
Abstract
Objective
Biopharmaceutics Classification System says that simvastatin (SMV) is a Class II drug with low bioavailability (5%). This is because it dissolves slowly and is broken down a lot in the first pass. Simvastatin transdermal patches were produced as part of this study's aim to treat hyperlipidemia. The Box–Behnken design (three-factor, three-level) was selected for optimization of patches.
Methodology
The optimization design involved 15 runs with independent factors hydroxypropyl methylcellulose K100, Eudragit L100, and polyethylene glycol 400 percentage, and dependent factors including folding endurance and in-vitro drug release.
Results
The results showed that the concentration of hydroxypropyl methylcellulose K-100 positively impacted the patch’s folding endurance. The fact that factor C was the only scenario where the p-value was less than 0.05 and the coefficient value was higher in the in vitro drug release model means that it has a greater influence on the release of medicines. The patches were also evaluated for drug content, swelling, moisture uptake, moisture content, etc. The optimized patch shows an in vitro drug release of 55.3% in up to 24 h. In vivo antihyperlipidemic activity was evaluated in albino Wistar rats. In the standard treatment (simvastatin oral) groups, there is a decrease in cholesterol (132.76 ± 0.35) and triglyceride level (139.80 ± 76) whereas in the test formulation group or test group, there is also a decrease in cholesterol (169.65 ± 0.21 mg/dL) and triglyceride level (151.20 ± 31 mg/dL) level.
Conclusion
Based on in-vitro and in-vivo results it can be concluded that simvastatin patches can be an alternative to traditional therapy.
Similar content being viewed by others


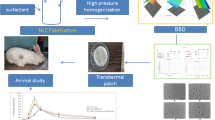
Background
There are considerable changes in plasma concentration when widely used dosage forms such as tablets, capsules, and oral liquids are administered in repeated doses. This fluctuation may lead to various adverse effects. Therefore, different modified release systems were successfully tested to minimize the fluctuation. There are several administration methods, including as oral, mucosal, inhalation, transdermal, and intravenous injection, depending on the delivery route. A promising method that can imitate the plasma concentration profile of intravenous infusion is the transdermal drug delivery system (TDDS), until the drug is present in the system [1]. In comparison to other routes of administration, the skin has many benefits, including the avoidance of gastric irritation, emptying rate effects and pH, hepatic first-pass metabolism, reduction of systemic side effects, sustained drug release, faster termination of therapy, reduced fluctuations in plasma levels, and avoidance of injection-related pain [2].
According to Zondek, when used topically as a 30 percent lanolin ointment, the external disinfectant chloroxylenol may be successful in treating urogenital infections [3,4,5] and open a new way to deliver the drug systemically through topical route. Over the past 40 years, a great deal of research has been done on transdermal administration of active pharmacological ingredients (API) [6]. A transdermal patch is a device that allows the API contained in it to travel through the skin and deliver medications to specific cells or organs [7]. The market for transdermal medication administration is about to shift into a new stage where pain relief isn't the primary driver [8]. Hormones, cardiovascular drugs, and drugs for the central nervous system are also administered through the transdermal system [8]. With the use of more recent drug delivery techniques that are effective, predictable, and safe, continuous delivery of a biologically active moiety is possible either systemically or to a specific site. They can perform better than traditional administration methods by monitoring the intensity, location, and length of the pharmacological action. A patch that is placed on the skin and includes medication that is meant to be absorbed into the bloodstream through the skin is called a transdermal patch [9], also known as a skin patch. Transdermal drug delivery has many benefits, including fewer side effects, less repetitive administration to achieve the desired plasma concentration, prolonged drug delivery, a reduction in the first-pass effect, and enhanced patient compliance, interruption of therapy when necessary etc. [10] All statin drugs have poor aqueous solubility and low oral bioavailability [11]. Due to their history of efficacy in adults, statins are one of the first-line medications considered for use in the pediatric population with dyslipidemia [12]. Simvastatin (SIM) inhibits 3-hydroxy-3-methylglutaryl coenzyme-A (HMG-CoA) reductase [13] and is commonly prescribed for cardiovascular illnesses due to its effects on decreasing lipid levels. Simvastatin enhances capillary morphogenesis in vitro and inhibits apoptosis in lymphatic endothelial cells, suggesting that it has lymphangiogenic properties that aid in the healing of wounds [14]. According to the Biopharmaceutics Classification System (BCS), SIM is a Class II [15] medication with low bioavailability (5%) due to its slow rate of solubility and extensive first-pass metabolism. SIM's poor oral bioavailability must be improved, which requires an increase in solubility. Although SIM has a short half-life, around two hours and a low water solubility (aqueous solubility 0.03 mg/L), it is removed by cytochrome P3A, which undergoes substantial metabolism in the liver and the intestines [16]. Delivery methods include transdermal, buccal, rectal, and parenteral are among the strategies that could be used to prevent first-pass metabolism. SIM is an excellent choice for the creation of transdermal films because to its strong first-pass metabolism, low molecular weight (418.56), high lipid solubility, low melting point (129 °C), capacity to act at low plasma concentrations [17]. The aim of this work to prepare a simvastatin transdermal patch (SIM-TP) and study its antihyperlipidemic activity in an animal model.
