
Abstract
YT521-B homology domain family member 2 (YTHDF2) is an N6-methyladenosine (m6A)-binding protein that was originally found to regulate the stability of mRNA. Growing evidence has shown that YTHDF2 can participate in multifarious bioprocesses, including embryonic development, immune response, and tumor progression. Furthermore, YTHDF2 is closely associated with the proliferation, apoptosis, invasion, and migration of tumor cells, suggesting its significant role in cancers. YTHDF2 primarily relies on m6A modification to modulate signaling pathways in cancer cells. However, the expression and function of YTHDF2 in human malignancies remain controversial. Meanwhile, the underlying molecular mechanisms of YTHDF2 have not been elucidated. In this review, we principally summarized the biological functions and molecular mechanisms of YTHDF2 in tumors and discussed its prognostic and therapeutic values.
Similar content being viewed by others


Introduction
RNA epitranscriptomics has been found to play key roles in numerous cellular functions and has attracted increasing attention. Presently, there have been more than 100 types of chemical modifications of RNA found in various cells [1]. N6-methyladenosine (m6A) is considered to be the most prevalent and ample internal transcription modification in eukaryotic messenger RNAs (mRNAs), microRNAs (miRNAs), and long noncoding RNAs (lncRNAs) [2, 3].
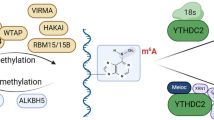
The methylation modification of m6A has been confirmed to be dynamic and reversible, involving methyltransferase “writers”, demethylase “erasers” and methylated reading protein “readers” [4]. For example, methyltransferase-like 3 (METTL3) and METTL14 can form a steady heterodimer complex. They shape the m6A methyltransferase complex (MTC) along with their accessory factors Wilms tumor 1-associated protein (WTAP), Vir like m6A methyltransferase associated (VIRMA/KIAA1429), RNA binding motif protein (RBM) 15/15b, zinc finger CCCH-Type containing 13 (ZC3H13), and HAKAI [5,6,7,8,9,10]. These factors act as m6A "writers" and collectively catalyze m6A modification. “Erasers”, including fat mass and obesity-associated protein (FTO) and alkB homolog 5 (ALKBH5), could dislodge the methyl code of m6A modification from target RNA [11, 12]. “Readers” include YT521-B homology (YTH) domain-containing protein, eukaryotic initiation factor 3 (eIF3), insulin-like growth factor 2 mRNA binding protein families (IGF2BPs), and heterogeneous nuclear ribonucleoprotein protein families (HNRNPs). They can recognize and bind to the site of the m6A modification and engender functional signals [13,14,15,16,17].
The YTH domain protein family, including YTHDF1, YTHDF2, YTHDF3, YTHDC1, and YTHDC2, has been validated as a direct m6A “reader” [18, 19]. Furthermore, YTHDF1, YTHDF2, and YTHDF3 primarily recognize and bind to the site of m6A modification in the cytoplasm, while YTHDC1 and YTHDC2 act in the nucleus [20, 21]. According to previous reports, YTHDF2, YTHDF3, and YTHDC2 functioned to accelerate the degradation of target mRNAs, YTHDF1, YTHDF3, and YTHDC2 increased the translation of target mRNAs, and YTHDC1 regulated the splicing and nuclear export of target mRNAs [22, 23]. The eIF3 protein is capable of binding to the site of m6A modification on the 5’-UTR of mRNA, thereby accelerating the translation of RNA [24]. The details of the currently known m6A modification mechanism are shown in Fig. 1.

The schematic of m6A modification mechanism. METTL3 and METTL14 shape the m6A methyltransferase complex along with WTAP, VIRMA, KIAA1429, RBM15/15b, ZC3H13, and HAKAI, serving as m6A "writers". FTO and ALKBH5 shape the m6A demethylase, serving as m6A "erasers". YTHDF1/2/3, eIF3, IGF2BP1/2/3, and HNRNPs shape the m6A modification binding protein, serving as m6A “readers”
Methylated m6A actively participates in many vital physiological processes, such as stem cell differentiation and pluripotency, embryonic development, circadian rhythm, and DNA damage response. With consecutive studies on the function and mechanism of m6A, it has been shown that the progression of several types of cancer can be affected by the abnormal expression of m6A methylation-related proteins [25,26,27,28]. Furthermore, methylated m6A is involved in the biological processes of cancer cells, including cell self-renewal and differentiation, the pluripotency of cancer stem cells (CSCs), cell proliferation, metastasis, and tumor immunity [29,30,31].
It is known that m6A modification can recruit particular “reader” proteins or alter the structure of mRNA to modulate the processing, stability, and translation of mRNA [32, 33]. Among them, YTHDF2, the binding protein of m6A, was the first discovered and most efficient m6A “reader” [29]. It was reported that YTHDF2 could regulate mRNA degradation and cell viability [16, 34]. The interaction binding site between YTHDF2 and m6A was usually located in the 3’-UTR of mRNA[29]. However, emerging evidence suggested that YTHDF2 specifically bound to mRNA bearing m6A methylation markers at the 5’-UTR, which subsequently facilitated protein translation [35]. YTHDF2 was reported to present dual functions in tumors by regulating the proliferation and migration of tumor cells [36, 37]. For example, YTHDF2 was upregulated and acted as an oncogene in multiple cancers, including acute myelocytic leukemia (AML), lung cancer, and gastric cancer [38,39,40]. In contrast, YTHDF2 was also found to be downregulated and served as a tumor suppressor in osteosarcoma and melanoma [31, 41].
Based on the controversial role of YTHDF2 in various cancers, we summarized its expression patterns and molecular mechanisms in tumorigenesis and discussed the potential prognostic and therapeutic value of YTHDF2 in malignant tumors.
The structure of YTHDF2
YTHDF2 occupies the full length of 579 amino acids (aa), has the regions localized to mRNA processing bodies (aa 2–384), and interacts with m6A-containing mRNAs (aa 385–579) that contains the YTH domain (aa 410–544), which includes the m6A binding site [16]. A previous study has indicated that the YTH domain of YTHDF2 is globularly folded with a central core consisting of eight β-strands (β1-β8), three α-helices (α1-α3), and two 310-helices. Furthermore, residues W486 in the β4-β5 loop, W432 in the β2 strand, and W491 in the β4-β5 loop form an aromatic cage containing m6A [42] (Fig. 2A). Interestingly, it was also proved that the YTH domain of YTHDF2 is a globular fold with a four-stranded β-sheet (β1-β4), four α-helices (α1-α4), and flanking regions on both sides. A hydrophobic pocket is formed by the aromatic residues Y418, W432, W486, and W491 [43, 44] (Fig. 2B).

Two hypotheses of YTHDF2 structure. A Eight β-strands (β1-β8), three α-helices (α1-α3) and two 310-helices in YTHDF2 domain, with W486, W432, and W491 form an aromatic cage containing m6A mononucleotides. B The structure of the YTH domain contains four β-sheets (β1–β4), four α-helices (α1–α4), and flanking regions on both sides. The aromatic residues Y418, W432, W486, and W491 form a hydrophobic pocket
Expression pattern and function of YTHDF2 in human cancers
The expression pattern of YTHDF2 has been confirmed in numerous studies, and the expression level of YTHDF2 has been found to vary in different types of cancer. In most cases, the expression of YTHDF2 is upregulated in tumor tissues in comparison with normal tissues, and YTHDF2 plays an oncogenic role in these types of cancers. Nevertheless, even within the same cancer type, several studies have yielded opposite results. The detailed expression levels of YTHDF2 in various cancers are shown in Table 1. The targets of YTHDF2 and their functions in cancers are shown in Table 2.
YTHDF2 in digestive system tumors
Gastrointestinal cancer
Gastric cancer (GC)
Yan et al. and Zhang et al. elucidated that YTHDF2 was upregulated in GC [39]. Conversely, recent studies demonstrated that YTHDF2 was downregulated in GC [45, 46]. Overexpression of YTHDF2 accelerated the degradation of phosphate and tension homology deleted on chromosome ten (PTEN) mRNA, a remarkable tumor suppressor, which considerably increased the proliferation, invasion, and migration of GC cells [39]. However, through Gene Set Enrichment Analysis (GSEA) and external experiments such as quantification of m6A methylation and western blot assay, YTHDF2 was found to be a potential tumor inhibitory factor, and high YTHDF2 expression was correlated with the prolonged survival time of GC patients [45]. Additionally, knockout of YTHDF2 significantly increased the expression of Forkhead box protein C2 (FOXC2), thereby suppressing the proliferation, invasion, and migration of GC cells [46].
