
Abstract
Background
Pectin methylesterase (PME, EC 3.1.1.11) is a hydrolytic enzyme that utilizes pectin as substrates, and plays a significant role in regulating pectin reconstruction thereby regulating plant growth. Pectin is one of the important components of the plant cell wall, which forms the main structural material of cotton fiber. In this research, cotton genome information was used to identify PMEs.
Results
We identified 80 (GaPME01-GaPME80) PME genes from diploid G. arboreum (A genome), 78 (GrPME01-GrPME78) PME genes from G. raimondii (D genome), and 135 (GhPME001-GhPME135) PME genes from tetraploid cotton G. hirsutum (AD genome). We further analyzed their gene structure, conserved domain, gene expression, and systematic evolution to lay the foundation for deeper research on the function of PMEs. Phylogenetic data indicated that members from the same species demonstrated relatively high sequence identities and genetic similarities. Analysis of gene structures showed that most of the PMEs genes had 2–3 exons, with a few having a variable number of exons from 4 to 6. There are nearly no differences in the gene structure of PMEs among the three (two diploid and one tetraploid) cotton species. Selective pressure analysis showed that the Ka/Ks value for each of the three cotton species PME families was less than one.
Conclusion
Conserved domain analysis showed that PMEs members had a relatively conserved C-terminal pectinesterase domain (PME) while the N-terminus was less conserved. Moreover, some of the family members contained a pectin methylesterase inhibitor (PMEI) domain. The Ka/Ks ratios suggested that the duplicated PMEs underwent purifying selection after the duplication events. This study provided an important basis for further research on the functions of cotton PMEs. Results from qRT-PCR indicated that the expression level of different PMEs at various fiber developmental stages was different. Moreover, some of the PMEs showed fiber predominant expression in secondary wall thickening indicating tissue-specific expression patterns.
Similar content being viewed by others


Background
Cotton (Gossypium spp.) is one of the most important natural fiber crops around the world. The improvement of cotton fiber quality is becoming increasingly important and is now a main focal point of cotton research [1, 2]. Pectin is an important component of cotton fiber and pectin metabolism may influence fiber quality. Previous studies showed that PMEs play an important role in the process of fiber development by influencing the chemical properties of pectin [1]. Process of cotton fiber cell develo** was purposely divided into four relative independent but overlap** stages: fiber initiation, elongation, secondary wall biosynthesis and maturation [3]. Fiber initiation and elongation are critical periods in which the number and lengths of fibers, secondary wall thickening (fiber strength), and other fiber quality traits are determined..The secondary wall thickening in cotton fibers starts 15–19 d after flowering and continues to thicken until 40–50d [4]. The increasing thickness of the fiber secondary wall gradually increases the strength of fibers.
A forward subtractive cDNA library constructed and sequenced from upland cotton (G. hirsutum) fibers during the secondary cell wall thickening stage. Computational analysis showed differentially expressed genes that may be involved in cell wall synthesis and modification of biological processes. Among them, several differentially expressed genes which encoded PMEs were identified. Thus, in order to elucidate the relationship between PMEs and fiber development, we analyzed identification, phylogeny expression of PMEs in G. arboreum, G. raimondii and G. hirsutum.
PMEs are widely present in plants and some microorganisms that possess a cell wall degradation function. PMEs catalyze the demethylesterification of pectin, which generates carboxyl groups during the release of methanol and hydrogen ions [5]. It plays an important role in cell wall composition modification and degradation if pectin in different development stages of plant, such as fruit maturity [6], pollen development and pollen tube growth [7], cambium cell differentiation, and other plant growth and so on. PMEs have a two-part influence on the cell wall. These produce carboxyl groups and combine with extracellular Ca2+ to form a calcium chain bridge between adjacent pectins, thereby hardening the cell wall and slowing cell diffuse growth [8]. And, the reaction of demethylesterification decreases the extracellular pH to increase the hydrolytic enzyme activities of enzymes such as poly-galacturonic acid and several pectin enzyme cleavage enzymes [9]. Pectin is subject to substantial degradation, causes cell wall structure relaxation, and enhances the growth of cell tips [10]. The activity of PMEs is regulated by pectin methylesterase inhibitors (PMEIs) [11] whose active site is the conserved PME domain. All members of PME family consist of a catalytically active zone PME domain; some also harbor a PMEI domain. Some proteins containing only one PMEI domain belong to the PMEI family. Therefore, the predicted proteins can be classified into two categories, type I, containing both PME and PMEI domains, and type II, consisting only a PME domain.
The PME belongs to a multigene family which was first described by Richard [12]. There are 66 PMEs in Arabidopsis [13], 16 in Phytophthora sojae [14], 43 in rice [15], 105 in flax [16], and 81 in G. raimondii [1].