Methods
Simvastatin was gift from Biodeal Pharmaceutical Pvt Ltd. Himachal Pradesh, India. Hydroxypropylmethyl cellulose (HPMC K100) was gift from Colorcon Asia Pvt Ltd, Goa, India and Eudragit RL 100 was obtained from EvoniK Roehm Pharma Polymers and all other ingredients are of analytical grade.
Preparation of SIM-TP
The formulation of SIM-TP is shown in Table 1. The solvent casting procedure was used to prepare the transdermal patch. Ethanol: dichloromethane (1:1) was used to dissolve the polymers Eudragit L-100 and HPMC K100. The polymeric dispersion was stirred continuously with a magnetic stirrer until a clear solution formed. Polyethylene glycol 400 (PEG 400) was used to dissolve the drug before it was added to the polymer solution. After that, the homogeneous polymer solution containing the drug was added to a petri dish that had been lined with aluminum foil and allowed to air dry for 24 h at room temperature. The petri dish was covered with an inverted funnel to stop the solvent from evaporating too quickly. Desiccators were used to store the dried patches for future study.
Optimization of SIM-TP
The response surface design The Box–Behnken design was utilized to optimize the simvastatin transdermal patch (SIM-TP). The three-factor, three-level design was selected. The three independent variables were HPMC K100, Eudragit RL100 (ERL100), and PEG400. Table 2 displays the variables and their corresponding levels. The Design-Expert 13 software proposed a 15-run trial for the optimization process. The proposed design is shown in Table 3.
Evaluation of transdermal patch
Physical appearance
Every transdermal patch was visually inspected for color, smoothness, and uniformity.
Thickness
A screw gauge was used to measure the patches' thickness. Each batch of films had its average and standard deviation calculated based on five readings.
Folding endurance
Careful consideration was given to the prepared films' capacity for multiple folds in one location. The patch strip (2 × 2 cm2) was folded repeatedly till it broke. The maximum simultaneous folds the film could withstand before failing were counted [18].
pH determination
The transdermal films were incubated in a glass petri dish for an hour in 0.5 ml of double-distilled water to give them time to swell. The glass electrode was allowed to acclimate before being brought near to the film surface. The surface pH was then measured with a digital pH meter, and the results were noted [19, 20].
Swelling index
The patch was weighed after being submerged in a petri dish containing 50 ml of pH 7.4 phosphate buffer every 10 min. The following formula was used to determine the swelling's intensity [21].
where the weight of the patch both before and after it absorbs moisture is represented by Xo and Xt.
Drug content
Transdermal films were divided into pieces with a predetermined area (1 cm2), placed in a volumetric flask of 100 mL, and then dissolved in methanol. After being sonicated, the mixture was left for 24 h. A 0.45-µm membrane was used to filter the fluid. Using a UV spectrophotometer (Labindia), the amount of medication in the solution was determined at 237 nm, the absorbance was measured, and data were collected [22].
Moisture content
The produced films were weighed individually until they reached equilibrium weight after being stored in desiccators with fused calcium chloride for 24 h at 40 °C. After reweighing the film 24 h later, the moisture content percentage was calculated using the procedure below [23].
Moisture uptake
The weighted films stored in a desiccator with a saturated potassium chloride solution for 24 h at 40 °C in order to maintain a Relative Humidity of 84%. The films must be reweighed after 24 h to determine the amount of moisture absorbed [23, 24].