Colorectal cancer (CRC)
It has been demonstrated that YTHDF2 is downregulated in CRC [47]. Moreover, Yang et al. detected that YTHDF2 inhibited the expression of X inactivate-specific transcript (XIST) in CRC cells, which could accelerate tumor growth and metastasis [47]. Similarly, Zhuang et al. demonstrated that YTHDF2 acted as a protective gene, which led to better overall survival (OS) in rectal cancer patients [48]. In summary, YTHDF2 may play an essential role in the prognosis of CRC. However, the protein levels of YTHDF2 were recently reported to be elevated in CRC tissues in comparison with adjacent normal tissues [49]. Overexpression of YTHDF2 facilitated the proliferation of CRC cells, suggesting that YTHDF2 may play a carcinogenic effect in CRC [49].
Hepatocellular carcinoma (HCC)
Hou et al. and Zhong et al. found decreased expression of YTHDF2 in both HCC tissues and HCC cells [36, 50]. Patients with low YTHDF2 expression presented higher TNM, advanced BCLC stage classification, lower OS and relapse-free survival (RFS) rates. Silencing of YTHDF2 accelerated tumor inflammation and vascular abnormalities, thereby promoting the tumor growth, metastasis, and vascular remodeling of liver cancer [50]. The positive expression of YTHDF2 could restrain cell proliferation and tumor growth in mouse xenografts [36]. Therefore, YTHDF2 may play a critical role in inhibiting tumorigenesis and prolonging survival time.
However, by analyzing data from the Cancer Genome Atlas (TCGA) and Gene Expression Omnibus (GEO) databases, Qu et al. demonstrated that most m6A-related genes, including YTHDF2, were drastically highly expressed in HCC tissues and hepatoblastoma cells compared with adjacent normal tissues [51,52,53]. Similarly, Chen et al. found that YTHDF2 was upregulated in liver cancer [54, 55]. Moreover, the overexpression of YTHDF2 resulted in shortened survival time and poor prognosis [51, 52]. Highly expressed YTHDF2 can promote the proliferation and migration of liver cancer cells [54, 55], increase the number of liver cancer stem cells (CSCs), and enhance the tumor burden and lung metastasis in vivo [53]. To conclude, the contrary results may be associated with the heterogeneity of cell lines and tumor tissues. Consequently, further studies are required to obtain an in-depth understanding of the factors affecting gene function in various cell backgrounds.
Pancreatic cancer (PC)
Chen et al. illustrated that the expression of YTHDF2 in PC tissues was upregulated, and found that YTHDF2 gradually increased with the elevation of clinical stage. Knockdown of YTHDF2 induced phase arrest of G1 and suppressed the proliferation of PC cells [56]. Nonetheless, YTHDF2 was observed to decrease the invasion, adhesion, migration, and EMT of pancreatic cancer cells [56]. Moreover, bioinformatics analysis and RNA immunoprecipitation (RIP) analysis revealed that YTHDF2 could bind to its target genes and then promote their degradation, resulting in increased or decreased growth of PC cells [57, 58]. Thus, it can be concluded that YTHDF2 acts as both a positive and negative factor in PC, and further investigations are warranted to improve our knowledge of the involved molecular mechanism.
YTHDF2 in respiratory tumors
Lung cancer
** et al. investigated the role of m6A-related genes in non-small-cell lung cancer (NSCLC) and discovered that YTHDF2 was downregulated in tumor tissues [59]. Sheng et al. found that YTHDF2 was highly expressed in lung cancer tissues compared with normal lung tissues [40]. Similarly, it was reported that the expression of YTHDF2 was upregulated in patients with lung adenocarcinoma and NSCLC through bioinformatic analysis [60,61,62]. Interestingly, the elevated expression of YTHDF2 was positively correlated with the OS and RFS of lung cancer patients, which was attributed to YTHDF2 promoting the enrichment of tumor-infiltrating lymphocytes and inhibiting the expression of PD-L1 [60, 61]. Moreover, the upregulation of YTHDF2 significantly increased the proliferation, migration, colony formation, metabolic defects, and pentose phosphate pathway (PPP) flux of lung cancer cells to promote lung cancer growth [40, 62]. However, YTHDF2 significantly suppressed cell proliferation, invasion, migration, and epithelial-mesenchymal transition (EMT) in NSCLC [59]. In conclusion, the function of YTHDF2 in lung cancer is controversial, and its specific role needs to be further clarified.
YTHDF2 in hematological malignancies
Acute myelocytic leukemia (AML)
Recent studies have investigated the expression patterns of YTHDF2 in primary AML patients [38, 63]. The results demonstrated that YTHDF2 was remarkably upregulated in all clinical AML subtypes and was essential to the initiation and dissemination of AML in both human and mouse models. YTHDF2 was found to abate the half-life of most m6A transcripts, which was conducive to the integrality of leukemic stem cell functions. In addition, knockdown of YTHDF2 in human AML cells markedly suppressed proliferation and promoted TNF-mediated apoptosis, while it did not influence loid differentiation and normal hematopoiesis [38, 63]. It was also proved that targeting YTHDF2 increased the number of hematopoietic stem cells and promoted marrow reconstitution. Meanwhile, knockout of YTHDF2 apparently prolonged the survival time in the AML mouse model compared with the control group [38].
Peripheral T-cell lymphoma (PTCL)
Recent studies have discovered repeated exacerbating deletions and mutations of a novel gene, YTHDF2, in peripheral T-cell lymphoma, not otherwise specified (PTCL-NOS), which may imply the functional importance of YTHDF2 in the pathogenesis of this disease[64]. PTCL-NOS were based on unique genetic profiles, including several discrete mature T cell tumor subtypes. These tumors also showed alterations in various low-frequency somatics, including YTHDF2 [64]. Hence, these findings may contribute to offering novel designs in molecular classification and patient stratification of PTCL-NOS.
YTHDF2 in urinary tumors
It was reported that YTHDF2 was overexpressed in both bladder cancer and prostate cancer (PCa) [65,66,67] and was remarkably downregulated in clear cell renal cell carcinoma (ccRCC) [68]. A lack of YTHDF2 could significantly decrease the migration rate and reduce the expression level of related proteins in bladder cancer cells, indicating that YTHDF2 acts as an oncogene in bladder cancer [65]. Moreover, the positive expression of YTHDF2 in prostate cancer patients manifested a high tumor grade [66]. With the knockdown of YTHDF2, the level of m6A in PCa cells was drastically increased. Concurrently, it remarkably suppressed cell proliferation, migration, and colony formation ability, and increased cell apoptosis [66, 67]. Therefore, YTHDF2 was found to act as a tumor-promoting factor in PCa. In ccRCC, YTHDF2 was uncovered to be a protective gene by univariate Cox regression analysis [68].
YTHDF2 in gynecological reproductive system tumors
Woo et al. and Niu et al. found no significant biological functions of YTHDF2 on ovarian cancer or breast cancer [69, 70]. However, recent studies showed that YTHDF2 was upregulated in ovarian cancer or triple-negative breast cancer (TNBC) [71]. Either overexpression or knockdown of YTHDF2 did not alter the expression level of tumor suppressor genes in breast cancer [69]. Woo et al. found that YTHDF2 was not the reader in the oncogenes of ovarian cancer and breast cancer and did not exert biological function [70]. However, expression of YTHDF2 was recently reported to be elevated in MYC-driven TNBC compared with hormone receptor-positive and human epidermal growth factor receptor 2 positive breast cancers [71]. The deficiency of YTHDF2 significantly reduced proliferation rates of TNBC cell lines, yet increased apoptosis, and G1 checkpoint arrest [71]. The study also indicated that YTHDF2 was crucial to the survival of TNBC cells, while it is dispensable for cells that were less dependent on high expression levels of MYC, suggesting the elusive role of YTHDF2 in breast cancer [71]. Moreover, YTHDF2 deficiency suppressed the proliferation, anchorage-independent growth, and colony-forming ability of ovarian cancer cell lines[72]. The diverse functions of YTHDF2 may depend on the different contexts of cancers or by modulating different target genes. In cervical cancer (CC), knockdown of YTHDF2 significantly increased the expression and stability of GAS5, a tumor suppressor gene, thereby inhibiting the proliferation, migration and invasion of CC cells in vitro and suppressing the tumor growth and metastasis of CC in vivo [73]. The above results revealed the critical role of YTHDF2-mediated epigenetic alterations in CC progression.
YTHDF2 in other cancers
YTHDF2 was also found to be upregulated in head and neck squamous cell carcinoma (HNSCC) [74], ocular melanoma [75], and glioblastoma (GBM) [76,77,78]. It was reported that YTHDF2 participated in regulating cell proliferation, migration, and invasion in vitro and in vivo, which indicated YTHDF2 as a carcinogenic gene in these tumors [75,76,77,78]. Other studies have shown that YTHDF2 is expressed at low levels in melanoma and osteosarcoma [31, 41], where YTHDF2 can directly combine and accelerate the degradation of other oncogenes. Low expression of YTHDF2 in these cancers was found to be linked to poor OS in patients, as well as increased tumor size, TNM stage, lymph node, and distant metastasis [31, 41].