Previous reports suggested that PMEs may play a part in cell wall development of cotton fibers [1]. At present, studies related to PME genes mainly focused on cloning, and functional analysis of single gene [17]; and few analysis had been carried out at the whole genome level [1]. In 2012, the genome of G. raimondii was completed [18, 19]. The genome map of cultivated cotton G. arboreum was available in 2014 [20]. And next year, the genome map of allotetraploid cultivated cotton (G. hirsutum cv TM-l) was completed [21, 22]. The whole genome sequencing of cotton species provides opportunities for comprehensive analysis and comparison of the PMEs. PMEs, and its homologous genes were analyzed using bioinformatics analysis on the cotton genome sequence. The results showed that sequence similarities and gene structures were highly conserved. In this study, the gene structure, expression, phylogenetic tree, collinearity of homologous genes and other corresponding analysis were examined systematically by employing the methods of bioinformatics. The results of this study will provide novel insights into research of synthesis mechanism of cotton fiber cell wall.
Results
Identification of cotton PMEs
From the three cotton genomes (AD, A, and D), we identified 135 full-length putative G. hirsutum PMEs (GhPME001-135), 80 full-length putative G. arboretum PMEs (GaPME01-GaPME80), and 78 full-length putative G. raimondii PMEs (GrPME01-GrPME78) (see Additional file 1: Table S1, and Fig. 1a, c). The family members were named according to their location and sequence on the chromosome.

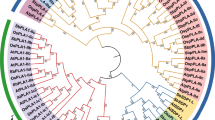
Phylogenetic relationship and gene structure of the G. arboreum and G. hirsutum PMEs. a A phylogenetic tree was constructed using MEGA 5.1 with the neighbor-joining (NJ) method with 1000 bootstrap replicates based on a multiple alignment of 135 amino acid sequences of PMEs from G. hirsutum. The eight major subfamilies are numbered I to VIII. b Exon/intron structure of PMEs from G. hirsutum. Exons and introns are represented by boxes and black lines, respectively. c A phylogenetic tree was constructed with MEGA 5.1 using the neighbor-joining (NJ) method with 1000 bootstrap replicates based on a multiple alignment of 80 amino acid sequences of PMEs from G. arboreum. The four major subfamilies are numbered I to IV. d Exon/intron structure of PMEs from G. arboreum. Exons and introns are represented by boxes and black lines, respectively
Gene structure and protein domain of PMEs in different species
The length of the PMEs between different cotton species was variable mainly due to large differences in the intron length of each gene. The length of the exons in PMEs ranged from 1045 bp to 13398 bp in G. arboreum, 1045 bp to 6730 bp in G. raimondii, and 964 bp to 4695 bp (with a majority between 1500 bp and 2500 bp) in G. hirsutum (Additional file 1: Table S1). The number of amino acid (AA) residues in the GaPMEs protein ranged from 301 to 1169, 316 to 1644 in GrPMEs, and 260 to 845 in GhPMEs (Additional file 1: Table S1). Asiatic cotton PMEs gene structure analysis results (Fig. 1d) showed that there were differences between different members. The members of the exon number ranged from 2 to 6, and the gene structure analysis showed that the gene structure of the family members was conserved. The gene structure could be mainly divided into three types. Type I has a typical of two exons and two introns; the differences in the first and second exon were highly conserved, but the length of introns was different. There were 37 such PMEs (46.25%) distributed in groups one, two, and three. Type II contained three exons, and there were 12 members (15%) in this group. Among the three exons in this group, the first two exons had significantly different length while the length of the third exon was highly conserved. Type III contained four to six exons with a shorter length than type I or II. These results suggested that the gene structures were similar between of G. hirsutum (Fig. 1b) and G. arboreum (Fig. 1d).
Eighty members of the PMEs family in G. arboreum had evolutionary tree clustering relations, and could be divided into four families (Fig. 1c). The analysis of the conserved sequence of the PMEs family members and domain analysis showed that all of them contain a PME domain. Most of the family members of PMEs contained both PME and PMEI. Only five proteins GaPME13 and GaPME46 in group 1, and GaPME40, GaPME47, and GaPME48 in group 2 included only the PME domain. Members of a fourth subfamily contained only a PME domain without a PMEI domain, and there was nearly no difference in G. hirsutum (see Additional file 1: Table S1).
Distribution of PMEs family members
We found 80 PMEs corresponding to the protein-coding genes in the Asiatic cotton database. These 80 genes were distributed on the 13 chromosomes (Additional file 2: Figure S1b), in which the most PMEs (11) were located on chromosome 1 and chromosome 10. Ten PMEs are mapped on chromosome 2, nine on chromosome 9, eight on chromosome 4, three on chromosome 6, and only one was mapped to chromosome 12. Each of the chromosomes 3, 5, 7, and 8 had four genes. Only one gene was not detected on the chromosome and was positioned on the scaffold. Eighty genes showed uneven distribution on the chromosomes. Some genes arised by tandem duplication. Seven genes (GaPME61- GaPME67) on chromosome 10 were located on the same block, which we named as cluster I. Five such clusters were located on chromosomes 1, 2, 9, 11, and 13; these clusters covered by 22.5% of PMEs. We found 135 PMEs in the cotton AD genome (Additional file 2: Figure S1a). Of these, all the 26 chromosomes except At_chr12, fourteen genes (10.4%) were located on chromosome Dt_chr9, 10 were mapped on chromosome At_chr9, and some genes appeared via tandem duplication on chromosomes in cotton AD and D genomes. We found 78 PMEs, of which were distributed to all the chromosomes except chromosomes 4, 12. Chromosomes 9, 8, 6, 7, 2, 1, had 16, 10, 8, 6, 2, and 3 chromosomes respectively. Chromosome 10 and 11 together had five genes; chromosome 5 and 13 had three while chromosomes 3 and 12 had one gene (Additional file 1: Table S1).
