
Abstract
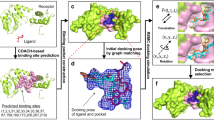
Protein–ligand blind docking is a widely used method for studying the binding sites and poses of ligands and receptors in pharmaceutical and biological research. Recently, our new blind docking server named CB-Dock2 has been released and is currently being utilized by researchers worldwide. CB-Dock2 outperforms state-of-the-art methods due to its accuracy in binding site identification and binding pose prediction, which are enabled by its knowledge-based docking engine. This highly automated server offers interactive and intuitive input and output web interfaces, making it an efficient and user-friendly tool for the bioinformatics and cheminformatics communities. This chapter provides a brief overview of the methods, followed by a detailed guide on using the CB-Dock2 server. Additionally, we present a case study that evaluates the performance of protein–ligand blind docking using this tool.
Access this chapter
Tax calculation will be finalised at checkout
Purchases are for personal use only
Similar content being viewed by others


References
Salentin S, Schreiber S, Haupt VJ et al (2015) PLIP: fully automated protein–ligand interaction profiler. Nucleic Acids Res 43:W443–W447
Jacob L, Vert JP (2008) Protein-ligand interaction prediction: an improved chemogenomics approach. Bioinformatics 24:2149–2156
Hassan NM, Alhossary AA, Mu Y et al (2017) Protein-ligand blind docking using QuickVina-W with inter-process Spatio-temporal integration. Sci Rep 7:15451
Hetényi C, van der Spoel D (2009) Efficient docking of peptides to proteins without prior knowledge of the binding site. Protein Sci 11:1729–1737
Hetényi C, van der Spoel D (2006) Blind docking of drug-sized compounds to proteins with up to a thousand residues. FEBS Lett 580(5):1447–1450
Sánchez-Linares I, Pérez-Sánchez H, Cecilia JM et al (2012) High-throughput parallel blind virtual screening using BINDSURF. BMC Bioinform 13(Suppl 14):S13
Lee HS, Zhang Y (2012) BSP-SLIM: a blind low-resolution ligand-protein docking approach using predicted protein structures. Proteins 80(1):93–110
Tunyasuvunakool K, Adler J, Wu Z et al (2021) Highly accurate protein structure prediction for the human proteome. Nature 596:590–596
Baek M, DiMaio F, Anishchenko I et al (2021) Accurate prediction of protein structures and interactions using a three-track neural network. Science 373:871–876
Rask-Andersen M, Almén MS, Schiöth HB (2011) Trends in the exploitation of novel drug targets. Nat Rev Drug Discov 10:579–590
Singh V, Mizrahi V (2017) Identification and validation of novel drug targets in mycobacterium tuberculosis. Drug Discov Today 22:503–509
Grosdidier A, Zoete V, Michielin O (2011) SwissDock, a protein-small molecule docking web service based on EADock DSS. Nucleic Acids Res 39:W270–W277
Qi W, Peng Z, Yang Zhang JY (2018) COACH-D: improved protein–ligand binding sites prediction with refined ligand-binding poses through molecular docking. Nucleic Acids Res 46:313–338
Zhang W, Bell EW, Yin M et al (2020) EDock: blind protein-ligand docking by replica-exchange Monte Carlo simulation. J Cheminform 12:37
Labbé CM, Rey J, Lagorce D et al (2015) MTiOpenScreen: a web server for structure-based virtual screening. Nucleic Acids Res 43:W448–W454
Liu Y, Yang X, Gan J et al (2022) CB-Dock2: improved protein–ligand blind docking by integrating cavity detection, docking and homologous template fitting. Nucleic Acids Res 50:W159–W164
Liu Y, Grimm M, Dai WT, Hou MC, **ao ZX, Cao Y (2020) CB-Dock: a web server for cavity detection-guided protein–ligand blind docking. Acta Pharmacol Sin 41:138–144
Cao Y, Dai W, Miao Z (2018) Evaluation of protein–ligand docking by Cyscore. Methods Mol Biol 1762:233–243
Cao Y, Li L (2014) Improved protein-ligand binding affinity prediction by using a curvature-dependent surface-area model. Bioinformatics 30:1674–1680
Trott O, Olson A (2010) Autodock vina: improving the speed and accuracy of docking. J Comput Chem 31(2):455–461
Huey R, Morris GM, Forli S (2012) Using AutoDock 4 and AutoDock Vina with AutoDockTools: a tutorial. The Scripps Research Institute Molecular 32
Trott O, Olson AJ (2010) Software news and update AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem 31:455–461
Yang X, Liu Y, Gan J et al (2022) FitDock: protein-ligand docking by template fitting. Brief Bioinform 23(3):bbac087
Hartshorn MJ, Verdonk ML, Chessari G et al (2007) Diverse, high-quality test set for the validation of protein-ligand docking performance. J Med Chem 50(4):726–741
Bienfait B, Ertl P (2013) JSME: a free molecule editor in JavaScript. J Cheminform 5:24
Liu JL, Miao ZC, Li L et al (2016) DRSP: a structural database for single residue substitutions in PDB. Prog Biochem Biophys 43:810–816
Dolinsky TJ, Czodrowski P, Li H et al (2007) PDB2PQR: expanding and upgrading automated preparation of biomolecular structures for molecular simulations. Nucleic Acids Res 35:W522–W525
Cao Y, Song L, Miao Z et al (2011) Improved side-chain modeling by coupling clash-detection guided iterative search with rotamer relaxation. Bioinformatics 27:785–790
Perola E, Herman L, Weiss J (2012) Development of a rule-based method for the assessment of protein Druggability. J Chem Inf Model 52:1027–1038
Liu N, Xu Z (2019) Using LeDock as a docking tool for computational drug design. IOP Conf Ser Earth Environ Sci 218:012143
Burley SK, Bhikadiya C, Bi C et al (2021) RCSB Protein Data Bank: powerful new tools for exploring 3D structures of biological macromolecules for basic and applied research and education in fundamental biology, biomedicine, biotechnology, bioengineering and energy sciences. Nucleic Acids Res 49:D437–D451
O’Boyle NM, Banck M, James CA et al (2011) Open Babel: an open chemical toolbox. J Cheminform 3:33
Krucinska J, Lombardo MN, Erlandsen H et al (2022) Structure-guided functional studies of plasmid-encoded dihydrofolate reductases reveal a common mechanism of trimethoprim resistance in Gram-negative pathogens. Commun Biol 5:459
Klon AE, Héroux A, Ross LJ et al (2002) Atomic structures of human Dihydrofolate reductase complexed with NADPH and two lipophilic Antifolates at 1.09Å and 1.05Å resolution. J Mol Biol 320:677–693
Acknowledgments
This work was supported by the National Natural Science Foundation of China under Grant [81973243].
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2024 The Author(s), under exclusive license to Springer Science+Business Media, LLC, part of Springer Nature
About this protocol

Cite this protocol
Liu, Y., Cao, Y. (2024). Protein–Ligand Blind Docking Using CB-Dock2. In: Gore, M., Jagtap, U.B. (eds) Computational Drug Discovery and Design. Methods in Molecular Biology, vol 2714. Humana, New York, NY. https://doi.org/10.1007/978-1-0716-3441-7_6
Download citation
DOI: https://doi.org/10.1007/978-1-0716-3441-7_6
Published:
Publisher Name: Humana, New York, NY
Print ISBN: 978-1-0716-3440-0
Online ISBN: 978-1-0716-3441-7
eBook Packages: Springer Protocols