
Abstract
Quantum mechanics (QM) methods provide a fine description of receptor-ligand interactions and of chemical reactions. Their use in drug design and drug discovery is increasing, especially for complex systems including metal ions in the binding sites, for the design of highly selective inhibitors, for the optimization of bi-specific compounds, to understand enzymatic reactions, and for the study of covalent ligands and prodrugs. They are also used for generating molecular descriptors for predictive QSAR/QSPR models and for the parameterization of force fields. Thanks to the continuous increase of computational power offered by GPUs and to the development of sophisticated algorithms, QM methods are becoming part of the standard tools used in computer-aided drug design (CADD). We present the most used QM methods and software packages, and we discuss recent representative applications in drug design and drug discovery.
Access this chapter
Tax calculation will be finalised at checkout
Purchases are for personal use only
Similar content being viewed by others
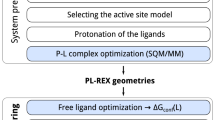

References
Szabo A, Ostlund NS (1996) Modern quantum chemistry. Dover Publishing, Mineola, NY
Cramer CJ (2006) Essentials of computational chemistry: theories and models. Wiley, Chichester, UK
Jensen F (2007) Introduction to computational chemistry. Wiley, Chichester, UK
Pople JA, Beveridge DL (1970) Approximate molecular orbital theory. McGraw-Hill, New York
Dewar MJS, Zoebisch EG, Healy EF, Stewart JJP (1985) AM1: a new general purpose quantum mechanical molecular model. J Am Chem Soc 107:3902–3909. https://doi.org/10.1021/ja00299a024
Stewart JJP (1989) Optimization of parameters for semiempirical methods I. Method. J Comput Chem 10:209–220. https://doi.org/10.1002/jcc.540100208
Dewar MJS, Jie C, Yu J (1993) SAM1; the first of a new series of general purpose quantum mechanical molecular models. Tetrahedron 49:5003–5038. https://doi.org/10.1016/S0040-4020(01)81868-8
Repasky MP, Chandrasekhar J, Jorgensen WL (2002) PDDG/PM3 and PDDG/MNDO: improved semiempirical methods. J Comput Chem 23:1601–1622. https://doi.org/10.1002/jcc.10162
Stewart JJ (2007) Optimization of parameters for semiempirical methods V: modification of NDDO approximations and application to 70 elements. J Mol Model 13:1173–1213. https://doi.org/10.1007/s00894-007-0233-4
Řezáč J, Fanfrlík J, Salahub D, Hobza P (2009) Semiempirical quantum chemical PM6 method augmented by dispersion and H-bonding correction terms reliably describes various types of noncovalent complexes. J Chem Theory Comput 5:1749–1760. https://doi.org/10.1021/ct9000922
Dral PO, Wu X, Spörkel L, Koslowski A, Weber W, Steiger R, Scholten M, Thiel W (2016) Semiempirical quantum-chemical orthogonalization-corrected methods: theory, implementation, and parameters. J Chem Theory Comput 12:1082–1096. https://doi.org/10.1021/acs.jctc.5b01046
Dral PO, Wu X, Thiel W (2019) Semiempirical quantum-chemical methods with orthogonalization and dispersion corrections. J Chem Theory Comput 15:1743–1760. https://doi.org/10.1021/acs.jctc.8b01265
Kříž K, Řezáč J (2019) Reparametrization of the COSMO solvent model for semiempirical methods PM6 and PM7. J Chem Inf Model 59:229–235. https://doi.org/10.1021/acs.jcim.8b00681
Hehre WJ, Ditchfield R, Pople JA (1972) Self-consistent molecular orbital methods. 12. Further extensions of Gaussian-type basis sets for use in molecular-orbital studies of organic-molecules. J Chem Phys 56:2257. https://doi.org/10.1063/1.1677527
Jensen F (2013) Atomic orbital basis sets. WIREs Comput Mol Sci 3:273–295. https://doi.org/10.1002/wcms.1123
Dunning TH (1989) Gaussian basis sets for use in correlated molecular calculations. I. The atoms boron through neon and hydrogen. J Chem Phys 90:1007–1023. https://doi.org/10.1063/1.456153
Kendall RA, Dunning TH, Harrison RJ (1992) Electron affinities of the first-row atoms revisited. Systematic basis sets and wave functions. J Chem Phys 96:6796–6806. https://doi.org/10.1063/1.462569
Weigend F, Ahlrichs R (2005) Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: design and assessment of accuracy. Phys Chem Chem Phys 7:3297–3305. https://doi.org/10.1039/b508541a
Møller C, Plesset MS (1934) Note on an approximation treatment for many-electron systems. Phys Rev 46:618–622. https://doi.org/10.1103/PhysRev.46.618
Shavitt I, Bartlett RJ (2009) Many-body methods in chemistry and physics: MBPT and coupled-cluster theory. Cambridge University Press, Cambridge
Ramabhadran RO, Raghavachari K (2013) Extrapolation to the gold-standard in quantum chemistry: computationally efficient and accurate CCSD(T) energies for large molecules using an automated thermochemical hierarchy. J Chem Theory Comput 9:3986–3994. https://doi.org/10.1021/ct400465q