Based on the results of collinearity analysis between G. raimondii and G. arboreum, 61 homologous gene pairs were distributed in 36 collinearity blocks (Fig. 2a, Additional file 3: Table S2). Among them, one syntonic block contained 19 homologous gene pairs in G. arboreum chromosome 10. We identified 57 homologous gene pairs between G. hirsutum (Fig. 2b, Additional file 4: Table S3) and G. arboreum, and 50 homologous gene pairs between G. hirsutum and G. raimondii (Fig. 2b, Additional file 4: Table S3). Some genes were not shown in Fig. 2 because they were not positioned on the chromosome (Additional file 4: Table S3).

CIRCOS figure of PME homologous genes pairs of G. raimondii and G. arboreum. a CIRCOS figure of PME homologous genes pairs of G. raimondii and G. arboreum. Lines represent homologous genes that are distributed in syntenic blocks between G. raimondii and G. arboreum chromosomes. b CIRCOS figure of PME homologous genes pairs of G. raimondii and G. hirsutum, G. arboreum and G. hirsutum. Lines represent homologous genes that are distributed in syntenic blocks between G. raimondii, G. arboreum and G. hirsutum chromosomes
Phylogenetic analysis
Phylogenetic analysis indicated that PMEs of the same species shared the highest similarities and had relatively close genetic relationships. In order to analyze the evolutionary relationships among the predicted GhPMEs, GaPMEs, and GrPMEs based on amino acid sequence, we aligned cotton amino acid sequences with 458 predicted PMEs from eight sequenced plants such as A. thaliana, rice, rice, grape, poplar, soybean, cocoa, papaya, and castor bean. Finally, phylogenetic trees were constructed by using MEGA with the neighbor-joining model. We found that the PMEs family could be divided into 10 subfamilies according to cluster analysis (Fig. 3a). The PMEs had a close genetic relationship within the same species and with cocoa. However, these genes were distant to other species on the evolutionary scale. These data indicated that the PMEs might evolve along with different directions for various species. Meanwhile, to examine the evolutionary relationship of PMEs in G. arboreum, G. raimondii, and G. hirsutum, the phylogenetic tree was built with 293 PMEs in which were divided into eight families (Fig. 3b).

Phylogenetic tree of PMEs. a Phylogenetic tree of PMEs from 11 species. The phylogenetic tree is based on a sequence alignment of the C-terminal PME domains of 751 PMEs protein sequences from 11 genomes, G. hirsutum, G. arboreum, G. raimondii, A. thaliana, O. sativa, V. vinifera, P. trichocarpa, G. max, T. cacao, C. papaya, and R. communis. The PME proteins are grouped into 10 distinct clades (I–X). b Phylogenetic tree of PME domain containing proteins from G. raimondii, G. arboreum, and G. hirsutum. The phylogenetic tree is based on a sequence alignment of the C-terminal PME domains of 293 PME protein sequences from three genomes, G. arboreum, G. raimondii, and G. hirsutum. The tree was generated with MEGA 5.1 using the neighbor-joining method. Bootstrap values from 1000 replicates are indicated at each node. The PME proteins are grouped into 8 distinct clades (I–VIII)
The value of the nonsynonymous substitution rate (Ka) to the synonymous substitution rate (Ks) substitutions (Ka/Ks) can be used as an indicator which could reflect selection pressure of a gene or a gene region during evolution. To infer the influence of selection on the evolution of the three cotton species versus cocoa, we estimated Ka/Ks values for all of them (Additional file 5: Table S4). Our results suggested that all of the three cotton species evolved mainly under the influence of stabilizing selection.
Transcriptome analysis
All of the identified PMEs of G. hirsutum were verified by transcriptome data. Expression of 75.56% (102 of 135 genes) in G. hirsutum (Fig. 4a) can be detected 0 from 15 day post anthesis (DPA) during fiber development. We detected the expression of 82.5% (66 of 80) PMEs of G. arboreum (Fig. 4b) and 71.8% (56 of 78) PMEs of G. raimondii (Fig. 4c) during fiber development (0–15 DPA). We found that 11 PMEs of G. hirsutum (Fig. 4d) and five PMEs of G. arboreum were predominantly expressed in fiber development at 15 DPA (belong to the period of secondary wall thickening) (Fig. 4e). However, only three genes in G. raimondii showed higher expression at 15 DPA (Fig. 4f).