In-vitro drug release
The in-vitro release study was conducted using a Franz diffusion (FD) cell with a dialysis membrane (HIMEDIA LA 395–10 MT). Phosphate buffer pH 7.4 with sodium lauryl sulfate (SLS) (0.5% w/v) served as the dissolving media. A phosphate buffer was placed into the diffusion cell's receptor compartment. The dialysis membrane received a buffer rinse before the experiment. Between the donor and receiver chambers of the diffusion cell, the membrane was securely clamped in place. A constant 37 °C was maintained while the diffusion cell was continuously stirred at a speed of 50 rpm. Samples were taken periodically to maintain the sink condition, and each time, an equivalent volume of phosphate buffer was added to the receptor compartment. Using a UV–visible spectrophotometer, the samples' absorbance at 237 nm was measured, and the cumulative percent drug release was computed [25].
Scanning electron microscopy (SEM)
To analyze their surface topography and morphology, a prepared patch was coated with gold–palladium under an argon atmosphere at ambient temperature. The surface morphology can then be examined using SEM. SEM can be used to assess the ultra-structure of a broken micro sponge particle.
In-vivo activity (animal study)
Antihyperlipidemic activity
The animals used were albino rats (150–250 g, either sex). The group and their treatment were shown in Table 4. Prior to the experiment, each animal was fasted for 12 h. Blood samples were then collected from each animal's retro-orbital sinus using capillary tubes while they were all under diethyl ether anesthesia to determine their lipid profiles. After being extracted, the serum was centrifuged for 10 min at 5000 rpm before being stored at − 20 °C for analysis. Prior to the test, the abdomen's hair was plucked with a depilatory cream and then rinsed with distilled water. The animals were given anesthesia before being restrained to a supine position on the day of the trial. On the area of the belly that was hair-free, the pressure-sensitive adhesive transdermal patch (2.5 cm2) was applied. An oral feeding needle was used to provide the medicine solution (10 mg/kg). Following oral and transdermal medication, blood samples of around 0.5 ml were collected via a retro-orbital puncture at intervals of 0.5, 2, 4, 8, 12, 24, and 48 h. A glass tube containing heparin and the anticoagulant ammonium oxalate was filled with blood from a retro-orbital puncture through capillaries (1 percent solution). After being rapidly separated by microcentrifugation at 5000 rpm, the plasma was stored at − 20 °C. An autoanalyzer was used to analyze the samples [26].
Skin irritation studies
The ability of the patches to irritate or sensitize the skin of rats was evaluated. 150–250 g albino rats of either sex were used in the experiment. To minimize collateral damage, the dorsal surface of the rat was carefully groomed. The rat's skin was shaved, and the optimized patch was applied, secured with tape. The animal was observed for 24 h to look for any signs of erythema or edema [27, 28].
Results
Discussion
Formulation optimization
The experiment's design called for 15 runs for the set of variables. All the runs recommended by the DOE were developed and tested for responses like folding endurance and drug release. Table 5 displays the design of experimental runs along with their responses.
Folding endurance
Folding endurance is the ability of any patch to withstand pressure applied in the form of folding at the same spot without breaking. This is an important need for patches since activity causes the skin at the application site to stretch and contract repeatedly, and the patch needs to have the best folding endurance during those times. The folding endurance equation is represented by Eq. 1, and the ANOVA table is represented as Table 6.
The Model F-value of 3.24 indicates that there is a 6.43% possibility that noise may result in an F-value this significant. Lack of fit is not significant when compared to pure error, as indicated by the F-value of 0.48 for the lack of fit. A negligible lack of fit is advantageous for the model.
For model terms to be deemed significant, the P-value needs to be less than 0.05. In cases where the value exceeds 0.1, model terms are not meaningful. Folding endurance was significantly impacted by Factor A, or HPMC K-100 concentration (p-value 0.0151). According to Eq. 1, Factor A has the highest coefficient and is preceded by a positive sign, indicating that Folding endurance improved with increasing HPMC concentration. This means that patches with greater HPMC K100 percentages tend to have higher folding endurance. Although increasing the concentration of PEG and ERL100 has a effect on folding endurance, there is a slight increase in folding endurance due to this effect. Therefore, there is a correlation between higher polymer concentration and increased folding endurance. Figure 1 displays the 3D response surface diagram of folding endurance.