Hohenberg P, Kohn W (1964) Inhomogeneous electron gas. Phys Rev 136:B864–B871
Kohn W, Sham LJ (1965) Self-consistent equations including exchange and correlation effects. Phys Rev 140:A1133–A1138. https://doi.org/10.1103/PhysRev.140.A1133
Perdew JP (1986) Density-functional approximation for the correlation energy of the inhomogeneous electron gas. Phys Rev B Condens Matter 33:8822–8824. https://doi.org/10.1103/physrevb.33.8822
Lee CT, Yang WT, Parr RG (1988) Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys Rev B Condens Matter Mater Phys 37:785–789. https://doi.org/10.1103/PhysRevB.37.785
Becke AD (1993) Density-functional thermochemistry. III. The role of exact exchange. J Chem Phys 98:5648–5652. https://doi.org/10.1063/1.464913
Perdew JP, Burke K, Ernzerhof M (1996) Generalized gradient approximation made simple. Phys Rev Lett 77:3865–3868. https://doi.org/10.1103/PhysRevLett.77.3865
Zhao Y, Truhlar DG (2008) The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor Chem Accounts 120:215–241. https://doi.org/10.1007/s00214-007-0310-x
Liao C, Sitzmann M, Pugliese A, Nicklaus MC (2011) Software and resources for computational medicinal chemistry. Future Med Chem 3(8):1057–1085. https://doi.org/10.4155/fmc.11.63
Kitchen DB (2017) Computer-aided drug discovery research at a global contract research organization. J Comput Aided Mol Des 31(3):309–318. https://doi.org/10.1007/s10822-016-9991-3
Muegge I, Bergner A, Kriegl JM (2017) Computer-aided drug design at Boehringer Ingelheim. J Comput Aided Mol Des 31(3):275–285. https://doi.org/10.1007/s10822-016-9975-3
Nitsche MA, Ferreria M, Mocskos EE, González Lebrero MC (2014) GPU accelerated implementation of density functional theory for hybrid QM/MM simulations. J Chem Theory Comput 10(3):959–967. https://doi.org/10.1021/ct400308n
Cavasotto CN, Adler NS, Aucar MG (2018) Quantum chemical approaches in structure-based virtual screening and lead optimization. Front Chem 6:188. https://doi.org/10.3389/fchem.2018.00188
Shi M, Xu D, Zeng J (2018) GPU accelerated quantum virtual screening: application for the natural inhibitors of New Delhi metalloprotein (NDM-1). Front Chem 6:564. https://doi.org/10.3389/fchem.2018.00564
Borbulevych O, Martin RI, Westerhoff LM (2018) High-throughput quantum-mechanics/molecular-mechanics (ONIOM) macromolecular crystallographic refinement with PHENIX/DivCon: the impact of mixed Hamiltonian methods on ligand and protein structure. Acta Cryst D74:1063–1077. https://doi.org/10.1107/S2059798318012913
Rode BM, Hofer TS, Randolf BR, Schwenk CF, Xenides D, Vchirawongkwin V (2006) Ab initio quantum mechanical charge field (QMCF) molecular dynamics: a QM/MM – MD procedure for accurate simulations of ions and complexes. Theor Chem Accounts 115:77–85. https://doi.org/10.1007/s00214-005-0049-1
Senn HM, Thiel W (2009) QM/MM methods for biomolecular systems. Angew Chem Int Ed Engl 48(7):1198–1229. https://doi.org/10.1002/anie.200802019
Cornell WD, Cieplak P, Bayly CI, Kollman PA (1993) Application of RESP charges to calculate conformational energies, hydrogen bond energies, and free energies of solvation. J Am Chem Soc 115:9620–9631. https://doi.org/10.1021/ja00074a030
Kotev M, Pascual R, Almansa C, Guallar V, Soliva R (2018) Pushing the limits of computational structure-based drug design with a cryo-EM structure: the Ca2+ channel α2δ-1 subunit as a test case. J Chem Inf Model 58(8):1707–1715. https://doi.org/10.1021/acs.jcim.8b00347
Bekker GJ, Araki M, Oshima K, Okuno Y, Kamiya N (2019) Dynamic docking of a medium-sized molecule to its receptor by multicanonical MD simulations. J Phys Chem B 123(11):2479–2490. https://doi.org/10.1021/acs.jpcb.8b12419
Wang J, Zhao C, Tu J, Yang H, Zhang X, Lv W, Zhai H (2019) Design of novel quinoline-aminopiperidine derivatives as Mycobacterium tuberculosis (MTB) GyrB inhibitors: an in silico study. J Biomol Struct Dyn 37(11):2913–2925. https://doi.org/10.1080/07391102.2018.1498806
Ryde U, Söderhjelm P (2016) Ligand-binding affinity estimates supported by quantum-mechanical methods. Chem Rev 116(9):5520–5566. https://doi.org/10.1021/acs.chemrev.5b00630
Arodola OA, Soliman ME (2017) Quantum mechanics implementation in drug-design workflows: does it really help? Drug Des Devel Ther 11:2551–2564. https://doi.org/10.2147/DDDT.S126344
Ganesan A, Coote ML, Barakat K (2017) Molecular dynamics-driven drug discovery: lea** forward with confidence. Drug Discov Today 22(2):249–269. https://doi.org/10.1016/j.drudis.2016.11.001
Awoonor-Williams E, Walsh AG, Rowley CN (2017) Modeling covalent-modifier drugs. Biochim Biophys Acta Proteins Proteom 1865(11 Pt B):1664–1675. https://doi.org/10.1016/j.bbapap.2017.05.009
Kulik HJ, Zhang J, Klinman JP, Martínez TJ (2016) How large should the QM region be in QM/MM calculations? The case of catechol O-methyltransferase. J Phys Chem B 120(44):11381–11394. https://doi.org/10.1021/acs.jpcb.6b07814