Expression patterns of the PMEs family in G. raimondii, G. arboreum and G. hirsutum. a Heatmap showing the clustering of 135 PMEs of G. hirsutum across five tissues (ovules at 0 DPA, 3 DPA, 6 DPA, 10 DPA, and 15 DPA; mentioned at the top of each lane). b Heatmap showing the clustering of 80 PMEs of G. arboreum across five tissues (ovules at 0, 3, 6, 10, and 15 DPA; mentioned at the top of each lane). c Heatmap showing the clustering of 78 PMEs of G. raimondii across five tissues (ovules at 0, 3, 6, 10, and 15 DPA; mentioned at the top of each lane). d Expression of 11 (G. hirsutum) PMEs is predominantly expressed at 15 DPA. e Expression of 5 (G. arboreum) PMEs is predominantly expressed at 15 DPA. f Expression of 3 (G. raimondii) PMEs is predominantly expressed at 15 DPA. The color scale at the bottom of the dendrogram shows the relative expression levels. RNA-seq data under the series accession number SRA180756 was obtained from the NCBI Sequence Read Archive (SRA) database
To examine the differential expression of homologous gene pairs among the three cotton species, PMEs of G. arboreum predominantly expressed in 15 DPA and its homologous genes were selected for phylogenetic analysis (Fig. 5a). Based on above results, four homologous gene pairs were chose for further studies. The data showed that expression patterns of homologous genes pairs were significantly affected. Moreover, the expression levels of almost all genes in the A genome of cotton were higher than the genes in the D genome, and lower than the genes in the AD genome of cotton (Fig. 5b), for example, the expression of GrPME23 in fiber development (at 15 days) was 12.28,007, whereas the expression of GaPME17 was 25.40,384. The expression of GhPME037 in fiber development (15 DPA) was 46.41,517. The expression level of GaPME34 was higher than the expression of its orthologous genes in the AD genome of cotton.

Analysis of PMEs predominantly expressed in fiber. a The phylogenetic tree was constructed with MEGA 5.1. b Heatmap showing the clustering of PMEs across five tissues (ovules at 0, 3, 6, 10, and 15 DPA; mentioned at the top of each lane). The color scale at the bottom of the dendrogram shows the relative expression levels. c Exon/intron structures of PMEs predominantly expressed in fibers. Exons and introns are represented by boxes and black lines, respectively. d PME domain of the PMEs protein. e Motif of the PME protein
To survey on mechanism of the differences among orthologous gene pairs, we compared their gene structure (Fig. 5c), protein domain conservation (Fig. 5d), and sequence motifs (Fig. 5e). The results showed that orthologous gene pairs have minimal to negligible effect on the structure of the genes. The length of the first exon affected the structure of these genes. GaPME17, GhPME037, and GrPME23, GaPME04 and GhPME089, GaPME34 and GhPME085 were all different in only this one exon. The conserved domain of the protein between the genes did not differ significantly. Protein of orthologous gene pairs varied only on the position of the conserved domain and the length of the non-conserved region (Fig. 5d).
Analysis of putative cis-element motifs of PMEs homologous genes pairs of G. arboreum and G. hirsutum showed significant differences between their promoter regions (Additional file 6: Figure S2). Thus, we speculated that structure variation in promoter region might affect expression levels of homologous gene pairs.
qRT-PCR analysis for PMEs homologous genes pairs
To verify the alteration of expression patterns of four PMEs homologous gene pairs in G. hirsutum and G. arboreum, qRT-PCR was employed in this study. The results (Fig. 6) showed that the expression of PMEs peaked in Asiatic cotton at 20 DPA and 25 DPA in upland cotton. The average performance of upland cotton was higher than the Asiatic cotton fiber development at 25 DPA suggested that the expression level of Asiatic cotton PMEs was decreased in the late development of cotton fiber. However, in upland cotton, the PMEs expression level continued to increasing. This probably caused the thickening of fibers in the secondary wall, de-esterification of the pectin in the cell wall, reinforcement of the cotton fiber cell wall; thus, increasing the strength and imparting high quality to the upland cotton fiber.

Expression analysis of 4 selected PMEs homologous genes pairs using RT-qPCR. The relative mRNA abundance of 4 selected PMEs was normalized to the reference gene histone 3 in different tissues. The bars show the standard deviation of three technical replications
PMEs activity
There are differences in PMEs activity in different cotton fiber development periods. Of the increasing in Asiatic cotton fiber development and PMEs activity gradually increased from fiber development at 10 DPA to 25 DPA. However, the PMEs activity decreased at 30 DPA. In upland cotton, PMEs activity continued to increasing (Additional file 7: Figure S3). The reason might be that the Asiatic cotton prematurely ended the secondary wall of the fiber growth causing feedback regulation by the cellulose content and accumulation of pectin.