Molecular mechanisms of YTHDF2 in tumorigenesis
As stated above, YTHDF2 is linked to multiple functions of human cancer cells and acts as an oncogene or tumor suppressor gene in different cancers. Here, the associated mechanisms of YTHDF2 in human cancers are listed and separated by its different expression patterns in various tumors. As presented in Fig. 3, the underlying mechanisms of YTHDF2 as an oncogene in various malignancies are gathered and summarized. YTHDF2 is also diminished and acts as a tumor suppressor in other malignant tumors. The underlying molecular mechanisms of YTHDF2 as a tumor suppressor were collected and are shown in Fig. 4.

The underlying mechanisms of YTHDF2 in promoting cancer progression. YTHDF2 plays a significant role in tumor proliferation, invasion, migration, metabolism, and apoptosis in an m6A-dependent manner. The fundamental mechanisms are shown as follow: A the pentose phosphate pathway, B tumor necrosis factor (TNF) signaling, C the PI3K/AKT signaling pathway, D Wnt/β-catenin pathway, E miRNAs modulate YTHDF2 expression, F YTHDF2 modulates the expression of tumor suppressors in an m6A-dependent manner

Roles of YTHDF2 in inhibition of cancer progression. YTHDF2 plays an essential role in tumor proliferation, invasion, migration, and metastasis. The fundamental mechanisms are shown as follow: A the PI3K/AKT signaling pathway, B the MAPK/ERK pathway, C Hippo/YAP pathway, D the inflammatory pathway, E YTHDF2 regulates the degradation of oncogenes in an m6A-dependent manner, F YTHDF2 directly interacts with YAP to enhance its degradation
YTHDF2 regulated the PPP
The pentose phosphate pathway (PPP) plays a significant role in regulating the growth of tumor cells by providing cells with ribose-5-phosphate and NADPH with the help of its key regulatory enzyme glucose-6-phosphate dehydrogenase (G6PD) and its rate-limiting enzyme 6-phosphogluconate dehydrogenase (6PGD) [79]. Sheng et al. identified that YTHDF2 could directly bind to the m6A-modified site of the 3'-UTR of 6PGD and accelerate the translation of 6PGD mRNA in lung cancer cells. Consequently, the expression of 6PGD in epigenetics was increased to enhance the flux of PPP and promote the cellular metabolism and tumor growth of lung cancer (Fig. 3A).
YTHDF2 suppressed TNF signaling pathway
As was reported, silencing of YTHDF2 could increase the half-life of the m6A levels of mRNA, indicating that YTHDF2 accelerated the degradation of m6A mRNA in leukemia [38]. Interestingly, the role of YTHDF2 was also found to be related to inhibition of tumor necrosis factor (TNF), which subsequently suppressed apoptosis of tumor cells. Specifically, knockdown of YTHDF2 was found to lead to the upregulation of TNF receptor superfamily member 1b (TNFRSF1B), which encodes TNF receptor 2 (TNF-R2), by extending the half-life of m6A-modified TNFRSF1B transcripts in leukemia cells [38, 63]. Previous studies indicated that TNFR2 binds to its ligand TNF, to mediate the binding of some adaptor proteins, which in turn initiate signal transduction to regulate cell death [80]. Thus, YTHDF2 may take part in inhibiting cell apoptosis by TNF signaling (Fig. 3B).
YTHDF2 regulated the PI3K/AKT signaling pathway
Activated phosphoinositide-3-kinase (PI3K) triggers AKT activation, leading to the activation of mammalian target of rapamycin (mTOR) and other signaling pathways that promote cell survival [81, 82], which is inhibited by PTEN [83]. Analysis of the RIP assay indicated that YTHDF2 could recognize and bind to PTEN mRNA to promote its degradation [39]. Therefore, the decreased expression of PTEN promotes the activation of PI3K/AKT signaling, thereby contributing to tumorigenesis (Fig. 3C). PI3K and PTEN modulated the downstream AKT and other effectors by acting contrary roles as ‘on–off’ switches [84]. Phosphoinositide-3-kinase catalytic beta (PI3KCB), a catalytic subunit of PI3K, was reported to be modified by m6A resulting in its degradation, which was caused by YTHDF2, and subsequently inhibited the activation of PI3K/AKT signaling pathway to limit tumor progression (Fig. 4A) [57].
YTHDF2 regulated the Wnt/β-catenin pathway
The Wnt/β-catenin pathway is highly conserved, and its abnormal activation facilitates cancer progression by enhancing cell proliferation and metastasis [85, 86]. Glycogen synthase kinase 3 beta (GSK3β) is a crucial component of the Wnt/β-catenin pathway, and its inactivation causes β-catenin to concentrate in the cell and transfer to the nucleus to enhance the progression of tumors [87, 88]. YTHDF2 was reported to recognize and bind m6A-modified GSK3β mRNA to promote its degradation, which subsequently decreased the phosphorylation of β-catenin to enhance the stability of the β-catenin protein, thereby promoting CRC cell proliferation [49]. Additionally, AXIN1, encoding a negative regulator of the Wnt/β-catenin pathway, was identified as a direct target of YTHDF2. Specifically, a remarkable enrichment of m6A in AXIN1 mRNA was detected, and the interaction between YTHDF2 and AXIN1 was determined by RIP-qPCR. Overexpression of YTHDF2 shortened the half-life of AXIN1 to decrease its expression to promote Wnt/β-catenin signaling, thus enhancing the progression of lung cancer (Fig. 3D) [62].
YTHDF2 acted as the target gene of miRNAs
It has been reported that miRNA is a significant bioactive molecule that induces posttranscriptional gene regulation in eukaryotes [89]. Yang et al. elaborated that miR-145 could modulate the levels of m6A by targeting the 3'-untranslated region (3'- UTR) of YTHDF2 mRNA in HCC cells [55]. Moreover, YTHDF2, which was negatively linked to miR-145, could reduce the levels of m6A in HCC cells, thereby promoting the proliferation of HCC cells [55]. Similarly, YTHDF2 was found to be the direct target gene of miR-495 by the dual-luciferase reporter assay. Overexpressed YTHDF2 could reverse the inhibitory role of miR-495 and decreased the m6A levels of mps one binder kinase activator 3B (MOB3B) to promote the proliferation and migration of prostate cancer cells[67]. Moreover, miR-6125 could inhibit the proliferative ability of CRC cells by targeting the YTHDF2 mRNA [49]. In summary, YTHDF2 could act as a target of miRNA to participate in the progression of cancer. However, the relationship between YTHDF2 and other miRNAs in human malignancies requires further investigation (Fig. 3E).
YTHDF2 inhibited the MAPK/ERK pathway
The mitogen-activated protein kinase (MAPK) / extracellular regulated kinase (ERK) signaling pathway is known to transmit signals from receptors on the cell surface to DNA in the nucleus, thereby promoting the proliferation and differentiation of cells [90, 91]. It was demonstrated that high expression of YTHDF2 in liver cancer cells could significantly reduce the phosphorylation levels of ERK and mitogen-activated protein kinase kinase (MAPKK/MEK) [36]. Furthermore, epidermal growth factor receptor (EGFR), one of the most important upstream targets of the MAPK/ERK pathway, is reported to be closely linked to cancer progression [36]. YTHDF2 was indicated to significantly suppress the expression of EGFR. Therefore, YTHDF2 could display a negative effect in regulating the stability of EGFR and then affect the MAPK/ERK pathway, which inhibits the growth of cancer (Fig. 4B) [36].
YTHDF2 inhibited Hippo/YAP pathway
The Hippo signaling pathway acts a significant role in mediating cell division, proliferation, differentiation, and apoptosis. YES-associated protein (YAP) is an effector of this pathway, which could initiate the gene transcription and translation of the Hippo pathway [92, 93]. YTHDF2 was found to reduce the levels of the classical Hippo signal transduction factors, including YAP, LATS1 and Mob1 [56]. Besides, YAP was found to be frequently linked to EMT [94]. Knockdown of YTHDF2 could decrease the expression of E-cadherin to increase the expression of vimentin and Snail, which are associated with EMT [95]. Furthermore, YAP was positively correlated with vimentin levels and negatively correlated with E-cadherin in the TCGA database [96]. Taken together, YTHDF2 could modulate the Hippo/YAP signaling pathway to inhibit EMT, thereby suppressing tumor migration and invasion in some cancers (Fig. 4C).
YTHDF2 regulated inflammatory cancer progression
Clinically, the stability of hypoxia-inducible factor (HIF) is linked to poor survival in various cancers [97, 98]. It has been reported that the activity of the YTHDF2 promoter could be reduced in hypoxia and reversed by the application of a HIF-2α inhibitor, which indicated the relationship between YTHDF2 and HIF-2α [50]. Moreover, silencing of YTHDF2 induced by HIF-2α could enhance the phosphorylation of signal transducer and activator of transcription 3 (STAT3) and the expression of interleukin-11 (IL-11) and serpin peptidase inhibitor clade E member 2 (Serpin E2) [50]. IL-11 could promote STAT3 activation and inflammatory cancer progression in an autocrine manner, and Serpin E2 could promote the progression of invasion and metastasis by reprogramming the tumor vascular system [99]. The results revealed that HIF-2α could induce hypoxia to reduce the expression of YTHDF2. In addition, YTHDF2 could inhibit the phosphorylation of STAT3 and the expression of Serpin E2, thereby reducing tumor growth and angiogenesis and resisting the occurrence of inflammatory cancer progression (Fig. 4D).