Lodola A, De Vivo M (2012) The increasing role of QM/MM in drug discovery. Adv Protein Chem Struct Biol 87:337–362. https://doi.org/10.1016/B978-0-12-398312-1.00011-1
Barbault F, Maurel F (2015) Simulation with quantum mechanics/molecular mechanics for drug discovery. Expert Opin Drug Discov 10(10):1047–1057. https://doi.org/10.1517/17460441.2015.1076389
Nascimento ÉCM, Oliva M, Świderek K, Martins JBL, Andrés J (2017) Binding analysis of some classical acetylcholinesterase inhibitors: insights for a rational design using free energy perturbation method calculations with QM/MM MD simulations. J Chem Inf Model 57(4):958–976. https://doi.org/10.1021/acs.jcim.7b00037
Ribeiro AJM, Santos-Martins D, Russo N, Ramos MJ, Fernandes PA (2015) Enzymatic flexibility and reaction rate: a QM/MM study of HIV-1 protease. ACS Catal 5:5617–5626. https://doi.org/10.1021/acscatal.5b00759
Chen J, Wang J, Zhang Q, Chen K, Zhu W (2015) A comparative study of trypsin specificity based on QM/MM molecular dynamics simulation and QM/MM GBSA calculation. J Biomol Struct Dyn 33(12):2606–2618. https://doi.org/10.1080/07391102.2014.1003146
Schirmeister T, Kesselring J, Jung S, Schneider TH, Weickert A, Becker J, Lee W, Bamberger D, Wich PR, Distler U, Tenzer S, Johé P, Hellmich UA, Engels B (2016) Quantum chemical-based protocol for the rational design of covalent inhibitors. J Am Chem Soc 138(27):8332–8335. https://doi.org/10.1021/jacs.6b03052
Cavalli A, Carloni P, Recanatini M (2006) Target-related applications of first principles quantum chemical methods in drug design. Chem Rev 106(9):3497–3519. https://doi.org/10.1021/cr050579p
Chung LW, Sameera WM, Ramozzi R, Page AJ, Hatanaka M, Petrova GP, Harris TV, Li X, Ke Z, Liu F, Li HB, Ding L, Morokuma K (2015) The ONIOM method and its applications. Chem Rev 115(12):5678–5796. https://doi.org/10.1021/cr5004419
Shen L, Yang W (2018) Molecular dynamics simulations with quantum mechanics/molecular mechanics and adaptive neural networks. J Chem Theory Comput 14(3):1442–1455. https://doi.org/10.1021/acs.jctc.7b01195
Cao Y, Romero J, Aspuru-Guzik A (2018) Potential of quantum computing for drug discovery. IBM J Res Dev 62(6):1–6. https://doi.org/10.1147/JRD.2018.2888987
Harder E, Damm W, Maple J, Wu C, Reboul M, **ang JY, Wang L, Lupyan D, Dahlgren MK, Knight JL, Kaus JW, Cerutti DS, Krilov G, Jorgensen WL, Abel R, Friesner RA (2016) OPLS3: a force field providing broad coverage of drug-like small molecules and proteins. J Chem Theory Comput 12:281–296. https://doi.org/10.1021/acs.jctc.5b00864
Wang J, Wolf RM, Caldwell JW, Kollman PA, Case DA (2004) Development and testing of a general AMBER force field. J Comput Chem 25:1157–1174. https://doi.org/10.1002/jcc.20035
Vanommeslaeghe K, Hatcher E, Acharya C, Kundu S, Zhong S, Shim J, Darian E, Guvench O, Lopes P, Vorobyov I, Mackerell AD Jr (2010) CHARMM general force field: a force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields. J Comput Chem 31:671–690. https://doi.org/10.1002/jcc.21367
Halgren TA (1999) MMFF VI. MMFF94s option for energy minimization studies. J Comput Chem 20:720–729. https://doi.org/10.1002/(SICI)1096-987X(199905)20:73.0.CO;2-X
Maier JA, Martinez C, Kasavajhala K, Wickstrom L, Hauser KE, Simmerling C (2015) ff14SB: improving the accuracy of protein side chain and backbone parameters from ff99SB. J Chem Theory Comput 11:3696–3713. https://doi.org/10.1021/acs.jctc.5b00255
Brooks BR, Brooks CL III, Mackerell AD Jr, Nilsson L, Petrella RJ, Roux B, Won Y, Archontis G, Bartels C, Boresch S, Caflisch A, Caves L, Cui Q, Dinner AR, Feig M, Fischer S, Gao J, Hodoscek M, Im W, Kuczera K, Lazaridis T, Ma J, Ovchinnikov V, Paci E, Pastor RW, Post CB, Pu JZ, Schaefer M, Tidor B, Venable RM, Woodcock HL, Wu X, Yang W, York DM, Karplus M (2009) CHARMM: the biomolecular simulation program. J Comput Chem 30:1545–1614. https://doi.org/10.1002/jcc.21287
Ivani I, Dans PD, Noy A, Pérez A, Faustino I, Hospital A, Walther J, Andrio P, Goñi R, Balaceanu A, Portella G, Battistini F, Gelpí JL, González C, Vendruscolo M, Laughton CA, Harris SA, Case DA, Orozco M (2016) Parmbsc1: a refined force field for DNA simulations. Nat Methods 13:55–58. https://doi.org/10.1038/nmeth.3658
Sztuba-Solinska J, Chavez-Calvillo G, Cline SE (2019) Unveiling the druggable RNA targets and small molecule therapeutics. Bioorg Med Chem 27(10):2149–2165. https://doi.org/10.1016/j.bmc.2019.03.057
Zhang C, Lu C, **g Z, Wu C, Piquemal JP, Ponder JW, Ren P (2018) AMOEBA polarizable atomic multipole force field for nucleic acids. J Chem Theory Comput 14(4):2084–2108. https://doi.org/10.1021/acs.jctc.7b01169