Discussion
We used bioinformatics analysis to identify 135 GhPME genes from AD genomes, 80 GaPME genes from A genomes, and 78 GrPME genes from D genomes. Cotton PMEs could be divided into four clades in two diploid species and eight groups in the tetraploid species, and all their last subfamilies were restricted to PME without a PMEI domain. We speculated the common hypothesis that PMEs that both PME and PMEI domains appear relatively late in the evolutionary process [23], similar to the species of that observed in A. thaliana PMEs [24]. Our analysis showed amount of reduction number of genes (from 81 to 78) as compared with Liu’s study (based on 81 sequences), mainly due to using a more stringent screening criteria. Phylogenetic analysis indicated that these could be divided into four subfamilies (Additional file 8: Figure S4), and the fourth subfamily only contained a PME domain (Additional file 1: Table S1) [1]. Previous studies identified 66, 59, and 89 PMEs coding genes in A. thaliana [24], O. sativa [25], and P. trichocarpa [26], respectively. The number of the PMEs varied greatly in different species. Previous studies had shown that the whole genome duplication (WGD) and tandem repeats were the main reasons of gene family expansion during the process of plant genome evolution [27, 28]. Plants have a higher rate of gene duplication compared to other eukaryotes [39]. The fiber quality of the parents and the line 69307 was described in Zhang’s report [40]. In the day of flowering, flower buds were tagged as zero DPA. The bolls were collected of each sample (5, 10, 15, 20, 25, 30 DPA) in the morning. The fibers were separated from the ovules, frozen in liquid nitrogen. Before RNA extraction, all the samples were stored at a refrigerator with −80 °C.
RNA-seq analysis
CTAB method was used to isolated the subsequent total RNA samples from 3 g of cotton fiber (G. hirsutum TM-1, G. arboreum Shixiya I and G. raimondii, 0, 3, 6, 10, and 15 DPA) [41]. A Nucleospin® RNA clean-up kit (MACHEREY-NAGEL, Düren, Germany) was used to purified the total RNA. An Agilent Bio-analyzer (Agilent Technologies, Santa Clara, CA, USA) was used to assess the quality of the RNA sample. After sequencing libraries were prepared following the manufacturer’s standard instructions, and all RNA samples were sequenced on an Illumina HiSeq 2500 platform (Illumina, Inc., San Diego, CA, USA). The CLC Genomics Workbench software 4 (http://www.clcbio.com) was used to analyze the transcriptome data with default parameters. MeV program was used to draw the heat map of the expression data [51]. Phylogenetic trees were constructed by employing MEGA software with neighbor joining model, and bootstrap values (1000 replicates) are indicated at each node [52]. Exons and introns were displayed by using The Gene Structure Display Server (GSDS, http://gsds.cbi.pku.edu.cn) [53]. Conserved domains prediction were performed using the SMART Package (http://smart.embl-heidelberg.de) program [54]. Motif analysis was conducted using online tools (maximum number of motifs, three; minimum motif width, six; and maximum motif width, 50) (http://meme.nbcr.net/meme) [55]. The cis-acting elements prediction was performed using an online tool PlantCARE (http://bioinformatics.psb.ugent.be/webtools/plantcare/html) [56].
PMEs activity assay
Based on the Hagerman with some modifications [57], total PMEs enzyme activity was measured. Briefly, 1 g of a fiber sample was taken in a prechilled mortar and 5.0 mL 8.8% (w/v) pre-cooled NaCl was added. The samples were centrifuged for 10 min; the supernatant was collected, and adjusted to pH =7.5 with 0.1 mol/L NaOH to obtain a crude enzyme solution. Four milliliters of 0.5% (w/v) pectin solution and 0.3 mL 0.01% (w/v) bromophenol blue were added to a test-tube, followed by adding 300 uL of an enzyme solution. After 2 min, the absorbance value, and the ΔA620/min∙g expressed enzyme activity, of each sample was analyzed in triplicate [57].
Abbreviations
- AA:
-
Amino acid
- DPA:
-
Day post anthesis
- Ga :
-
Gossypium arboreum
- Gb :
-
Gossypium barbadense
- Gh :
-
Gossypium hirsutum
- Gr :
-
Gossypium raimondii
- Ka:
-
Nonsynonymous substitution rate
- Ks:
-
Synonymous substitution rate
- LEA :
-
Late embryogenesis abundant
- MAPK :
-
Mitogen aetivated protein kinase
- MAPKKK :
-
Mitogen activated protein kinase kinase kinase
- PME:
-
Pectin methylesterase
- PMEI:
-
Pectin methylesterase inhibitor
- qRT-PCR:
-
Quantitative real-time polymerase chain reaction
- WGD:
-
Whole genome duplication
References
Liu Q, Talbot M, Llewellyn DJ. Pectin methylesterase and pectin remodelling differ in the fibre walls of two gossypium species with very different fibre properties. PLoS One. 2013;8(6):e65131.
Jamshed M, Jia F, Gong J, Palanga KK, Shi Y, Li J, Shang H, Liu A, Chen T, Zhang Z. Identification of stable quantitative trait loci (QTLs) for fiber quality traits across multiple environments in Gossypium hirsutum recombinant inbred line population. BMC Genomics. 2016;17(1):1.
Basra AS, Malik C. Development of the cotton fiber. Int Rev Cytol. 1984;89(1):65–113.
Wilkins TA, Arpat AB. The cotton fiber transcriptome. Physiol Plant. 2005;124(3):295–300.
Jolie RP, Duvetter T, Van Loey AM, Hendrickx ME. Pectin methylesterase and its proteinaceous inhibitor: a review. Carbohydr Res. 2010;345(18):2583–95.
