Luciferase Reporter Assay System (Promega). Each experiment was repeated in triplicate.
The wild-type and m6A-mutant IGFBP2 3′ untranslated region (3′-UTR) were directly synthesized and inserted into the psiCHECK2 vector (Promega) by Vigene Biosciences, Inc. Briefly, 400 ng wild-type or mutant psiCHECK2-IGFBP2-3′-UTR and 100 ng FTO-expressing vector (i.e., pCMV6-wtFTO, pCMV6-mutFTO, or pCMV6-mock) were cotransfected into HEK-293T cells in 24-well plates with the SuperFect® Transfection Reagent (Qiagen). Relative luciferase activity was assessed using the Dual-Luciferase Reporter Assay System (Promega) 48 h posttransfection. Each experiment was repeated in triplicate.
Gene-specific m6A-qPCR
The Magna MeRIP m6A Kit (17-10499, Millipore) was used to estimate the relative m6A levels in individual transcripts. Briefly, poly(A) mRNA was enriched from total RNA using the GenElute™ mRNA Miniprep Kit (Sigma-Aldrich), one-tenth of which was preserved as the input. Subsequently, mRNA was incubated overnight at 4 °C with protein A/G magnetic beads preconjugated with m6A antibody (MABE1006) or normal mouse IgG antibody (CS200621). The bead- immunoprecipitated methylated mRNAs were eluted by free m6A competition and purified using the RNeasy MiniElute Cleanup kit (#74204, Qiagen). Purified immunoprecipitated mRNAs and input sample were evaluated by qPCR using the primers listed in Additional file 2: Table S3.
RNA immunoprecipitation (RIP) assay
The Magna RIP RNA-Binding Protein Immunoprecipitation Kit (17–700, Millipore) was used to perform RIP assays following the manufacturer’s instructions. Briefly, cell lysates were incubated at 4 °C overnight with magnetic beads preconjugated with antibodies against FTO (#31687, Cell Signaling Technology), YTHDF1 (#57530, Cell Signaling Technology), YTHDF2 (24744-1-AP, Proteintech), YTHDF3 (sc-377119, Santa Cruz Biotechnology), or mouse IgG. One-tenth of cell lysate was saved as an input sample. RNAs were eluted using proteinase K at 55 °C for 30 min. Eluted RNAs were purified using the RNeasy MiniElute Cleanup Kit (#74204 Qiagen). qPCR was performed to evaluate the interaction between YTHDF2 and mRNA transcripts. Primers used are listed in Additional file 2: Table S3.
RNA stability
Cells were incubated with actinomycin D (final concentration 5 mg/mL) and harvested after 1, 1.5, or 3 h of incubation. Total RNA was extracted using the TRIzol reagent (Invitrogen), followed by qPCR. The mRNA degradation rate was estimated in accordance with previous studies [18] using the formula: In(C/C0) = − Kdecayt. The mRNA half-life of the decay t1/2 was calculated as follows: t1/2 = In2/Kdecay.
RNA-seq and m6A-seq
Wild-type FTO-overexpressing and control Kasumi-1 cell lines were used in triplicate to conduct RNA-seq (RNA sequencing) and m6A-seq (m6A sequencing). RNA-seq and m6A-seq were performed by LC-BIO Bio-tech Ltd. (Hangzhou, China). Briefly, total RNA was extracted from wild-type FTO-overexpressing (wtFTO) and control (mock) cells using the TRIzol reagent. The RNA integrity number (RIN) of total RNA was assessed using an Agilent Bioanalyzer 2100 with an RNA 6000 Nano LabChip Kit (Agilent) with RIN ≥ 7. Poly(A) mRNA was enriched from total RNA using poly-T oligo-attached magnetic beads (Invitrogen) and then fragmented using divalent cations at elevated temperatures. Fragmented mRNAs were incubated at 4 °C with m6A antibody (#202003, Synaptic Systems) for 2 h in IP buffer, and then incubated with protein-A beads for another 2 h at 4 °C. The mixture was eluted with elution buffer (6.7 mM m6A in IP buffer) and precipitated using 75% ethanol. The eluted IP and input mRNAs were used to construct a cDNA library with strand-specific library preparation using the dUTP method. Then 2 × 150 bp paired-end sequencing was performed on an Illumina Novaseq 6000 platform (Illumina). The accession number of these data in the Sequence Read Archive (SRA) database is PRJNA876028.