Other m6A-dependent mechanisms of YTHDF2
YTHDF2 could enhance the degradation of oncogenes or tumor suppressor genes in an m6A-dependent manner, thereby influencing the development of tumors [23]. Chen et al. noted that YTHDF2 could bind to plasmacytoma variant translocation 1 (PVT1), a well-known oncogenic long noncoding RNA (lncRNA) in osteosarcoma. Furthermore, knockdown of YTHDF2 in cancer cells was shown to attenuate the upregulation of PVT1 and reversed the half-life of PVT1, indicating that YTHDF2 was vital to the stability of PVT1 and affected the progression of tumors [41]. It was also reported that high expression of YTHDF2 increased the decay of programmed death 1 (PD-1) mRNA, which played a significant role in melanoma. In addition, YTHDF2 could inhibit the progression of melanoma by suppressing the autophagy/NF-κB/FTO axis (Fig. 4E) [31]. In contrast, Chen et al. noted that in HCC, the 3'-end of SOCS2 transcript could directly bind to YTHDF2 [54]. Downregulation of YTHDF2 increased the expression of SOCS2 and negatively regulated the JAK/STAT signaling pathway, which suppressed the phosphorylation of STAT5 and inhibited the growth of cancer cells [54, 100]. Interestingly, luciferase assays and polysome profiling found that YTHDF2 retained the m6A methylation of the 5'-UTR of OCT4 mRNA, resulting in enhanced protein expression, thereby promoting liver cancer progression. Thus, YTHDF2 may function by regulating the expression of target genes to influence tumor development (Fig. 3F).
m6A-independent manners of YTHDF2 in cancers
YTHDF2 could modulate the degradation of some mRNAs containing m6A [34]. As reported by ** et al., YTHDF1 and YTHDF2 competitively interact with YTHDF3 to regulate the expression of YAP in lung cancer in a manner independent of m6A [59]. Moreover, YTHDF2 could also accelerate the degradation of YAP mRNA through the Argonaute 2 (AGO2) system, inhibiting the growth and metastasis of tumor cells to diminish disease progression (Fig. 4F) [59].
Conclusions
In this review, we summarized the expression of YTHDF2 in human malignancies and generalized its relevant biological functions. More importantly, the underlying molecular mechanism and the clinical prognostic and therapeutic value of YTHDF2 in several cancers were also discussed. YTHDF2 was found to be highly expressed in multiple tumor tissues and cells, thereby acting as a carcinogenic factor [38, 54]. However, contrary conclusions were reported in melanoma, osteosarcoma, and CRC, where YTHDF2 acted as a tumor suppressor [31, 41, 47]. YTHDF2 was also found to be both upregulated and downregulated in lung cancer, GC, and liver cancer, which indicated that YTHDF2 might play a dual role as both an oncogene and tumor suppressor [39, 40, 45, 50, 54, 59]. The outcomes of the diversity may be linked to the extracellular microenvironment, heterogeneity of tumor tissues, and the related upstream or downstream regulators [101,102,103,104]. Additionally, the possible explanation may also be the interaction and the functioning site between YTHDF2 and the target genes. YTHDF2 could accelerate tumor growth by combining with tumor suppressors to trigger a downstream cascade, while it could exert the opposite effect by interacting with oncogenes [41, 65]. Therefore, further investigations are still needed to clarify the discrepancies to obtain better identifications of the effects and the underlying mechanisms of YTHDF2 in different cancers.
YTHDF2 can be involved in the progression of multiple cancers in an m6A-dependent manner, which is associated with various molecules and pathways [64]. MiR-145 and miR-495 could directly target YTHDF2 to affect the development of malignant tumors [55, 67]. Dysregulation of YTHDF2 in cancer could also regulate EMT, glucose metabolism, and apoptosis, which have been considered as significant factors in the progression of cancer [38, 40, 59]. Additionally, YTHDF2 enhances the degradation of oncogenes or tumor suppressors, such as the MAPK/ERK and PI3K/AKT signaling pathways, in an m6A-dependent manner. YTHDF2 was also found to take effects in an m6A-independent manner by promoting the degradation of YAP mRNA by the AGO2 system [59]. These reports suggest that YTHDF2 has tremendous potential in clinical application as a new target of diagnosis, treatment, and prognosis in tumor patients. Still, the reverse effect of YTHDF2 in distinct cancers or even in identical cancer might be related to genes with contrary functions or distinct binding sites, and the specific mechanism remains to be further elucidated.
In recent years, the role of m6A methylation in the prophylaxis and treatment of malignant tumors has received growing attention [105]. m6A methylation and its regulatory proteins were found to have the potential to be prognostic markers and therapeutic targets [106]. Several studies have shown that m6A methylation inhibitors, such as an inhibitor of FTO, can provide beneficial effects on the treatment of cancer [107]. As a primary “reader” protein of m6A, YTHDF2 has been shown to play a crucial role in m6A methylation modification [108, 109]. Given the above investigations, we summarized the significant effects of YTHDF2 on the modification of m6A and cancer progression. It can be conjectured that the development of effective inhibitors of YTHDF2 may provide novel strategies for the treatment of a variety of cancers in the future. However, the development and therapeutic effects of YTHDF2-related products still need to be further explored in the direction of cancer treatment.
Availability of data and materials
Not applicable.
Abbreviations
- 6PGD:
-
6-Phosphogluconate dehydrogenase
- AGO2:
-
Argonaute 2
- ALKBH5:
-
AlkB homolog 5
- AML:
-
Acute myelocytic leukemia
- BMF:
-
Bcl2 modifying factor
- BNIP3:
-
BCL2 interacting protein 3
- CC:
-
Cervical cancer
- ccRCC:
-
Clear cell renal cell carcinoma
- CRC:
-
Colorectal cancer
- CSC:
-
Cancer stem cells
- CSF-1:
-
Colony stimulating factor 1
- CXCR4:
-
C-X-C motif chemokine receptor 4
- EGFR:
-
Epidermal growth factor receptor
- eIF3:
-
Eukaryotic initiation factor 3
- EMT:
-
Epithelial–mesenchymal transition
- ERK:
-
Extracellular regulated kinase
- FOXC2:
-
Forkhead box protein C2
- FTO:
-
Fat mass and obesity associated-protein
- G6PD:
-
Glucose-6-phosphate dehydrogenase
- GAS5:
-
Growth arrest specific 5
- GBM:
-
Glioblastoma
- GC:
-
Gastric cancer
- GEO:
-
Gene Expression Omnibus
- GSEA:
-
Gene Set Enrichment Analysis
- GSK3β:
-
Glycogen synthase kinase 3 beta
- HCC:
-
Hepatocellular carcinoma
- HIF:
-
Hypoxia-inducible factor
- HIVEP2:
-
HIVEP zinc finger 2
- HNRNPs:
-
Heterogeneous nuclear ribonucleoprotein protein families
- HNSCC:
-
Head and neck squamous cell carcinoma
- IGF2BPs:
-
Insulin-like growth factor 2 mRNA binding protein families
- IL-11:
-
Interleukin-11
- KIAA1429:
-
VIRMA, Vir like m6A methyltransferase associated
- KLF4:
-
Kruppel like factor 4
- LHPP:
-
Phospholysine phosphohistidine inorganic pyrophosphate phosphatase
- lncRNAs:
-
Long noncoding RNAs
- LXRA:
-
Liver X receptors A
- m6A:
-
N6-methyladenosine
- MAPK:
-
Mitogen-activated protein kinase
- MAPKK/MEK:
-
Mitogen-activated protein kinase kinase
- METTL:
-
Methyltransferase like
- miRNAs:
-
MicroRNAs
- MOB3B:
-
MOB kinase activator 3B
- mRNAs:
-
Messenger RNAs
- MTC:
-
Methyltransferase complex
- mTOR:
-
Mammalian target of rapamycin
- NKX3–1:
-
NK3 homeobox 1
- NSCLC:
-
Non-small-cell lung cancer
- OCT4 (POU5F1):
-
POU class 5 homeobox 1
- OS:
-
Overall survival
- PC:
-
Pancreatic cancer
- PCa:
-
Prostate cancer
- PD-1:
-
Programmed death 1
- PER1:
-
Period circadian regulator 1
- PI3K:
-
Phosphoinositide-3-kinase
- PIK3CB:
-
Phosphoinositide-3-kinase catalytic beta
- PPP:
-
Pentose phosphate pathway
- PRSS23:
-
Serine protease 23
- PTCL-NOS:
-
Peripheral T-cell lymphoma, not otherwise specified
- PTEN:
-
Phosphate and tension homology deleted on chromosome ten
- PVT1:
-
Plasmacytoma variant translocation 1
- RBM15/15b:
-
RNA binding motifprotein 15/15b
- RFS:
-
Relapse-free survival
- Serpin E2:
-
Serpin peptidase inhibitor clade E member 2
- SETD7:
-
SET domain containing 7
- SOCS2:
-
Suppressor of cytokine signaling 2
- SOX10:
-
SRY-Box transcription factor 10
- STAT:
-
Signal transducers and activators of transcription
- STAT3:
-
Signal transducer and activator of transcription 3
- TCGA:
-
The Cancer Genome Atlas
- TNBC:
-
Triple-negative breast cancer
- TNF:
-
Tumor necrosis factor
- TNF-R2:
-
TNF receptor 2
- TNFRSF1B:
-
TNF receptor superfamily member 1b
- TP53:
-
Tumor protein P53
- UBXN1:
-
UBX domain protein 1
- VEGFA:
-
Vascular endothelial growth factor A
- WTAP:
-
Wilms tumor 1-associated protein
- XIST:
-
X inactivate-specific transcript
- YAP:
-
YES-associated protein
- YTHDF:
-
YT521-B homology domain family
- ZC3H13:
-
Zinc finger CCCH-type containing 13
References
Merkurjev D, Hong WT, Iida K, Oomoto I, Goldie BJ, Yamaguti H, Ohara T, Kawaguchi SY, Hirano T, Martin KC, Pellegrini M, Wang DO. Synaptic N(6)-methyladenosine (m(6)A) epitranscriptome reveals functional partitioning of localized transcripts. Nat Neurosci. 2018;21(7):1004–14.