Kagan-Zur V, Tieman DM, Marlow SJ, Handa AK. Differential regulation of polygalacturonase and pectin methylesterase gene expression during and after heat stress in ripening tomato (Lycopersicon esculentum Mill.) fruits. Plant Mol Biol. 1995;29(6):1101–10.
Bosch M, Cheung AY, Hepler PK. Pectin methylesterase, a regulator of pollen tube growth. Plant Physiol. 2005;138(3):1334–46.
Qin Y-M, Zhu Y-X. How cotton fibers elongate: a tale of linear cell-growth mode. Curr Opin Plant Biol. 2011;14(1):106–11.
Wen F, Zhu Y, Hawes MC. Effect of pectin methylesterase gene expression on pea root development. Plant Cell. 1999;11(6):1129–40.
Catoire LC L, Pierron M, Morvan C, du Penhoat CH, Goldberg R. Investigation of the action patterns of pectinmethylesterase isoforms through kinetic analyses and NMR spectroscopy - Implications in cell wall expansion. J Biol Chem. 1998;50:33150–6.
BALESTRIERI C, CASTALDO D, GIOVANE A, QUAGLIUOLO L, SERVILLO L. A glycoprotein inhibitor of pectin methylesterase in kiwi fruit (Actinidia chinensis). Eur J Biochem. 1990;193(1):183–7.
Richard L, Qin L-X, Gadal P, Goldberg R. Molecular cloning and characterisation of a putative pectin methylesterase cDNA in Arabidopsis thaliana (L.). FEBS Lett. 1994;355(2):135–9.
Louvet R, Cavel E, Gutierrez L, Guénin S, Roger D, Gillet F, Guerineau F, Pelloux J. Comprehensive expression profiling of the pectin methylesterase gene family during silique development in Arabidopsis thaliana. Planta. 2006;224(4):782–91.
Horowitz BB, Ospina-Giraldo MD. The pectin methylesterase gene complement of phytophthora sojae: structural and functional analyses, and the evolutionary relationships with its oomycete homologs. PLoS One. 2015;10(11):e0142096.
Jeong HY, Nguyen HP, Lee C. Genome-wide identification and expression analysis of rice pectin methylesterases: Implication of functional roles of pectin modification in rice physiology. J Plant Physiol. 2015;183:23–9.
Pinzón-Latorre D, Deyholos MK. Characterization and transcript profiling of the pectin methylesterase (PME) and pectin methylesterase inhibitor (PMEI) gene families in flax (Linum usitatissimum). BMC Genomics. 2013;14(1):742.
Tingting C, Xukai L, Ruyi W, Liangcai P, Tao X. Cloning and expression analysis of GhPME 1 and GhPME 2 in cotton. J China Agric Univ. 2012;17(005):7–14.
Wang K, Wang Z, Li F, Ye W, Wang J, Song G, Yue Z, Cong L, Shang H, Zhu S, et al. The draft genome of a diploid cotton Gossypium raimondii. Nat Genet. 2012;44(10):1098–103.
Paterson AH, Wendel JF, Gundlach H, Guo H, Jenkins J, ** D, Llewellyn D, Showmaker KC, Shu S, Udall J. Repeated polyploidization of Gossypium genomes and the evolution of spinnable cotton fibres. Nature. 2012;492(7429):423–7.
Li F, Fan G, Wang K, Sun F, Yuan Y, Song G, Li Q, Ma Z, Lu C, Zou C. Genome sequence of the cultivated cotton Gossypium arboreum. Nat Genet. 2014;46(6):567–72.
Li F, Fan G, Lu C, **ao G, Zou C, Kohel RJ, Ma Z, Shang H, Ma X, Wu J, et al. Genome sequence of cultivated Upland cotton (Gossypium hirsutum TM-1) provides insights into genome evolution. Nat Biotechnol. 2015;33(5):524–30.
Zhang T, Hu Y, Jiang W, Fang L, Guan X, Chen J, Zhang J, Saski CA, Scheffler BE, Stelly DM, et al. Sequencing of allotetraploid cotton (Gossypium hirsutum L. acc. TM-1) provides a resource for fiber improvement. Nat Biotechnol. 2015;33(5):531–7.
Micheli F. Pectin methylesterases: cell wall enzymes with important roles in plant physiology. Trends Plant Sci. 2001;6(9):414–9.
Hongo S, Sato K, Yokoyama R, Nishitani K. Demethylesterification of the Primary Wall by PECTIN METHYLESTERASE35 Provides Mechanical Support to the Arabidopsis Stem. Plant cell. 2012;24:2624–34.
Wang M, Yuan D, Gao W, Li Y, Tan J, Zhang X. A comparative genome analysis of PME and PMEI families reveals the evolution of pectin metabolism in plant cell walls. PLoS One. 2013;8(8):1–12.
Markovič O, Janeček Š. Pectin methylesterases: sequence-structural features and phylogenetic relationships. Carbohydr Res. 2004;339(13):2281–95.
Chothia C, Gough J, Vogel C, Teichmann SA. Evolution of the protein repertoire. Science. 2003;300(5626):1701–3.