RNA pull-down and mass spectrometry analysis
Biotin-labelled RNA oligonucleotides containing m6A or adenosine were directly synthesized by Sangon Biotech. The Pierce Magnetic RNA-Protein Pull-Down Kit (20164, Thermo Fisher Scientific) was used to perform RNA pull-down assays, following the manufacturer’s instructions. Briefly, up to 50 pmol of biotin-labelled RNAs was mixed with 50 μL streptavidin beads at 25 °C for 30 min and then incubated together with up to 200 μg of protein lysates with rotation for 1 h at 4 °C. After washing thrice, streptavidin beads were boiled, separated by 10% SDS-PAGE, and detected by western blot analysis or silver staining. Mass spectrometry analysis was performed by Qinglian Biotech Co. Ltd. Briefly, in-gel proteins were digested with trypsin. Peptides were then used for LC–MS/MS analysis on an EASY nLC 1200 ultra-high-pressure system (Thermo Fisher Scientific) with a nano-electrospray ion source attached to a quadrupole Orbitrap mass spectrometer (Q Exactive HF-X, Thermo Fisher Scientific).
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the iProX partner repository [Full size image
Considering the m6A "eraser" role of FTO, we only regarded the mRNA transcripts that carried m6A peaks with decreasing abundance, namely m6A hypomethylated peaks, upon FTO overexpression as potential targets of FTO. Combined with RNA-seq data, we found 372 hypomethylated peaks in FTO-overexpressing cells relative to control cells, among which 255 mRNA transcripts were significantly upregulated (hypo-up, fold change ≥ 1.2; p < 0.05), whereas 117 were downregulated (hypo-down, fold change ≤ 0.83; p < 0.05, Fig. 5D), suggesting that the majority of m6A hypomethylated mRNA transcripts (68.5%) were associated with upregulated mRNA levels in FTO-overexpressing cells. GSEA analysis [41] indicated that the hypo-up transcripts were significantly enriched with target genes of SOX2 and NANOG, BRCA1 and CHEK2 network, and many important biological processes, such as cell cycle, immune system and RNA metabolism (Additional file 1: Figure S5C). Hypo-down transcripts were significantly enriched with genes involving the BRCA1 network (Additional file 1: Figure S5D).
We further analyzed the expression correlations between FTO and the m6A hypomethylated genes in AML patient samples from the TCGA dataset. We found that 11 hypo-up (CEP57, FTX, ZC3H11A, TERF2, PAX6, IGFBP2, BTBD3, NOA1, VKORC1L1, GCC2, and YAF2) and 7 hypo-down (SSU72, FBXW5, SERF2, Rab3A, TCEA3, TSPAN14, and GGT1) genes demonstrated a significantly positive and negative correlation in expression, respectively, with FTO in TCGA AML datasets (Fig. 5E and Additional file 2: Table S2). In agreement, gene-specific m6A qPCR assays confirmed that among the 7 m6A-hypo genes (PAX6, TERF2, TCEA3, Rab3A, CEP57, YAF2, and FBXWS), 6 were associated with a significant reduction in the m6A levels (86%), whereas 2 non-m6A-hypo genes (DNMT1 and DNMT3A) in m6A-seq data did not demonstrate a significant decrease in m6A abundance (Fig. 5F). This verifies the reliability of our m6A-seq data.