3D response surface diagram of folding endurance
In-vitro drug release
The formulation of various drug batches and their related in-vitro release percentages are displayed in Table 5. The formulation's key ingredients are HPMC K100, ERL 100, and PEG, which are altered to produce various release profiles. The amount of medication released from the formulation during a specific time under simulated physiological conditions is measured by the in-vitro release percentage. To achieve the appropriate release profile for the medicine, the formulation can be optimized using the data. Table 7 displays a summary of the model fitting. The linear model for in-vitro drug release was anticipated by the software. Based on its Model F-value of 4.76, the model is deemed significant. Only 2.30 percent of the time is there a probability that noise may result in an F-value this large. The F-value of 1.0 for the lack of fit indicates that it is not significant when compared to the pure error. A non-significant lack of fit is advantageous for the model.
The in-vitro drug release ANOVA table is shown in Table 8. The in-vitro drug release model's p-value of 0.0230, which is less than 0.05, shows that it is significant.
The in-vitro drug release model's ANOVA table reveals that factor C, or PEG400 concentration, is the only scenario where the p-value is less than 0.05. PEG400 was used as a permeation enhancer in this optimization design. The increased PEG400 concentration will result in increased in-vitro drug release. Equation 2 represented in terms of coded factors allows one to predict the results for specific values of each variable. The relative importance of the variables can be determined by comparing the factor coefficients using the coded equation. Factors A and B are preceded by a negative sign in the equation, and factor C is preceded by a positive sign. An increase in the value of factor C leads to an increase in the release of drugs in vitro, which is what the positive sign denotes as a synergistic effect. The fact that C has a higher coefficient value means that it has a greater influence on the release of medicines.
The optimized value for factors and their response were predicted by software and shown in Table 9. Based on criteria, the software predicted that the optimized patch's levels of factors A, B, and C would be 1.80, 0.1, and 50, respectively, and that its responses to folding endurance and in-vitro drug release would be 266.36 and 53.17, respectively. An optimized patch of the specified level of factors was constructed, and its performance was assessed.
Thickness and physical appearance
Table 9 displays the thicknesses of all formulations. The resulting patches were homogenous, opaque, pliable, and smooth at varying polymer concentrations. The thickness of the films ranged from 0.52 ± 0.76 to 0.625 ± 0.85 mm.
Swelling index
The formulation and related swelling index of various samples are shown in Table 10 (F1–F15). The swelling index calculates how much the samples swell in a certain solvent. Sample F12 had the highest swelling index of 62.38 ± 0.69%, while sample F2 had the lowest swelling index of 43.45 ± 0.86%. The research indicates that different formulations may cause varying levels of edema and emphasizes the significance of formulating products that are best suited for a given application.
Moisture content
Each formulation had a different moisture content ranging from 5.1 ± 0.43% to 8.9 ± 0.51%. All formulas had a mean moisture content of 6.6%, with a standard deviation of 1.27%. Because of the hydrophilic characteristic of HPMC, it was found that high concentrations of HPMC had higher % moisture contents.
Moisture uptake
With a standard deviation of 0.49–0.72, each formulation has a distinct moisture uptake value that spans from 6.2 to 11.6 percent. Patches with high HPMC content exhibited high moisture absorption.
Drug content
The percentages of drugs present in each formulation, designated as F1 through F15, are shown in the Table 9. These percentages show how many active medication components there are in each formulation (measured in mass). The F4 and F14 contain the lowest percentages of drug content, while the F11 and F12 have the highest amounts. The medication concentration in the other formulations is between 90.76 and 98.75%. Pharmaceutical companies employ formulations to enhance the efficacy and delivery of their products (Figs. 2, 3).

3D response surface diagram of in-vitro drug release

Overlay plot of the design predicting the values of individual factors and their response based on input criteria
In-vitro drug release
The in vitro release profile is a vital tool for predicting how drugs will behave in vivo. Release analyzes are necessary to forecast the repeatability of the rate and duration of pharmaceutical activity. Figure 4 displays the optimized transdermal patch's percentage medication release over the course of 24 h. Over time, the cumulative proportion of medication release gradually rises, reaching its peak at 24 h (55.3%). The variation in drug release characteristics among all patch formulations could perhaps be attributed to the presence of polymeric chain cross-linking networks. Different polymeric mixtures used to make transdermal patches have different diffusion pathways, which affect the delivery and intensity of the dispersion.