Li Y, Liu Z, Li J, Han L, Liu J, Zhao Z, Wang R (2014) Comparative assessment of scoring functions on an updated benchmark: 2. Evaluation methods and general results. J Chem Inf Model 54(6):1700–1716. https://doi.org/10.1021/ci500080q
Zhou T, Caflish A (2010) High-throughput virtual screening using quantum mechanical probes: discovery of selective kinase inhibitors. ChemMedChem 5(7):1007–1014. https://doi.org/10.1002/cmdc.201000085
Lu J, Zhang Z, Ni Z, Shen H, Tu Z, Liu H, Lu R (2014) QM/MM-PB/SA scoring of the interaction strength between Akt kinase and apigenin analogues. Comput Biol Chem 52:25–33. https://doi.org/10.1016/j.compbiolchem.2014.07.002
Mazanetz MP, Ichihara O, Law RJ, Whittaker M (2011) Prediction of cyclin-dependent kinase 2 inhibitor potency using the fragment molecular orbital method. J Chem 3:2. https://doi.org/10.1186/1758-2946-3-2
Heifetz A, Aldeghi M, Chudyk EI, Fedorov DG, Bodkin MJ, Biggin PC (2016) Using the fragment molecular orbital method to investigate agonist-orexin-2 receptor interactions. Biochem Soc Trans 44(2):574–581. https://doi.org/10.1042/BST20150250
Hsieh TJ, Lin HY, Tu Z, Lin TC, Wu SC, Tseng YY, Liu FT, Hsu ST, Lin CH (2016) Dual thio-digalactoside-binding modes of human galectins as the structural basis for the design of potent and selective inhibitors. Scientific Rep 6:29457. https://doi.org/10.1038/srep29457
Chudyk EI, Sarrat L, Aldeghi M, Fedorov DG, Bodkin MJ, James T, Southey M, Robinson R, Morao I, Heifetz A (2018) Exploring GPCR-ligand interactions with the Fragment Molecular Orbital (FMO) method. Methods Mol Biol 1705:179–195. https://doi.org/10.1007/978-1-4939-7465-8_8
Zou Y, Wang F, Wang Y, Guo W, Zhang Y, Xu Q, Lai Y (2017) Systematic study of imidazoles inhibiting IDO1 via the integration of molecular mechanics and quantum mechanics calculations. Eur J Med Chem 131:152–170. https://doi.org/10.1016/j.ejmech.2017.03.021
Borbulevych O, Martin RI, Tickle IJ, Westerhoff LM (2016) XModeScore: a novel method for accurate protonation/tautomer-state determination using quantum-mechanically driven macromolecular X-ray crystallographic refinement. Acta Cryst D72:586–598. https://doi.org/10.1107/S2059798316002837
Zhou T, Huang D, Caflisch A (2010) Quantum mechanical methods for drug design. Curr Top Med Chem 10(1):33–45. https://doi.org/10.2174/156802610790232242
Hargis JC, Vankayala SL, White JK, Woodcock HL (2014) Identification and characterization of noncovalent interactions that drive binding and specificity in DD-peptidases and β-lactamases. J Chem Theory Comput 10(2):855–864. https://doi.org/10.1021/ct400968v
Avgy-David HH, Senderowitz H (2015) Toward focusing conformational ensembles on bioactive conformations: a molecular mechanics/quantum mechanics study. J Chem Inf Model 55:2154–2167. https://doi.org/10.1021/acs.jcim.5b00259
Chen J, Wang J, Zhang Q, Chen K, Zhu W (2015) Probing origin of binding difference of inhibitors to MDM2 and MDMX by polarizable molecular dynamics simulation and QM/MM-GBSA calculation. Sci Rep 5:17421. https://doi.org/10.1038/srep17421
MacDonald CA, Boyd RJ (2015) Computational insights into the suicide inhibition of Plasmodium falciparum Fk506-binding protein 35. Bioorg Med Chem Lett 25(16):3221–3225. https://doi.org/10.1016/j.bmcl.2015.05.079
McCormick MC, Keijzer K, Polavarapu A, Schultz FA, Baik MH (2014) Understanding intrinsically irreversible, non-Nernstian, two-electron redox processes: a combined experimental and computational study of the electrochemical activation of platinum(IV) antitumor prodrugs. J Am Chem Soc 136(25):8992–9000. https://doi.org/10.1021/ja5029765
Lence E, van der Kamp MW, González-Bello C, Mulholland AJ (2018) QM/MM simulations identify the determinants of catalytic activity differences between type II dehydroquinase enzymes. Org Biomol Chem 16(24):4443–4455. https://doi.org/10.1039/c8ob00066b
Kuhn B, Jacobsen W, Christians U, Benet LZ, Kollman PA (2001) Metabolism of sirolimus and its derivative everolimus by cytochrome P450 3A4: insights from docking, molecular dynamics, and quantum chemical calculations. J Med Chem 44(12):2027–2034. https://doi.org/10.1021/jm010079y
Sun H, Scott DO (2010) Structure-based drug metabolism predictions for drug design. Chem Biol Drug Des 75:3–17. https://doi.org/10.1111/j.1747-0285.2009.00899.x
Tyzack JD, Kirchmair J (2019) Computational methods and tools to predict cytochrome P450 metabolism for drug discovery. Chem Biol Drug Des 93(4):377–386. https://doi.org/10.1111/cbdd.13445
Bobovská A, Tvaroška I, Kóňa J (2016) Using DFT methodology for more reliable predictive models: design of inhibitors of Golgi α-mannosidase II. J Mol Graph Model 66:47–57. https://doi.org/10.1016/j.jmgm.2016.03.004
Sullivan DJ Jr, Kaludov N, Martinov MN (2011) Discovery of potent, novel, non-toxic anti-malarial compounds via quantum modelling, virtual screening and in vitro experimental validation. Malar J 10:274. https://doi.org/10.1186/1475-2875-10-274