IGFBP2 is directly targeted by FTO via its m6A demethylase activity
We further found that the high expression of IGFBP2 (insulin-like growth factor binding protein 2), which was listed in Fig. 5E, was significantly upregulated in patients with t(8;21) AML (n = 30) compared with normal control (n = 9, GSE30285 and GSE34814, Additional file 1: Fig. S5E). Moreover, IGFBP2 was associated with significantly worse event-free survival and overall survival in patients with AML (GSE6891, n = 344, log-rank test, p < 0.001 and p = 0.006, respectively; Additional file 1: Fig. S5F) and t(8;21) AML (n = 26, log-rank test, p = 0.045 and p = 0.061, respectively; Fig. 5G). The primary BM cells from AML patients at diagnosis with poor response to Ara-C had a significantly higher level of IGFBP2 mRNA compared with high responders (GSE97393; Fig. 5H). Moreover, IGFBP2 has been shown to support the migration and survival of AML cells [42]. We observed that IGFBP2 manifested a significant positive association with the expression of FTO in AML cohorts from the TCGA database (Additional file 1: Fig. S5G) and our in-house t(8;21) AML cohorts (Fig. 5I). Our m6A-seq and RNA-seq data indicated that overexpression of FTO caused a significant reduction of m6A levels in the 3′ untranslated region (3′-UTR) of IGFBP2 mRNA (fold change = 0.72, p = 0.001, Fig. 5J) and significant increase level of IGFBP2 transcripts (fold change = 2.22, p < 0.001; Fig. 5K). We thus aimed at IGFBP2 as a potential target of FTO for further investigation.
Overexpression of wild-type FTO upregulated the expression of IGFBP2 at both the mRNA and protein levels compared with those in control and mutant FTO cells, whereas silencing the endogenous expression of FTO resulted, as expected, to an opposite pattern (Fig. 6A–D). In addition, treatment with FB23-2 also resulted in a dose-dependent reduction in the expression of IGFBP2 (Fig. 6E, F). RIP assay demonstrated the direct binding of FTO to IGFBP2 transcripts (Fig. 6G). Gene-specific m6A qPCR assays further confirmed that forced expression of wild-type FTO substantially reduced the levels of m6A in IGFBP2 mRNA transcripts (Fig. 6H); opposite results were shown when the endogenous FTO was knocked down (Fig. 6I). To further validate that FTO targets the 3′-UTR of IGFBP2 mRNA transcripts, we created reporter vectors baring the coding sequence of IGFBP2 3′-UTR or corresponding m6A consensus mutant sequences (Additional file 1: Fig. S6A) and found that compared with the empty and mutant FTO vectors, luciferase activity was significantly promoted by wild-type FTO when cotransfected with the reporter vector containing the wild-type IGFBP2 3′-UTR fragment with intact m6A consensus, whereas this effect was abrogated when cotransfected with reporter vectors containing mutant m6A consensuses (Fig. 6J).

FTO promotes the expression of IGFBP2 in an m6A-dependent manner. A, B Expression of IGFBP2 assessed by qPCR (A) and western blotting (B) in SKNO-1 or Kasumi-1 cells transduced lentivirally with wild-type FTO (wt-FTO), mutant FTO (mut-FTO), or mock vectors. C, D Decreased expression of IGFBP2 detected by qPCR (C) and western blotting (D) in SKNO-1 and Kasumi-1 cells after knockdown of FTO by shRNAs. E, F Expression of IGFBP2 analyzed by qPCR (E) and western blotting (F) in SKNO-1 or Kasumi-1 cells after FB23-2 treatment for 72 h. G RIP-qPCR showing the interaction between IGFBP2 mRNA transcripts and FTO protein in SKNO-1 and Kasumi-1 cells. H, I The levels of m6A in IGFBP2 transcripts assessed by gene-specific m6A qPCR assay in SKNO-1 and Kasumi-1 cells transduced with wt-FTO, mut-FTO, or control vector (H); or with FTO shRNA or shNS vectors (I). J Dual luciferase reporter assays for the effect of wild-type or mutant FTO on the relative luciferase activity of psiCHECK2-IGFBP2-3′-UTR with either wild-type or mutant m6A sites. K, L The mRNA half-life (t1/2) of IGFBP2 transcripts in SKNO-1 and Kasumi-1 cells transduced with FTO shRNA or shNS vectors (K) or treated with DMSO or FB23-2 for 72 h (L). M–O RIP-qPCR showing the interaction between IGFBP2 mRNA transcripts and YTHDF1 (M), YTHDF2 (N) and YTHDF3 (O) protein in SKNO-1 and Kasumi-1 cells. P, Q Increased expression of IGFBP2 after YTHDF1, YTHDF2 and YTHDF3 knockdown analyzed by qPCR (P) and western blotting (Q) in SKNO-1 and Kasumi-1 cells
RNA stability assays indicated that FTO knockdown noticeably shortened the half-life of IGFBP2 mRNA transcripts in SKNO-1 and Kasumi-1 cells (Fig. 6K). Likewise, the half-life of IGFBP2 mRNA transcripts manifested a dose-dependent reduction when the two t(8;21) AML cells were treated with FB23-2 (Fig. 6L), suggesting that upregulation of IGFPB2 is at least partially attributed to the increased stability of its transcripts mediated by FTO-induced m6A demethylation.