Chen T, Hao YJ, Zhang Y, Li MM, Wang M, Han W, Wu Y, Lv Y, Hao J, Wang L, Li A, Yang Y, ** KX, Zhao X, Li Y, ** XL, Lai WY, Wu LG, Jiang G, Wang HL, Sang L, Wang XJ, Yang YG, Zhou Q. m(6)A RNA methylation is regulated by microRNAs and promotes reprogramming to pluripotency. Cell Stem Cell. 2015;16(3):289–301.
Mauer J, Luo X, Blanjoie A, Jiao X, Grozhik AV, Patil DP, Linder B, Pickering BF, Vasseur JJ, Chen Q, Gross SS, Elemento O, Debart F, Kiledjian M, Jaffrey SR. Reversible methylation of m(6)A(m) in the 5’ cap controls mRNA stability. Nature. 2017;541(7637):371–5.
Meyer KD, Jaffrey SR. Rethinking m(6)A Readers, Writers, and Erasers. Annu Rev Cell Dev Biol. 2017;33:319–42.
Liu J, Yue Y, Han D, Wang X, Fu Y, Zhang L, Jia G, Yu M, Lu Z, Deng X, Dai Q, Chen W, He C. A METTL3-METTL14 complex mediates mammalian nuclear RNA N6-adenosine methylation. Nat Chem Biol. 2014;10(2):93–5.
Chen XY, Zhang J, Zhu JS. The role of m(6)A RNA methylation in human cancer. Mol Cancer. 2019;18(1):103.
Deng X, Su R, Weng H, Huang H, Li Z, Chen J. RNA N(6)-methyladenosine modification in cancers: current status and perspectives. Cell Res. 2018;28(5):507–17.
Wen J, Lv R, Ma H, Shen H, He C, Wang J, Jiao F, Liu H, Yang P, Tan L, Lan F, Shi YG, He C, Shi Y, Diao J. Zc3h13 Regulates Nuclear RNA m(6)A Methylation and Mouse Embryonic Stem Cell Self-Renewal. Mol Cell. 2018;69(6):1028-1038.e1026.
Lan Q, Liu PY, Haase J, Bell JL, Hüttelmaier S, Liu T. The Critical Role of RNA m(6)A Methylation in Cancer. Cancer Res. 2019;79(7):1285–92.
Cai Y, Feng R, Lu T, Chen X, Zhou X, Wang X. Novel insights into the m(6)A-RNA methyltransferase METTL3 in cancer. Biomark Res. 2021;9(1):27.
Huang Y, Yan J, Li Q, Li J, Gong S, Zhou H, Gan J, Jiang H, Jia GF, Luo C, Yang CG. Meclofenamic acid selectively inhibits FTO demethylation of m6A over ALKBH5. Nucleic Acids Res. 2015;43(1):373–84.
Huang J, Shao Y, Gu W. Function and clinical significance of N6-methyladenosine in digestive system tumours. Exp Hematol Oncol. 2021;10(1):40.
Choe J, Lin S, Zhang W, Liu Q, Wang L, Ramirez-Moya J, Du P, Kim W, Tang S, Sliz P, Santisteban P, George RE, Richards WG, Wong KK, Locker N, Slack FJ, Gregory RI. mRNA circularization by METTL3-eIF3h enhances translation and promotes oncogenesis. Nature. 2018;561(7724):556–60.
Huang H, Weng H, Sun W, Qin X, Shi H, Wu H, Zhao BS, Mesquita A, Liu C, Yuan CL, Hu YC, Hüttelmaier S, Skibbe JR, Su R, Deng X, Dong L, Sun M, Li C, Nachtergaele S, Wang Y, Hu C, Ferchen K, Greis KD, Jiang X, Wei M, Qu L, Guan JL, He C, Yang J, Chen J. Recognition of RNA N(6)-methyladenosine by IGF2BP proteins enhances mRNA stability and translation. Nat Cell Biol. 2018;20(3):285–95.
Zhou KI, Shi H, Lyu R, Wylder AC, Matuszek Ż, Pan JN, He C, Parisien M, Pan T. Regulation of Co-transcriptional pre-mrna splicing by m(6)a through the low-complexity protein hnRNPG. Mol Cell. 2019;76(1):70-81.e79.
Wang X, Lu Z, Gomez A, Hon GC, Yue Y, Han D, Fu Y, Parisien M, Dai Q, Jia G, Ren B, Pan T, He C. N6-methyladenosine-dependent regulation of messenger RNA stability. Nature. 2014;505(7481):117–20.
Liu N, Zhou KI, Parisien M, Dai Q, Diatchenko L, Pan T. N6-methyladenosine alters RNA structure to regulate binding of a low-complexity protein. Nucleic Acids Res. 2017;45(10):6051–63.
Liao S, Sun H, Xu C. YTH Domain: A Family of N(6)-methyladenosine (m(6)A) Readers. Genomics Proteomics Bioinform. 2018;16(2):99–107.
Wang T, Kong S, Tao M, Ju S. The potential role of RNA N6-methyladenosine in Cancer progression. Mol Cancer. 2020;19(1):88.
Ries RJ, Zaccara S, Klein P, Olarerin-George A, Namkoong S, Pickering BF, Patil DP, Kwak H, Lee JH, Jaffrey SR. m(6)A enhances the phase separation potential of mRNA. Nature. 2019;571(7765):424–8.
Zhao T, Sun D, Zhao M, Lai Y, Liu Y, Zhang Z. N(6)-methyladenosine mediates arsenite-induced human keratinocyte transformation by suppressing p53 activation. Environ Pollut. 2020;259:113908.
Hsu PJ, Zhu Y, Ma H, Guo Y, Shi X, Liu Y, Qi M, Lu Z, Shi H, Wang J, Cheng Y, Luo G, Dai Q, Liu M, Guo X, Sha J, Shen B, He C. Ythdc2 is an N(6)-methyladenosine binding protein that regulates mammalian spermatogenesis. Cell Res. 2017;27(9):1115–27.
Li M, Zhao X, Wang W, Shi H, Pan Q, Lu Z, Perez SP, Suganthan R, He C, Bjørås M, Klungland A. Ythdf2-mediated m(6)A mRNA clearance modulates neural development in mice. Genome Biol. 2018;19(1):69.
Meyer KD, Patil DP, Zhou J, Zinoviev A, Skabkin MA, Elemento O, Pestova TV, Qian SB, Jaffrey SR. 5’ UTR m(6)A promotes cap-independent translation. Cell. 2015;163(4):999–1010.
Liu J, Eckert MA, Harada BT, Liu SM, Lu Z, Yu K, Tienda SM, Chryplewicz A, Zhu AC, Yang Y, Huang JT, Chen SM, Xu ZG, Leng XH, Yu XC, Cao J, Zhang Z, Liu J, Lengyel E, He C. m(6)A mRNA methylation regulates AKT activity to promote the proliferation and tumorigenicity of endometrial cancer. Nat Cell Biol. 2018;20(9):1074–83.
Zaccara S, Ries RJ, Jaffrey SR. Reading, writing and erasing mRNA methylation. Nat Rev Mol Cell Biol. 2019;20(10):608–24.
Li T, Hu PS, Zuo Z, Lin JF, Li X, Wu QN, Chen ZH, Zeng ZL, Wang F, Zheng J, Chen D, Li B, Kang TB, **e D, Lin D, Ju HQ, Xu RH. METTL3 facilitates tumor progression via an m(6)A-IGF2BP2-dependent mechanism in colorectal carcinoma. Mol Cancer. 2019;18(1):112.