Sullivan DJ, Liu Y, Mott BT, Kaludov N, Martinov MN (2015) Discovery of novel liver-stage antimalarials through quantum similarity. PLoS One 10(5):e0125593. https://doi.org/10.1371/journal.pone.0125593
Świderek K, Tuñón I, Moliner V, Bertran J (2015) Computational strategies for the design of new enzymatic functions. Arch Biochem Biophys 582:68–79. https://doi.org/10.1016/j.abb.2015.03.013
Alexandrova AN, Jorgensen WL (2009) Origin of the activity drop with the E50D variant of catalytic antibody 34E4 for Kemp elimination. J Phys Chem B 113(2):497–504. https://doi.org/10.1021/jp8076084
Gong W, Wu R, Zhang Y (2015) Thiol versus hydroxamate as zinc binding group in HDAC inhibition: an ab initio QM/MM molecular dynamics study. J Comput Chem 36:2228–2235. https://doi.org/10.1002/jcc.24203
Hitzenberger M, Schuster D, Hofer TS (2017) The binding mode of the sonic hedgehog inhibitor robotnikinin, a combined docking and QM/MM MD study. Front Chem 5:76. https://doi.org/10.3389/fchem.2017.00076
Steinmann C, Olsson MA, Ryde U (2018) Relative ligand-binding free energies calculated from multiple short QM/MM MD simulations. J Chem Theory Comput 14:3228–3237. https://doi.org/10.1021/acs.jctc.8b00081
Zhu T, **ao X, Ji C, Zhang JZ (2013) A new quantum calibrated force field for zinc-protein complex. J Chem Theory Comput 9(3):1788–1798. https://doi.org/10.1021/ct301091z
**ong X, Chen Z, Cossins BP, Xu Z, Shao Q, Ding K, Zhu W, Shi J (2015) Force fields and scoring functions for carbohydrate simulation. Carbohydr Res 401:73–81. https://doi.org/10.1016/j.carres.2014.10.028
Cole DJ, Vilseck JZ, Tirado-Rives J, Payne MC, Jorgensen WL (2016) Biomolecular force field parameterization via atoms-in-molecule electron density partitioning. J Chem Theory Comput 12(5):2312–2323. https://doi.org/10.1021%2Facs.jctc.6b00027
Visscher KM, Geerke DP (2019) Deriving force-field parameters from first principles using a polarizable and higher order dispersion model. J Chem Theory Comput 15(3):1875–1883. https://doi.org/10.1021/acs.jctc.8b01105
Hsiao YW, Sanchez-Garcia E, Doerr M, Thiel W (2010) Quantum refinement of protein structures: implementation and application to the red fluorescent protein DsRed.M1. J Phys Chem B 114(46):15413–15423. https://doi.org/10.1021/jp108095n
Li X, Hayik SA, Merz KM Jr (2010) QM/MM X-ray refinement of zinc metalloenzymes. J Inorg Biochem 104(5):512–522. https://doi.org/10.1016/j.**orgbio.2009.12.022
Fu Z, Li X, Miao Y, Merz KM Jr (2013) Conformational analysis and parallel QM/MM X-ray refinement of protein bound anti-Alzheimer drug donepezil. J Chem Theory Comput 9(3):1686–1693. https://doi.org/10.1021/ct300957x
Dittrich B, Lübben J, Mebs S, Wagner A, Luger P, Flaig R (2017) Accurate bond lengths to hydrogen atoms from single-crystal X-ray diffraction by including estimated hydrogen ADPs and comparison to neutron and QM/MM benchmarks. Chemistry 23(19):4605–4614. https://doi.org/10.1002/chem.201604705
Chaskar P, Zoete V, Röhrig UF (2014) Toward on-the-fly quantum mechanical/molecular mechanical (QM/MM) docking: development and benchmark of a scoring function. J Chem Inf Model 54(11):3137–3152. https://doi.org/10.1021/ci5004152
Wichapong K, Rohe A, Platzer C, Slynko I, Erdmann F, Schmidt M, Sippl W (2014) Application of docking and QM/MM-GBSA rescoring to screen for novel Myt1 kinase inhibitors. J Chem Inf Model 54(3):881–893. https://doi.org/10.1021/ci4007326
Zang P, Gong A, Zhang P, Yu J (2016) Targeting druggable enzymome by exploiting natural medicines: an in silico-in vitro integrated approach to combating multidrug resistance in bacterial infection. Pharm Biol 54(4):604–618. https://doi.org/10.3109/13880209.2015.1068338
Khandelwal A, Balaz S (2007) QM/MM linear response method distinguishes ligand affinities for closely related metalloproteins. Proteins 69(2):326–339. https://doi.org/10.1002/prot.21500
Sawada T, Fedorov DG, Kitaura K (2010) Role of the key mutation in the selective binding of avian and human influenza hemagglutinin to sialosides revealed by quantum-mechanical calculations. J Am Chem Soc 132(47):16862–16872. https://doi.org/10.1021/ja105051e
Rathore RS, Sumakanth M, Reddy MS, Reddanna P, Rao AA, Erion MD, Reddy MR (2013) Advances in binding free energies calculations: QM/MM-based free energy perturbation method for drug design. Curr Pharm Des 19(26):4674–4686. https://doi.org/10.2174/1381612811319260002
Otsuka T, Okimoto N, Taiji M (2015) Assessment and acceleration of binding energy calculations for protein-ligand complexes by the fragment molecular orbital method. J Comput Chem 36(30):2209–2218. https://doi.org/10.1002/jcc.24055