To screen specific m6A readers targeting the 3′-UTR of IGFBP2, we performed an RNA pull-down assay using 4 pairs of m6A methylated or unmethylated single-stranded RNAs carrying part of the 3′-UTR sequence of IGFBP2 (~ 20 bp in length) as baits, followed by mass spectrometry analysis. Each pair of RNA baits contained one of the 4 putative m6A consensus motifs in the 3′-UTR of IGFBP2 (Additional file 1: Fig. S6B). We detected that the m6A reader YTHDF2 was dramatically enriched in all 4 m6A-modified RNA baits, YTHDF1 and YTHDF3 were markedly enriched in 3 of the 4 m6A-methylated baits compared with the unmethylated control, whereas other known readers were enriched in no more than 1 bait (YTHDC2, IGF2BP2/3, HNRNPA2B1 and HNRNPC were enriched in only 1 baits and YTHDC1/3, IGF2BP1 were enriched in none of the 4 baits; Additional file 1: Fig. S6C). RIP assays further confirmed that YTHDF1, YTHDF2 and YTHDF3 bound to IGFBP2 transcripts directly (Fig. 6M–O). The expression of IGFBP2 was barely altered when either YTHDF2 or YTHDF3 was knocked down (Additional file 1: Fig. S6D and S6E). However, when the three YTHDF proteins were simultaneously knockdown by shRNA, the expression of IGFBP2 were greatly increased (Fig. 6P, Q), suggesting that the YTHDFs proteins work together to mediate the decay of IGFBP2 mRNA.
IGFBP2 is a functionally important target gene of FTO in t(8;21) AML
The pathological role of IGFPB2 in t(8;21) AML was then investigated. When endogenous IGFBP2 was knocked down in the two t(8;21) AML cell lines (Fig. 7A), we observed a substantial repression on cell proliferation (Fig. 7B) and colony-forming capacity (Additional file 1: Fig. S7A) and a promotion on G1 cell cycle arrest (Additional file 1: Fig. S7B), cell apoptosis (Fig. 7C and Additional file 1: Fig. S7C) and cell differentiation (Fig. 7D, E), which was largely similar to the effect of the FTO knockdown. Moreover, IGFBP2 knockdown notably decreased the IC50 value of Ara-C in the two Ara-C resistant cell lines (Fig. 7F).