Zhou X, Zhan L, Huang K, Wang X. The functions and clinical significance of circRNAs in hematological malignancies. J Hematol Oncol. 2020;13(1):138.
Du H, Zhao Y, He J, Zhang Y, ** H, Liu M, Ma J, Wu L. YTHDF2 destabilizes m(6)A-containing RNA through direct recruitment of the CCR4-NOT deadenylase complex. Nat Commun. 2016;7:12626.
Weng H, Huang H, Wu H, Qin X, Zhao BS, Dong L, Shi H, Skibbe J, Shen C, Hu C, Sheng Y, Wang Y, Wunderlich M, Zhang B, Dore LC, Su R, Deng X, Ferchen K, Li C, Sun M, Lu Z, Jiang X, Marcucci G, Mulloy JC, Yang J, Qian Z, Wei M, He C, Chen J. METTL14 Inhibits Hematopoietic Stem/Progenitor Differentiation and Promotes Leukemogenesis via mRNA m(6)A Modification. Cell Stem Cell. 2018;22(2):191-205.e199.
Yang S, Wei J, Cui YH, Park G, Shah P, Deng Y, Aplin AE, Lu Z, Hwang S, He C, He YY. m(6)A mRNA demethylase FTO regulates melanoma tumorigenicity and response to anti-PD-1 blockade. Nat Commun. 2019;10(1):2782.
Musselman CA, Lalonde ME, Côté J, Kutateladze TG. Perceiving the epigenetic landscape through histone readers. Nat Struct Mol Biol. 2012;19(12):1218–27.
Wojtas MN, Pandey RR, Mendel M, Homolka D, Sachidanandam R, Pillai RS. Regulation of m(6)A transcripts by the 3’→5’ RNA helicase YTHDC2 is essential for a successful meiotic program in the mammalian germline. Mol Cell. 2017;68(2):374-387.e312.
Li Z, Qian P, Shao W, Shi H, He XC, Gogol M, Yu Z, Wang Y, Qi M, Zhu Y, Perry JM, Zhang K, Tao F, Zhou K, Hu D, Han Y, Zhao C, Alexander R, Xu H, Chen S, Peak A, Hall K, Peterson M, Perera A, Haug JS, Parmely T, Li H, Shen B, Zeitlinger J, He C, et al. Suppression of m(6)A reader Ythdf2 promotes hematopoietic stem cell expansion. Cell Res. 2018;28(9):904–17.
Zhou J, Wan J, Gao X, Zhang X, Jaffrey SR, Qian SB. Dynamic m(6)A mRNA methylation directs translational control of heat shock response. Nature. 2015;526(7574):591–4.
Zhong L, Liao D, Zhang M, Zeng C, Li X, Zhang R, Ma H, Kang T. YTHDF2 suppresses cell proliferation and growth via destabilizing the EGFR mRNA in hepatocellular carcinoma. Cancer Lett. 2019;442:252–61.
Chen YG, Chen R, Ahmad S, Verma R, Kasturi SP, Amaya L, Broughton JP, Kim J, Cadena C, Pulendran B, Hur S, Chang HY. N6-methyladenosine modification controls circular RNA immunity. Mol Cell. 2019;76(1):96-109.e109.
Paris J, Morgan M, Campos J, Spencer GJ, Shmakova A, Ivanova I, Mapperley C, Lawson H, Wotherspoon DA, Sepulveda C, Vukovic M, Allen L, Sarapuu A, Tavosanis A, Guitart AV, Villacreces A, Much C, Choe J, Azar A, van de Lagemaat LN, Vernimmen D, Nehme A, Mazurier F, Somervaille TCP, Gregory RI, O’Carroll D, Kranc KR. Targeting the RNA m(6)A Reader YTHDF2 selectively compromises cancer stem cells in acute myeloid leukemia. Cell Stem Cell. 2019;25(1):137–48.
Yan J, Huang X, Zhang X, Chen Z, Ye C, **ang W, Huang Z. LncRNA LINC00470 promotes the degradation of PTEN mRNA to facilitate malignant behavior in gastric cancer cells. Biochem Biophys Res Commun. 2020;521(4):887–93.
Sheng H, Li Z, Su S, Sun W, Zhang X, Li L, Li J, Liu S, Lu B, Zhang S, Shan C: YTH domain family 2 promotes lung cancer cell growth by facilitating 6-phosphogluconate dehydrogenase mRNA translation. Carcinogenesis 2019.
Chen S, Zhou L, Wang Y. ALKBH5-mediated m(6)A demethylation of lncRNA PVT1 plays an oncogenic role in osteosarcoma. Cancer Cell Int. 2020;20:34.
Li F, Zhao D, Wu J, Shi Y. Structure of the YTH domain of human YTHDF2 in complex with an m(6)A mononucleotide reveals an aromatic cage for m(6)A recognition. Cell Res. 2014;24(12):1490–2.
Zhu T, Roundtree IA, Wang P, Wang X, Wang L, Sun C, Tian Y, Li J, He C, Xu Y. Crystal structure of the YTH domain of YTHDF2 reveals mechanism for recognition of N6-methyladenosine. Cell Res. 2014;24(12):1493–6.
Wang JY, Lu AQ. The biological function of m6A reader YTHDF2 and its role in human disease. Cancer Cell Int. 2021;21(1):109.
Zhang C, Zhang M, Ge S, Huang W, Lin X, Gao J, Gong J, Shen L. Reduced m6A modification predicts malignant phenotypes and augmented Wnt/PI3K-Akt signaling in gastric cancer. Cancer Med. 2019;8(10):4766–81.
Shen X, Zhao K, Xu L, Cheng G, Zhu J, Gan L, Wu Y, Zhuang Z. YTHDF2 Inhibits Gastric Cancer Cell Growth by Regulating FOXC2 Signaling Pathway. Front Genet. 2020;11:592042.
Yang X, Zhang S, He C, Xue P, Zhang L, He Z, Zang L, Feng B, Sun J, Zheng M. METTL14 suppresses proliferation and metastasis of colorectal cancer by down-regulating oncogenic long non-coding RNA XIST. Mol Cancer. 2020;19(1):46.
Zhuang J, Lin C, Ye J: m(6) A RNA methylation regulators contribute to malignant progression in rectal cancer. J Cell Physiol 2020.
Li H, Zhang N, Jiao X, Wang C, Sun W, He Y, Ren G, Huang S, Li M, Chang Y, ** Z, **e Q, Zhang X, Huang H, ** H. Downregulation of microRNA-6125 promotes colorectal cancer growth through YTHDF2-dependent recognition of N6-methyladenosine-modified GSK3beta. Clin Transl Med. 2021;11(10):e602.
Hou J, Zhang H, Liu J, Zhao Z, Wang J, Lu Z, Hu B, Zhou J, Zhao Z, Feng M, Zhang H, Shen B, Huang X, Sun B, He C, **a Q. YTHDF2 reduction fuels inflammation and vascular abnormalization in hepatocellular carcinoma. Mol Cancer. 2019;18(1):163.
Cui X, Wang Z, Li J, Zhu J, Ren Z, Zhang D, Zhao W, Fan Y, Zhang D, Sun R. Cross talk between RNA N6-methyladenosine methyltransferase-like 3 and miR-186 regulates hepatoblastoma progression through Wnt/β-catenin signalling pathway. Cell Prolif. 2020;53(3):e12768.
Qu N, Qin S, Zhang X, Bo X, Liu Z, Tan C, Wen G, Jiang H. Multiple m(6)A RNA methylation modulators promote the malignant progression of hepatocellular carcinoma and affect its clinical prognosis. BMC Cancer. 2020;20(1):165.
Zhang C, Huang S, Zhuang H, Ruan S, Zhou Z, Huang K, Ji F, Ma Z, Hou B, He X: YTHDF2 promotes the liver cancer stem cell phenotype and cancer metastasis by regulating OCT4 expression via m6A RNA methylation. Oncogene 2020.
Chen M, Wei L, Law CT, Tsang FH, Shen J, Cheng CL, Tsang LH, Ho DW, Chiu DK, Lee JM, Wong CC, Ng IO, Wong CM. RNA N6-methyladenosine methyltransferase-like 3 promotes liver cancer progression through YTHDF2-dependent posttranscriptional silencing of SOCS2. Hepatology. 2018;67(6):2254–70.
Yang Z, Li J, Feng G, Gao S, Wang Y, Zhang S, Liu Y, Ye L, Li Y, Zhang X. MicroRNA-145 Modulates N(6)-Methyladenosine Levels by Targeting the 3’-Untranslated mRNA Region of the N(6)-Methyladenosine Binding YTH Domain Family 2 Protein. J Biol Chem. 2017;292(9):3614–23.
Chen J, Sun Y, Xu X, Wang D, He J, Zhou H, Lu Y, Zeng J, Du F, Gong A, Xu M. YTH domain family 2 orchestrates epithelial-mesenchymal transition/proliferation dichotomy in pancreatic cancer cells. Cell Cycle. 2017;16(23):2259–71.