Ohno S, Wolf U, Atkin NB. Evolution from fish to mammals by gene duplication. Hereditas. 1968;59(1):169–87.
Wei K, Wang Y, **e D. Identification and expression profile analysis of the protein kinase gene superfamily in maize development. Mol Breed. 2014;33(1):155–72.
Salnikov VV, Grimson MJ, Seagull RW, Haigler CH. Localization of sucrose synthase and callose in freeze-substituted secondary-wall-stage cotton fibers. Protoplasma. 2003;221(3–4):175–84.
Ruan Y-L, Llewellyn DJ, Furbank RT. Suppression of sucrose synthase gene expression represses cotton fiber cell initiation, elongation, and seed development. Plant Cell. 2003;15(4):952–64.
Zhang L, Xue J, Yu H, Li Q, Li R. The expression and function study of pectin methylesterase genes which regulate and control the petal falling in Arabidopsis. Plant Physiol J. 2012;48(4):350–8.
Yeom HW, Streaker CB, Zhang QH, Min DB. Effects of pulsed electric fields on the quality of orange juice and comparison with heat pasteurization. J Agric Food Chem. 2000;48(10):4597–605.
Ohno S. Evolution by gene duplication. Department of Biology City of Hope Medical Center Duarte. New York: Springer Science & Business Media; 2013.
Hughes AL. The evolution of functionally novel proteins after gene duplication. Proc R Soc Lond B Biol Sci. 1994;256(1346):119–24.
Moore RC, Purugganan MD. The evolutionary dynamics of plant duplicate genes. Curr Opin Plant Biol. 2005;8(2):122–8.
Zheng X, Zhu J, Kapoor A, Zhu JK. Role of Arabidopsis AGO6 in siRNA accumulation, DNA methylation and transcriptional gene silencing. EMBO J. 2007;26(6):1691–701.
Levesque-Tremblay G, Müller K, Mansfield SD, Haughn GW. HIGHLY METHYL ESTERIFIED SEEDS is a pectin methyl esterase involved in embryo development. Plant Physiol. 2015;167(3):725–37.
Sun F-D, Zhang J-H, Wang S-F, Gong W-K, Shi Y-Z, Liu A-Y, Li J-W, Gong J-W, Shang H-H, Yuan Y-L. QTL map** for fiber quality traits across multiple generations and environments in upland cotton. Mol Breed. 2011;30(1):569–82.
Zhang Z, Li J, Muhammad J, Cai J, Jia F, Shi Y, Gong J, Shang H, Liu A, Chen T, et al. High resolution consensus map** of quantitative trait loci for fiber strength, length and micronaire on chromosome 25 of the upland cotton (Gossypium hirsutum L.). PLoS One. 2015;10(8):e0135430.
Ji SJ, Lu YC, Feng JX, Wei G, Li J, Shi YH, Fu Q, Liu D, Luo JC, Zhu YX. Isolation and analyses of genes preferentially expressed during early cotton fiber development by subtractive PCR and cDNA array. Nucleic Acids Res. 2003;31(10):2534–43.
Cao J, Schneeberger K, Ossowski S, Gunther T, Bender S, Fitz J, Koenig D, Lanz C, Stegle O, Lippert C, et al. Whole-genome sequencing of multiple Arabidopsis thaliana populations. Nat Genet. 2011;43(10):956–63.
Goff SA, Ricke D, Lan T-H, Presting G, Wang R, Dunn M, Glazebrook J, Sessions A, Oeller P, Varma H, et al. A draft sequence of the rice genome (Oryza sativa L. ssp. Japonica). Science. 2002;296:79–99.
Jaillon O, Aury JM, Noel B, Policriti A, Clepet C, Casagrande A, Choisne N, Aubourg S, Vitulo N, Jubin C, et al. The grapevine genome sequence suggests ancestral hexaploidization in major angiosperm phyla. Nature. 2007;449(7161):463–7.
Tuskan GA, DiFazio S, Jansson S, Bohlmann J, Grigoriev, Hellsten, Putnam N, Ralph S, Rombauts, Salamov A, et al. The genome of black cottonwood, Populus trichocarpa (Torr. & Gray). Science. 2006;313:1596–604.
Schmutz J, Cannon SB, Schlueter J, Ma J, Mitros T, Nelson W, Hyten DL, Song Q, Thelen JJ, Cheng J, et al. Genome sequence of the palaeopolyploid soybean. Nature. 2010;463(7278):178–83.
Argout X, Salse J, Aury JM, Guiltinan MJ, Droc G, Gouzy J, Allegre M, Chaparro C, Legavre T, Maximova SN, et al. The genome of Theobroma cacao. Nat Genet. 2011;43(2):101–8.
Ming R, Hou S, Feng Y, Yu Q, Dionne-Laporte A, Saw JH, Senin P, Wang W, Ly BV, Lewis KL, et al. The draft genome of the transgenic tropical fruit tree papaya (Carica papaya Linnaeus). Nature. 2008;452(7190):991–6.