IGFBP2 promotes leukemogenesis and resistance to Ara-C. A Confirmation of IGFBP2 knockdown by shRNAs in SKNO-1 and Kasumi-1 cells using western blotting. B Effects of IGFBP2 knockdown on the proliferation of SKNO-1 and Kasumi-1 cells by CCK8 assays. C Effect of silencing IGFBP2 on cell apoptosis in SKNO-1 and Kasumi-1 cells 72 h after siRNA transfection. D The effect of IGFBP2 knockdown on differentiation of SKNO-1 and Kausmi-1 cells. The percentage of CD11b+ cells was quantified (right panel). E Wright-Giemsa staining of SKNO-1 and Kausmi-1 cells with or without IGFBP2 knockdown. F CCK-8 assays for SKNO-1 and Kasumi-1 cells transfected with IGFBP2 shRNA or scramble vectors treated with varying concentrations of Ara-C for 48 h. G Western blot analysis for knockdown of Igfbp2 in AML1-ETO9a-driven AML cells. H Kaplan–Meier survival curves of AML1-ETO9a-driven AML mice (n = 10 for each group) after Igfbp2 knockdown. I Wright-Giemsa staining of bone marrow and H&E staining of livers and spleens for Igfbp2-knockdown AML1-ETO9a-driven AML mice. J–O Flow cytometric analysis of the percentage of GFP+ AML1-ETO9a AML cells (J–L) and distribution of anti-CD11b-stained GFP + AML cells (M–O) in PB, BM, and SP of AML1-ETO9a-driven AML mice with and without Igfbp2 knockdown 7 weeks after transplantation
In the AML1-ETO9a AML mouse model, we found that Igfbp2 knockdown prolonged the overall survival of recipient mice (log-rank test, p < 0.001; Fig. 7G, H). Leukemic infiltration in BM, liver and spleen was also significantly suppressed when Igfbp2 was knocked down (Fig. 7I and Additional file 1: Fig. S7D–E). Moreover, Igfbp2 knockdown decreased the abundance of leukemic cells (Fig. 7J–L) and the proportion of CD11b-stained leukemic cells (Fig. 7M–O) in PB, BM, and spleen samples.
Rescue experiments demonstrated that when IGFBP2 was forced to be overexpressed in FTO-knockdown t(8;21) AML cells (Fig. 8A), the restoration of cell proliferation (Fig. 8B), colony-forming capacity (Fig. 8C) and regain of Ara-C tolerance (Fig. 8D and Additional file 1: Fig. S8A) were observed. Rescue experiments conducted with AML1-ETO9a AML mouse model also revealed a significant restoration in the infiltration of leukemic cells in PB, BM, liver and spleen when Igfbp2 was overexpressed in Fto-knockdown cells (Fig. 8E–G and Additional file 1: Fig. S8B–C). Force expression of Igfbp2 greatly prevented differentiation of leukemic cells in PB, BM and spleen mediated by Fto knockdown (Additional file 1: Fig. S8D). Taken together, our data suggest that IGFBP2 is a functionally critical target of FTO and is essential for FTO-mediated t(8;21) AML progression and Ara-C resistance.

FTO regulates leukemogenesis and sensitivity of t(8;21) AML cells to Ara-C through IGFBP2. A Western blotting for the expression of IGFBP2 in SKNO-1 and Kasumi-1 cells transduced with shNS (pLKO.1-shNS + empty pTSB), shFTO (pLKO.1-shFTO#1 + empty pTSB), shFTO + IGFBP2 (pLKO.1-shFTO#1 + pTSB-IGFBP2-CDS), or IGFBP2 (pLKO.1-shNS + pTSB-IGFBP2-CDS). shNS, scramble shRNA. B CCK-8 assays for the effects of FTO knockdown and/or overexpression of IGFBP2 on the proliferation of SKNO-1 and Kasumi-1 cells. C Effects of FTO knockdown with IGFBP2 overexpression on colony-forming capacity of SKNO-1 and Kasumi-1 cells. D CCK-8 assays for the effects of FTO knockdown and/or overexpression of IGFBP2 on the sensitivity to Ara-C in SKNO-1 and Kasumi-1 cells. E–G Effects of FTO knockdown with IGFBP2 overexpression on AML1-ETO9a-driven AML mice. Western blot analysis for Igfbp2 and Fto (E), Wright-Giemsa staining of bone marrow and H&E staining of livers and spleens (F) and flow cytometric analysis of the abundance of GFP+ AML1-ETO9a AML cells in PB, BM and SP (G) were shown. H Proposed model depicting the regulatory interactions and role of FTO in leukemogenesis and Ara-C resistance in t(8;21) AML.