Tian J, Zhu Y, Rao M, Cai Y, Lu Z, Zou D, Peng X, Ying P, Zhang M, Niu S, Li Y, Zhong R, Chang J, Miao X. N(6)-methyladenosine mRNA methylation of PIK3CB regulates AKT signalling to promote PTEN-deficient pancreatic cancer progression. Gut. 2020;69(12):2180–92.
Guo X, Li K, Jiang W, Hu Y, **ao W, Huang Y, Feng Y, Pan Q, Wan R. RNA demethylase ALKBH5 prevents pancreatic cancer progression by posttranscriptional activation of PER1 in an m6A-YTHDF2-dependent manner. Mol Cancer. 2020;19(1):91.
** D, Guo J, Wu Y, Yang L, Wang X, Du J, Dai J, Chen W, Gong K, Miao S, Li X, Sun H. m(6)A demethylase ALKBH5 inhibits tumor growth and metastasis by reducing YTHDFs-mediated YAP expression and inhibiting miR-107/LATS2-mediated YAP activity in NSCLC. Mol Cancer. 2020;19(1):40.
Zhang Y, Liu X, Liu L, Li J, Hu Q, Sun R. Expression and prognostic significance of m6A-related genes in lung adenocarcinoma. Med Sci Monit. 2020;26:e919644.
Tsuchiya K, Yoshimura K, Inoue Y, Iwashita Y, Yamada H, Kawase A, Watanabe T, Tanahashi M, Ogawa H, Funai K, Shinmura K, Suda T, Sugimura H. YTHDF1 and YTHDF2 are associated with better patient survival and an inflamed tumor-immune microenvironment in non-small-cell lung cancer. Oncoimmunology. 2021;10(1):1962656.
Li Y, Sheng H, Ma F, Wu Q, Huang J, Chen Q, Sheng L, Zhu X, Zhu X, Xu M. RNA m(6)A reader YTHDF2 facilitates lung adenocarcinoma cell proliferation and metastasis by targeting the AXIN1/Wnt/beta-catenin signaling. Cell Death Dis. 2021;12(5):479.
Chen Z, Shao YL, Wang LL, Lin J, Zhang JB, Ding Y, Gao BB, Liu DH, Gao XN. YTHDF2 is a potential target of AML1/ETO-HIF1alpha loop-mediated cell proliferation in t(8;21) AML. Oncogene. 2021;40(22):3786–98.
Watatani Y, Sato Y, Miyoshi H, Sakamoto K, Nishida K, Gion Y, Nagata Y, Shiraishi Y, Chiba K, Tanaka H, Zhao L, Ochi Y, Takeuchi Y, Takeda J, Ueno H, Kogure Y, Shiozawa Y, Kakiuchi N, Yoshizato T, Nakagawa MM, Nanya Y, Yoshida K, Makishima H, Sanada M, Sakata-Yanagimoto M, Chiba S, Matsuoka R, Noguchi M, Hiramoto N, Ishikawa T, et al. Molecular heterogeneity in peripheral T-cell lymphoma, not otherwise specified revealed by comprehensive genetic profiling. Leukemia. 2019;33(12):2867–83.
**e H, Li J, Ying Y, Yan H, ** K, Ma X, He L, Xu X, Liu B, Wang X, Zheng X, **e L. METTL3/YTHDF2 m(6) A axis promotes tumorigenesis by degrading SETD7 and KLF4 mRNAs in bladder cancer. J Cell Mol Med. 2020;24(7):4092–104.
Li J, **e H, Ying Y, Chen H, Yan H, He L, Xu M, Xu X, Liang Z, Liu B, Wang X, Zheng X, **e L. YTHDF2 mediates the mRNA degradation of the tumor suppressors to induce AKT phosphorylation in N6-methyladenosine-dependent way in prostate cancer. Mol Cancer. 2020;19(1):152.
Du C, Lv C, Feng Y, Yu S. Activation of the KDM5A/miRNA-495/YTHDF2/m6A-MOB3B axis facilitates prostate cancer progression. J Exp Clin Cancer Res. 2020;39(1):223.
Chen J, Yu K, Zhong G, Shen W. Identification of a m(6)A RNA methylation regulators-based signature for predicting the prognosis of clear cell renal carcinoma. Cancer Cell Int. 2020;20:157.
Niu Y, Lin Z, Wan A, Chen H, Liang H, Sun L, Wang Y, Li X, **ong XF, Wei B, Wu X, Wan G. RNA N6-methyladenosine demethylase FTO promotes breast tumor progression through inhibiting BNIP3. Mol Cancer. 2019;18(1):46.
Woo HH, Chambers SK. Human ALKBH3-induced m(1)A demethylation increases the CSF-1 mRNA stability in breast and ovarian cancer cells. Biochim Biophys Acta Gene Regul Mech. 2019;1862(1):35–46.
Einstein JM, Perelis M, Chaim IA, Meena JK, Nussbacher JK, Tankka AT, Yee BA, Li H, Madrigal AA, Neill NJ, Shankar A, Tyagi S, Westbrook TF, Yeo GW. Inhibition of YTHDF2 triggers proteotoxic cell death in MYC-driven breast cancer. Mol Cell. 2021;81(15):3048–64.
Xu F, Li J, Ni M, Cheng J, Zhao H, Wang S, Zhou X, Wu X. FBW7 suppresses ovarian cancer development by targeting the N(6)-methyladenosine binding protein YTHDF2. Mol Cancer. 2021;20(1):45.
Wang X, Zhang J, Wang Y. Long noncoding RNA GAS5-AS1 suppresses growth and metastasis of cervical cancer by increasing GAS5 stability. Am J Transl Res. 2019;11(8):4909–21.
Zhou X, Han J, Zhen X, Liu Y, Cui Z, Yue Z, Ding L, Xu S. Analysis of genetic alteration signatures and prognostic values of m6A regulatory genes in head and neck squamous cell carcinoma. Front Oncol. 2020;10:718.
Yu J, Chai P, **e M, Ge S, Ruan J, Fan X, Jia R. Histone lactylation drives oncogenesis by facilitating m(6)A reader protein YTHDF2 expression in ocular melanoma. Genome Biol. 2021;22(1):85.
Dixit D, Prager BC, Gimple RC, Poh HX, Wang Y, Wu Q, Qiu Z, Kidwell RL, Kim LJY, **e Q, Vitting-Seerup K, Bhargava S, Dong Z, Jiang L, Zhu Z, Hamerlik P, Jaffrey SR, Zhao JC, Wang X, Rich JN. The RNA m6A reader YTHDF2 maintains oncogene expression and is a targetable dependency in glioblastoma stem cells. Cancer Discov. 2021;11(2):480–99.
Fang R, Chen X, Zhang S, Shi H, Ye Y, Shi H, Zou Z, Li P, Guo Q, Ma L, He C, Huang S. EGFR/SRC/ERK-stabilized YTHDF2 promotes cholesterol dysregulation and invasive growth of glioblastoma. Nat Commun. 2021;12(1):177.
Chai RC, Chang YZ, Chang X, Pang B, An SY, Zhang KN, Chang YH, Jiang T, Wang YZ. YTHDF2 facilitates UBXN1 mRNA decay by recognizing METTL3-mediated m(6)A modification to activate NF-kappaB and promote the malignant progression of glioma. J Hematol Oncol. 2021;14(1):109.
Liu R, Li W, Tao B, Wang X, Yang Z, Zhang Y, Wang C, Liu R, Gao H, Liang J, Yang W. Tyrosine phosphorylation activates 6-phosphogluconate dehydrogenase and promotes tumor growth and radiation resistance. Nat Commun. 2019;10(1):991.
Grabinger T, Bode KJ, Demgenski J, Seitz C, Delgado ME, Kostadinova F, Reinhold C, Etemadi N, Wilhelm S, Schweinlin M, Hänggi K, Knop J, Hauck C, Walles H, Silke J, Wajant H, Nachbur U. Inhibitor of apoptosis protein-1 regulates tumor necrosis factor-mediated destruction of intestinal epithelial cells. Gastroenterology. 2017;152(4):867–79.
Ciruelos Gil EM. Targeting the PI3K/AKT/mTOR pathway in estrogen receptor-positive breast cancer. Cancer Treat Rev. 2014;40(7):862–71.
Luo X, Cao M, Gao F, He X. YTHDF1 promotes hepatocellular carcinoma progression via activating PI3K/AKT/mTOR signaling pathway and inducing epithelial-mesenchymal transition. Exp Hematol Oncol. 2021;10(1):35.
Zhao C, Tao T, Yang L, Qin Q, Wang Y, Liu H, Song R, Yang X, Wang Q, Gu S, **ong Y, Zhao D, Wang S, Feng D, Jiang WG, Zhang J, He J. Loss of PDZK1 expression activates PI3K/AKT signaling via PTEN phosphorylation in gastric cancer. Cancer Lett. 2019;453:107–21.