Chan AP, Crabtree J, Zhao Q, Lorenzi H, Orvis J, Puiu D, Melake-Berhan A, Jones KM, Redman J, Chen G, et al. Draft genome sequence of the oilseed species Ricinus communis. Nat Biotechnol. 2010;28(9):951–6.
Finn RD, Clements J, Eddy SR. HMMER web server: interactive sequence similarity searching. Nucleic Acids Res. 2011;39(Web Server issue):W29–37.
Voorrips RE. MapChart: software for the graphical presentation of linkage maps and QTLs. J Hered. 2002;93(1):77–8.
Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol. 2011;28(10):2731–9.
Hu B, ** J, Guo A-Y, Zhang H, Luo J, Gao G. GSDS 2.0: an upgraded gene feature visualization server. Bioinformatics. 2015;31(8):1296–7.
Letunic I, Doerks T, Bork P. SMART: recent updates, new developments and status in 2015. Nucleic Acids Res. 2015;43(D1):D257–60.
Bailey TL, Williams N, Misleh C, Li WW. MEME: discovering and analyzing DNA and protein sequence motifs. Nucleic Acids Res. 2006;34(Web Server issue):W369–73.
Lescot M, Dehais P, Thijs G, Marchal K, Moreau Y, Van de Peer Y, Rouze P, Rombauts S. PlantCARE, a database of plant cis-acting regulatory elements and a portal to tools for in silico analysis of promoter sequences. Nucleic Acids Res. 2002;30(1):325–7.
Hagerman AEA, Paul J. Continuous spectrophotometric assay for plant pectin methyl esterase. J Agric Food Chem. 1986;34:440–4.
Acknowledgments
Thank you to Dr. Daojie Wang at the Institute of Henan University for his insightful comments while preparing the manuscript.
Funding
This work was supported by National Natural Science Foundation of China (31471538), and the National High Technology Research and Development Program of China (2013AA102601).
Availability of data and materials
G. hirsutum, G. arboreum, and G. raimondii (http://cgp.genomics.org.cn), A. thaliana (http://www.arabidopsis.org), O. sativa (http://rapdb.dna.affrc.go.jp), V. vinifera (http://www.genoscope.cns.fr/spip/Vitis-vinifera-e.html), P. s trichocarpa (http://www.phytozome.net/poplar), G. max (http://www.phytozome.net/soybean), T. cacao L. (http://cocoagendb.cirad.fr), C. papaya (http://asgpb.mhpcc.hawaii.edu), and castor bean (http://castorbean.jcvi.org). The phylogenetic data was uploaded to Treebase under Accession URL: http://purl.org/phylo/treebase/phylows/study/TB2:S20141.
Authors’ contributions
YLY, HHS, and QG designed the experiments, WJL conducted gene expression analyses and drafted the manuscript. HHS, and CSZ provided the RNA-seq data. JC and SMF participated in the sequence alignment and PMEs activity test. ZZ, XYD, YNT, WWS, PTL, QWL, PKK, and MJ performed phylogenetic analyses of PMEs. DJW, WKG, YZS, TTC, and JWL helped to draft the manuscript. JWG and AYL prepared the cotton. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Not applicable.
Ethics approval and consent to participate
Not applicable.
Author information
Authors and Affiliations
Corresponding author
Additional files
Additional file 1: Table S1.
PMEs genes identified from the three cotton species. (XLSX 36 kb)
Additional file 2: Figure S1.
Chromosomal location of PMEs. a. Chromosomal location of 135 GhPME genes. A total of 104 genes are located on normal chromosomes, whereas the other 31 are located on scaffolds. b. Chromosomal location of 80 GaPME genes. A total of 79 genes are located on normal chromosomes, whereas the other one is located on scaffolds. (TIF 1445 kb)
Additional file 3: Table S2.
Orthologous PMEs gene pairs of G. arboreum and G. raimondii. (XLSX 14 kb)
Additional file 4: Table S3.
Orthologous PMEs gene pairs of the three cotton species. (XLSX 23 kb)
Additional file 5: Table S4.
Selective pressure between T. cacao L. and the three cotton species paralogous PMEs gene pairs. (XLSX 23 kb)
Additional file 6: Figure S2.
Analysis of putative cis-element motifs of PME homologous genes pairs of G. arboreum and G. hirsutum promoter. cis-element motifs are represented by boxes. (TIF 612 kb)
Additional file 7: Figure S3.
Cotton fiber proteins were isolated and used for PME activity assay. Error bars represent the SE of three biological replicates. (TIF 173 kb)
Additional file 8: Figure S4.
A phylogenetic tree was constructed with MEGA 5.1 using the neighbor-joining (NJ) method with 1000 bootstrap replicates based on a multiple alignment of 78 amino acid sequences of PMEs from G. raimondii. The four major subfamilies are numbered I to IV. (TIF 1228 kb)
Additional file 9: Table S5.
Primer pairs used in quantitative real-time PCR analysis. (DOCX 12 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Li, W., Shang, H., Ge, Q. et al. Genome-wide identification, phylogeny, and expression analysis of pectin methylesterases reveal their major role in cotton fiber development. BMC Genomics 17, 1000 (2016). https://doi.org/10.1186/s12864-016-3365-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-016-3365-z