Su PC, Tsai CC, Mehboob S, Hevener KE, Johnson ME (2015) Comparison of radii sets, entropy, QM methods, and sampling on MM-PBSA, MM-GBSA, and QM/MM-GBSA ligand binding energies of F. tularensis Enoyl-ACP reductase (FabI). J Comput Chem 36(25):1859–1873. https://doi.org/10.1002/jcc.24011
Ehrlich S, Göller AH, Grimme S (2017) Towards full quantum-mechanics-based protein-ligand binding affinities. ChemPhysChem 18(8):898–905. https://doi.org/10.1002/cphc.201700082
Caballero J, Alzate-Morales JH, Vergara-Jaque A (2011) Investigation of the differences in activity between hydroxycycloalkyl N1 substituted pyrazole derivatives as inhibitors of B-Raf kinase by using docking, molecular dynamics, QM/MM, and fragment-based de novo design: study of binding mode of diastereomer compounds. J Chem Inf Model 51(11):2920–2931. https://doi.org/10.1021/ci200306w
Sun TY, Wang Q, Zhang J, Wu T, Zhang F (2013) Trastuzumab-Peptide interactions: mechanism and application in structure-based ligand design. Int J Mol Sci 14(8):16836–16850. https://doi.org/10.3390/ijms140816836
De Colibus L, Wang X, Spyrou JAB, Kelly J, Ren J, Grimes J, Puerstinger G, Stonehouse N, Walter TS, Hu Z, Wang J, Li X, Peng W, Rowlands D, Fry EE, Rao Z, Stuart DI (2014) More powerful virus inhibitors from structure-based analysis of HEV71 capsid-binding molecules. Nat Struct Mol Biol 21(3):282–288. https://doi.org/10.1038/nsmb.2769
Zanatta G, Nunes G, Bezerra EM, da Costa RF, Martins A, Caetano EW, Freire VN, Gottfried C (2014) Antipsychotic haloperidol binding to the human dopamine D3 receptor: beyond docking through QM/MM refinement toward the design of improved schizophrenia medicines. ACS Chem Neurosci 5(10):1041–1054. https://doi.org/10.1021/cn500111e
Yu N, Hayik SA, Wang B, Liao N, Reynolds CH, Merz KM Jr (2006) Assigning the protonation states of the key aspartates in β-Secretase using QM/MM X-ray structure refinement. J Chem Theory Comput 2(4):1057–1069. https://doi.org/10.1021/ct0600060
Lee W, Luckner SR, Kisker C, Tonge PJ, Engels B (2011) Elucidation of the protonation states of the catalytic residues in mtKasA - implications for inhibitor design. Biochemistry 50(25):5743–5756. https://doi.org/10.1021/bi200006t
Vega-Teijido MA, El Chamy Maluf S, Bonturi CR, Sambrano JR, Ventura ON (2014) Theoretical insight into the mechanism for the inhibition of the cysteine protease cathepsin B by 1,2,4-thiadiazole derivatives. J Mol Model 20(6):2254. https://doi.org/10.1007/s00894-014-2254-0
Ma S, Vogt KA, Petrillo N, Ruhoff AJ (2015) Characterizing the protonation states of the catalytic residues in apo and substrate-bound human T-cell leukemia virus type 1 protease. Comput Biol Chem 56:61–70. https://doi.org/10.1016/j.compbiolchem.2015.04.002
Kocak A, Erol I, Yildiz M, Can H (2016) Computational insights into the protonation states of catalytic dyad in BACE1-acyl guanidine based inhibitor complex. J Mol Graph Model 70:226–235. https://doi.org/10.1016/j.jmgm.2016.10.013
Chakravorty DK, Wang B, Ucisik MN, Merz KM Jr (2011) Insight into the cation-π interaction at the metal binding site of the copper metallochaperone CusF. J Am Chem Soc 133(48):19330–19333. https://doi.org/10.1021/ja208662z
Zhou J, **e H, Liu Z, Luo HB, Wu R (2014) Structure-function analysis of the conserved tyrosine and diverse π-stacking among class I histone deacetylases: a QM (DFT)/MM MD study. J Chem Inf Model 54(11):3162–3171. https://doi.org/10.1021/ci500513n
Lenz SAP, Wetmore SD (2017) QM/MM study of the reaction catalyzed by alkyladenine DNA glycosylase: examination of the substrate specificity of a DNA repair enzyme. J Phys Chem B 121(49):11096–11108. https://doi.org/10.1021/acs.jpcb.7b09646
Zhou J, Wang YS (2017) Rational redesign of a cation···π···π stacking at cardiovascular Fbw7-Skp1 complex interface and its application for deriving self-inhibitory peptides to disrupt the complex interaction. J Mol Model 23(10):296. https://doi.org/10.1007/s00894-017-3456-z
Zhang L, Hao GF, Tan Y, ** Z, Huang MZ, Yang GF (2009) Bioactive conformation analysis of cyclic imides as protoporphyrinogen oxidase inhibitor by combining DFT calculations, QSAR and molecular dynamic simulations. Bioorg Med Chem 17(14):4935–4942. https://doi.org/10.1016/j.bmc.2009.06.003
Mandal M, Zhu Z, Cumming JN, Liu X, Strickland C, Mazzola RD, Caldwell JP, Leach P, Grzelak M, Hyde L, Zhang Q, Terracina G, Zhang L, Chen X, Kuvelkar R, Kennedy ME, Favreau L, Cox K, Orth P, Buevich A, Voigt J, Wang H, Kazakevich I, McKittrick BA, Greenlee W, Parker EM, Stamford AW (2012) Design and validation of bicyclic iminopyrimidinones as beta amyloid cleaving enzyme-1 (BACE1) inhibitors: conformational constraint to favor a bioactive conformation. J Med Chem 55(21):9331–9345. https://doi.org/10.1021/jm301039c
Arooj M, Sakkiah S, Kim S, Arulalapperumal V, Lee KW (2013) A combination of receptor-based pharmacophore modeling & QM techniques for identification of human chymase inhibitors. PLoS One 8(4):e63030. https://doi.org/10.1371/journal.pone.0063030