Hines MJ, Coffre M, Mudianto T, Panduro M, Wigton EJ, Tegla C, Osorio-Vasquez V, Kageyama R, Benhamou D, Perez O, Bajwa S, McManus MT, Ansel KM, Melamed D, Koralov SB. miR-29 sustains B cell survival and controls terminal differentiation via regulation of PI3K signaling. Cell Rep. 2020;33(9):108436.
Zhan T, Rindtorff N, Boutros M. Wnt signaling in cancer. Oncogene. 2017;36(11):1461–73.
Yu W, Zhang X, Zhang W, **ong M, Lin Y, Chang M, Xu L, Lu Y, Liu Y, Zhang J. 19-Hydroxybufalin inhibits non-small cell lung cancer cell proliferation and promotes cell apoptosis via the Wnt/beta-catenin pathway. Exp Hematol Oncol. 2021;10(1):48.
Cancer Genome Atlas N: Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012, 487(7407):330–337.
Hu S, Ren S, Cai Y, Liu J, Han Y, Zhao Y, Yang J, Zhou X, Wang X: Glycoprotein PTGDS promotes tumorigenesis of diffuse large B-cell lymphoma by MYH9-mediated regulation of Wnt-beta-catenin-STAT3 signaling. Cell Death Differ 2021.
Ha M, Kim VN. Regulation of microRNA biogenesis. Nat Rev Mol Cell Biol. 2014;15(8):509–24.
Henslee AB, Steele TA. Combination statin and chemotherapy inhibits proliferation and cytotoxicity of an aggressive natural killer cell leukemia. Biomark Res. 2018;6:26.
Harder A. MEK inhibitors - novel targeted therapies of neurofibromatosis associated benign and malignant lesions. Biomark Res. 2021;9(1):26.
Lin Z, Zhou P, von Gise A, Gu F, Ma Q, Chen J, Guo H, van Gorp PR, Wang DZ, Pu WT. Pi3kcb links Hippo-YAP and PI3K-AKT signaling pathways to promote cardiomyocyte proliferation and survival. Circ Res. 2015;116(1):35–45.
Zhang J, He X, Wan Y, Zhang H, Tang T, Zhang M, Yu S, Zhao W, Chen L. CD44 promotes hepatocellular carcinoma progression via upregulation of YAP. Exp Hematol Oncol. 2021;10(1):54.
Park J, Kim DH, Shah SR, Kim HN. Kshitiz, Kim P, Quiñones-Hinojosa A, Levchenko A: Switch-like enhancement of epithelial-mesenchymal transition by YAP through feedback regulation of WT1 and Rho-family GTPases. Nat Commun. 2019;10(1):2797.
Li W, Zong S, Shi Q, Li H, Xu J, Hou F. Hypoxia-induced vasculogenic mimicry formation in human colorectal cancer cells: Involvement of HIF-1a, Claudin-4, and E-cadherin and Vimentin. Sci Rep. 2016;6:37534.
Zhou X, Chen N, Xu H, Zhou X, Wang J, Fang X, Zhang Y, Li Y, Yang J, Wang X. Regulation of Hippo-YAP signaling by insulin-like growth factor-1 receptor in the tumorigenesis of diffuse large B-cell lymphoma. J Hematol Oncol. 2020;13(1):77.
Chen F, Chen J, Yang L, Liu J, Zhang X, Zhang Y, Tu Q, Yin D, Lin D, Wong PP, Huang D, **ng Y, Zhao J, Li M, Liu Q, Su F, Su S, Song E. Extracellular vesicle-packaged HIF-1α-stabilizing lncRNA from tumour-associated macrophages regulates aerobic glycolysis of breast cancer cells. Nat Cell Biol. 2019;21(4):498–510.
Xu D, Wang Q, Jiang Y, Zhang Y, Vega-Saenzdemiera E, Osman I, Dai W. Roles of Polo-like kinase 3 in suppressing tumor angiogenesis. Exp Hematol Oncol. 2012;1(1):5.
Ollila S, Domènech-Moreno E, Laajanen K, Wong IP, Tripathi S, Pentinmikko N, Gao Y, Yan Y, Niemelä EH, Wang TC, Viollet B, Leone G, Katajisto P, Vaahtomeri K, Mäkelä TP. Stromal Lkb1 deficiency leads to gastrointestinal tumorigenesis involving the IL-11-JAK/STAT3 pathway. J Clin Invest. 2018;128(1):402–14.
de Bock CE, Demeyer S, Degryse S, Verbeke D, Sweron B, Gielen O, Vandepoel R, Vicente C, Vanden Bempt M, Dagklis A, Geerdens E, Bornschein S, Gijsbers R, Soulier J, Meijerink JP, Heinäniemi M, Teppo S, Bouvy-Liivrand M, Lohi O, Radaelli E, Cools J. HOXA9 cooperates with activated JAK/STAT signaling to drive leukemia development. Cancer Discov. 2018;8(5):616–31.
Roper N, Gao S, Maity TK, Banday AR, Zhang X, Venugopalan A, Cultraro CM, Patidar R, Sindiri S, Brown AL, Goncearenco A, Panchenko AR, Biswas R, Thomas A, Rajan A, Carter CA, Kleiner DE, Hewitt SM, Khan J, Prokunina-Olsson L, Guha U. APOBEC mutagenesis and copy-number alterations are drivers of proteogenomic tumor evolution and heterogeneity in metastatic thoracic tumors. Cell Rep. 2019;26(10):2651-2666.e2656.
Chu Y, Zhou X, Wang X. Antibody-drug conjugates for the treatment of lymphoma: clinical advances and latest progress. J Hematol Oncol. 2021;14(1):88.
Liu Y, Zhou X, Wang X. Targeting the tumor microenvironment in B-cell lymphoma: challenges and opportunities. J Hematol Oncol. 2021;14(1):125.
Zhou X, Fang X, Jiang Y, Geng L, Li X, Li Y, Lu K, Li P, Lv X, Wang X. Klotho, an anti-aging gene, acts as a tumor suppressor and inhibitor of IGF-1R signaling in diffuse large B cell lymphoma. J Hematol Oncol. 2017;10(1):37.
Ma S, Chen C, Ji X, Liu J, Zhou Q, Wang G, Yuan W, Kan Q, Sun Z. The interplay between m6A RNA methylation and noncoding RNA in cancer. J Hematol Oncol. 2019;12(1):121.
Lin X, Chai G, Wu Y, Li J, Chen F, Liu J, Luo G, Tauler J, Du J, Lin S, He C, Wang H. RNA m(6)A methylation regulates the epithelial mesenchymal transition of cancer cells and translation of Snail. Nat Commun. 2019;10(1):2065.
Huang Y, Su R, Sheng Y, Dong L, Dong Z, Xu H, Ni T, Zhang ZS, Zhang T, Li C, Han L, Zhu Z, Lian F, Wei J, Deng Q, Wang Y, Wunderlich M, Gao Z, Pan G, Zhong D, Zhou H, Zhang N, Gan J, Jiang H, Mulloy JC, Qian Z, Chen J, Yang CG. Small-Molecule Targeting of Oncogenic FTO Demethylase in Acute Myeloid Leukemia. Cancer Cell. 2019;35(4):677-691.e610.
Ivanova I, Much C, Di Giacomo M, Azzi C, Morgan M, Moreira PN, Monahan J, Carrieri C, Enright AJ, O’Carroll D. The RNA m(6)A reader YTHDF2 is essential for the post-transcriptional regulation of the maternal transcriptome and oocyte competence. Mol Cell. 2017;67(6):1059-1067.e1054.
Ma S, Yan J, Barr T, Zhang J, Chen Z, Wang LS, Sun JC, Chen J, Caligiuri MA, Yu J. The RNA m6A reader YTHDF2 controls NK cell antitumor and antiviral immunity. J Exp Med. 2021;218:8.
Acknowledgements
Not applicable.
Funding
This study was supported by National Natural Science Foundation (No.82170189, No.82070203, No.81800194, No.81770210, No.81473486 and No. 81270598); Key Research and Development Program of Shandong Province (No.2018CXGC1213); Development Project of Youth Innovation Teams in Colleges and Universities of Shandong Province (No.2020KJL006); China Postdoctoral Science Foundation (No.2021T140422, No.2020M672103); Technology Development Projects of Shandong Province (No.2017GSF18189); Translational Research Grant of NCRCH (No.2021WWB02, No.2020ZKMB01); Shandong Provincial Natural Science Foundation (ZR2021YQ51); Technology Development Project of **an City (No.202134034); Taishan Scholars Program of Shandong Province; Shandong Provincial Engineering Research Center of Lymphoma; Academic Promotion Programme of Shandong First Medical University (No.2019QL018, No.2020RC006).
Author information
Authors and Affiliations
Contributions
XZ and XC wrote and edited the manuscript. XC collected the related literature. XZ and XC finished the figures and tables. XW and XZ provided the feedback and guidance. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