Pasha FA, Neaz MM (2013) Molecular dynamics and QM/MM-based 3D interaction analyses of cyclin-E inhibitors. J Mol Model 19(2):879–891. https://doi.org/10.1007/s00894-012-1620-z
Bembenek SD, Keith JM, Letavic MA, Apodaca R, Barbier AJ, Dvorak L, Aluisio L, Miller KL, Lovenberg TW, Carruthers NI (2008) Lead identification of acetylcholinesterase inhibitors-histamine H3 receptor antagonists from molecular modeling. Bioorg Med Chem 16(6):2968–2973. https://doi.org/10.1016/j.bmc.2007.12.048
Remko M, Remková A, Broer R (2016) Theoretical study of molecular structure and physicochemical properties of novel factor Xa inhibitors and dual factor Xa and factor IIa inhibitors. Molecules 21(2):185. https://doi.org/10.3390/molecules21020185
Pardhi T, Vasu K (2018) Identification of dual kinase inhibitors of CK2 and GSK3β: combined qualitative and quantitative pharmacophore modeling approach. J Biomol Struct Dyn 36(1):177–194. https://doi.org/10.1080/07391102.2016.1270856
Spiegel K, Magistrato A (2006) Modeling anticancer drug-DNA interactions via mixed QM/MM molecular dynamics simulations. Org Biomol Chem 4(13):2507–2517. https://doi.org/10.1039/b604263p
Lodola A, Capoferri L, Rivara S, Chudyk E, Sirirak J, Dyguda-Kazimierowicz E, Andrzej Sokalski W, Mileni M, Tarzia G, Piomelli D, Mor M, Mulholland AJ (2011) Understanding the role of carbamate reactivity in fatty acid amide hydrolase inhibition by QM/MM mechanistic modelling. Chem Commun (Camb) 47(9):2517–2519. https://doi.org/10.1039/c0cc04937a
Schmidt TC, Welker A, Rieger M, Sahu PK, Sotriffer CA, Schirmeister T, Engels B (2014) Protocol for rational design of covalently interacting inhibitors. ChemPhysChem 15(15):3226–3235. https://doi.org/10.1002/cphc.201402542
Capoferri L, Lodola A, Rivara S, Mor M (2015) Quantum mechanics/molecular mechanics modeling of covalent addition between EGFR-cysteine 797 and N-(4-anilinoquinazolin-6-yl) acrylamide. J Chem Inf Model 55(3):589–599. https://doi.org/10.1021/ci500720e
Ren W, Pengelly R, Farren-Dai M, Shamsi Kazem Abadi S, Oehler V, Akintola O, Draper J, Meanwell M, Chakladar S, Świderek K, Moliner V, Britton R, Gloster TM, Bennet AJ (2018) Revealing the mechanism for covalent inhibition of glycoside hydrolases by carbasugars at an atomic level. Nat Commun 9(1):3243. https://doi.org/10.1038/s41467-018-05702-7
James C, Pettit GR, Nielsen OF, Jayakumar VS, Joe IH (2008) Vibrational spectra and ab initio molecular orbital calculations of the novel anti-cancer drug combretastatin A-4 prodrug. Spectrochim Acta A Mol Biomol Spectrosc 70(5):1208–1216. https://doi.org/10.1016/j.saa.2007.10.052
Karaman R (2011) Computational-aided design for dopamine prodrugs based on novel chemical approach. Chem Biol Drug Des 78(5):853–863. https://doi.org/10.1111/j.1747-0285.2011.01208.x
Karaman R, Fattash B, Qtait A (2013) The future of prodrugs - design by quantum mechanics methods. Expert Opin Drug Deliv 10(5):713–729. https://doi.org/10.1517/17425247.2013.786699
Arfeen M, Patel DS, Abbat S, Taxak N, Bharatam PV (2014) Importance of cytochromes in cyclization reactions: quantum chemical study on a model reaction of proguanil to cycloguanil. J Comput Chem 35(28):2047–2055. https://doi.org/10.1002/jcc.23719
Ponte F, Russo N, Sicilia E (2018) Insights from computations on the mechanism of reduction by ascorbic acid of PtIV prodrugs with asplatin and its chlorido and bromido analogues as model systems. Chemistry 24(38):9572–9580. https://doi.org/10.1002/chem.201800488
Van der Kamp MW, Chaudret R, Mulholland AJ (2013) QM/MM modelling of ketosteroid isomerase reactivity indicates that active site closure is integral to catalysis. FEBS J 280(13):3120–3131. https://doi.org/10.1111/febs.12158
Kaiyawet N, Lonsdale R, Rungrotmongkol T, Mulholland AJ, Hannongbua S (2015) High-level QM/MM calculations support the concerted mechanism for Michael addition and covalent complex formation in thymidylate synthase. J Chem Theory Comput 11(2):713–722. https://doi.org/10.1021/ct5005033
Kumari M, Kozmon S, Kulhánek P, Štepán J, Tvaroška I, Koča J (2015) Exploring reaction pathways for O-GlcNAc transferase catalysis. A string method study. J Phys Chem B 119(12):4371–4381. https://doi.org/10.1021/jp511235f
Fernandes HS, Ramos MJ, Cerqueira NMFSA (2017) The catalytic mechanism of the pyridoxal-5′-phosphate-dependent enzyme, histidine decarboxylase: a computational study. Chemistry 23(38):9162–9173. https://doi.org/10.1002/chem.201701375
Elsässer B, Zauner FB, Messner J, Soh WT, Dall E, Brandstetter H (2017) Distinct roles of catalytic cysteine and histidine in the protease and ligase mechanisms of human legumain as revealed by DFT-based QM/MM simulations. ACS Catal 7(9):5585–5593. https://doi.org/10.1021/acscatal.7b01505
Roy S, Kästner J (2017) Catalytic mechanism of salicylate dioxygenase: QM/MM simulations reveal the origin of unexpected regioselectivity of the ring cleavage. Chemistry 23(37):8949–8962. https://doi.org/10.1002/chem.201701286
Brás NF, Fernandes PA, Ramos MJ (2018) Understanding the rate-limiting step of glycogenolysis by using QM/MM calculations on human glycogen phosphorylase. ChemMedChem 13(15):1608–1616. https://doi.org/10.1002/cmdc.201800218
Lonsdale R, Houghton KT, Żurek J, Bathelt CM, Foloppe N, de Groot MJ, Harvey JN, Mulholland AJ (2013) Quantum mechanics/molecular mechanics modeling of regioselectivity of drug metabolism in cytochrome P450 2C9. J Am Chem Soc 135(21):8001–8015. https://doi.org/10.1021/ja402016p
Tyzack JD, Williamson MJ, Torella R, Glen RC (2013) Prediction of cytochrome P450 xenobiotic metabolism: tethered docking and reactivity derived from ligand molecular orbital analysis. J Chem Inf Model 53(6):1294–1305. https://doi.org/10.1021/ci400058s
Lonsdale R, Rouse SL, Sansom MS, Mulholland AJ (2014) A multiscale approach to modelling drug metabolism by membrane-bound cytochrome P450 enzymes. PLoS Comput Biol 10(7):e1003714. https://doi.org/10.1371/journal.pcbi.1003714
Putkaradze N, Kiss FM, Schmitz D, Zapp J, Hutter MC, Bernhardt R (2017) Biotransformation of prednisone and dexamethasone by cytochrome P450 based systems - identification of new potential drug candidates. J Biotechnol 242:101–110. https://doi.org/10.1016/j.jbiotec.2016.12.011
Ferreira AM, Krishnamurthy M, Moore BM II, Finkelstein D, Bashford D (2009) Quantitative structure-activity relationship (QSAR) for a series of novel cannabinoid derivatives using descriptors derived from semi-empirical quantum-chemical calculations. Bioorg Med Chem 7(6):2598–2606. https://doi.org/10.1016/j.bmc.2008.11.059
Güssregen S, Matter H, Hessler G, Müller M, Schmidt F, Clark T (2012) 3D-QSAR based on quantum-chemical molecular fields: toward an improved description of halogen interactions. J Chem Inf Model 52(9):2441–2453. https://doi.org/10.1021/ci300253z
Ginex T, Muñoz-Muriedas J, Herrero E, Gibert E, Cozzini P, Luque FJ (2016) Application of the quantum mechanical IEF/PCM-MST hydrophobic descriptors to selectivity in ligand binding. J Mol Model 22(6):136. https://doi.org/10.1007/s00894-016-2991-3
Hudson BD, Whitley DC, Ford MG, Swain M, Essex JW (2008) Pattern recognition based on color-coded quantum mechanical surfaces for molecular alignment. J Mol Model 14(1):49–57. https://doi.org/10.1007/s00894-007-0251-2
Vázquez J, Deplano A, Herrero A, Ginex T, Gibert E, Rabal O, Oyarzabal J, Herrero E, Luque FJ (2018) Development and validation of molecular overlays derived from three-dimensional hydrophobic similarity with PharmScreen. J Chem Inf Model 58(8):1596–1609. https://doi.org/10.1021/acs.jcim.8b00216
Cannizzaro CE, Ashley JA, Janda KD, Houk KN (2003) Experimental determination of the absolute enantioselectivity of an antibody-catalyzed Diels-Alder reaction and theoretical explorations of the origins of stereoselectivity. J Am Chem Soc 125(9):2489–2506. https://doi.org/10.1021/ja020879d
Smith AJT, Müller R, Toscano MD, Kast P, Hellinga HW, Hilvert D, Houk KN (2008) Structural reorganization and preorganization in enzyme active sites: comparisons of experimental and theoretically ideal active site geometries in the multistep serine esterase reaction cycle. J Am Chem Soc 130(46):15361–15373. https://doi.org/10.1021/ja803213p
Frushicheva MP, Cao J, Chu ZT, Warshel A (2010) Exploring challenges in rational enzyme design by simulating the catalysis in artificial kemp eliminase. Proc Natl Acad Sci U S A 107(39):16869–16874. https://doi.org/10.1073/pnas.1010381107
Singh MK, Chu ZT, Warshel A (2014) Simulating the catalytic effect of a designed mononuclear zinc metalloenzyme that catalyzes the hydrolysis of phosphate triesters. J Phys Chem B 118(42):12146–12152. https://doi.org/10.1021/jp507592g
Acknowledgments
The authors thank Christophe Iftner, Franck Taillez, and Emilie Pihan (Evotec (France) SAS, Toulouse, France) for valuable suggestions to the manuscript.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2020 Springer Science+Business Media, LLC, part of Springer Nature
About this protocol

Cite this protocol
Kotev, M., Sarrat, L., Gonzalez, C.D. (2020). User-Friendly Quantum Mechanics: Applications for Drug Discovery. In: Heifetz, A. (eds) Quantum Mechanics in Drug Discovery. Methods in Molecular Biology, vol 2114. Humana, New York, NY. https://doi.org/10.1007/978-1-0716-0282-9_15
Download citation
DOI: https://doi.org/10.1007/978-1-0716-0282-9_15
Published:
Publisher Name: Humana, New York, NY
Print ISBN: 978-1-0716-0281-2
Online ISBN: 978-1-0716-0282-9
eBook Packages: Springer Protocols