In-vitro drug release of optimized simvastatin patch
In-vivo study
Albino adult male and female in good health Wistar rats weighing between 110 and 240 g were used. Figure 5 depicts the impact of the simvastatin patch that has been optimized on total cholesterol levels. Rats were effectively exposed to Triton to cause hyperlipidemia, which was visible in the disease-control group. In comparison to normal controlled animals, disease-control animals have higher cholesterol and triglyceride levels (173.200.12 mg/dl and 189.02.68 mg/dl, respectively). The cholesterol level was seen to slightly decline in the test formulation. The levels of cholesterol and triglycerides are shown in the Fig. 5. The NC group has the lowest levels of both cholesterol and triglycerides, with average levels of 89.03 ± 0.89 mg/dL and 79.80 ± 0.28 mg/dL, respectively. The DC group rats treated with triton has the highest levels, with average levels of 173.20 ± 0.12 mg/dL for cholesterol and 189.02 ± 0.68 mg/dL for triglycerides, indicating the induction of hyperlipidemia and a higher risk for cardiovascular disease. In the standard treatment (simvastatin oral) groups, there is a decrease in the cholesterol (132.76 ± 0.35) and triglyceride level (139.80 ± 76) whereas, in the test formulation group or test group, there was also a decrease in cholesterol (169.65 ± 0.21 mg/dL) and triglyceride level of (151.20 ± 31 mg/dL) level. The triglyceride level in the standard treatment group and test group were comparable.

Effect of simvastatin patch on A Triglyceride B Cholesterol in triton-induced hyperlipidemia in rats
Scanning electron microscopy study
Any material's morphological or surface changes can be evaluated using scanning electron microscopy. Various magnifications of the SEM images of the optimized patches before and after drug release were displayed in Fig. 6. Prior to medication release, the optimized simvastatin patch may seem uniformly smooth in SEM pictures. At 10 µm magnification, the patch's surface exhibits a matrix structure with uniformly dispersed drug particles; nevertheless, at 50 µm magnification, the patch surface seems to be nearly smooth. To regulate the medication's release rate, thin film coatings might also be present on top of the drug matrix.

SEM images of optimized simvastatin patch before drug release (A, B) and after drug release (C, D)
The improved simvastatin patch's SEM pictures after drug release might reveal a rougher surface with fewer drug particles. Contrary to the previous photograph, the drug particles that were remaining on the surface were distributed unevenly. Drug elution may cause wear and tear on the patches, and drug release is confirmed by some porous forms on the surface of the patches, which demonstrate that the medication was gone out of the patches.
Conclusion
The formulation of SIM-TP was prepared by the solution casting method using the polymers Eudragit L-100 and HPMC K100. The Design-Expert 13 software proposed a 15-run trial for the optimization process. The three independent variables were HPMC K100, Eudragit RL100 (ERL100), and polyethylene glycol 400 (PEG400). Physical Appearance, Thickness, Folding Endurance, and pH Determination were evaluated. The results showed that With a p-value of 0.0151, factor A—the concentration of HPMC K-100—had a noteworthy effect on folding endurance. A higher concentration of polymers was associated with greater folding endurance. The linear model for in-vitro drug release is significant, as evidenced by the model's p-value of 0.0230, which is less than 0.05. Factor C, or PEG400 concentration, is the only scenario where the p-value is less than 0.05. The factor coefficients can be compared using the coded equation to get the relative significance of the variables. The design expert 13 program was used to predict the values of factors A, B, and C and their responses to folding endurance and in-vitro drug release. The optimized patch's levels of factors A, B, and C were 1.80, 0.1, and 50, respectively, and its responses to folding endurance and in-vitro drug release were 266.36 and 53.17%, respectively. In-vitro drug release, in-vivo antihyperlipidemic action, and scanning electron microscopy were used to assess the improved patch. The improved patch released 55.30 percent of the medication during a 24-h period, which corresponded to the projected value. Transdermal patches were evaluated for thickness, physical appearance, swelling index, moisture content, and moisture uptake. All formulations had a mean moisture content of 6.6 percent, with a standard deviation of 1.27. The in-vivo activity of the optimized simvastatin patch was observed in albino adult male and female in good health Wistar rats, with the NC group having the lowest levels of cholesterol and triglyceride levels. The DC group rats treated with triton had the highest cholesterol and triglyceride levels, while the standard treatment (simvastatin oral) groups had a decrease in cholesterol and triglyceride levels. Scanning electron microscopy showed that the optimized simvastatin patch had a rougher surface with fewer drug particles after drug release. Drug elution may cause wear and tear on the patches, and drug release is confirmed by some porous forms on the surface. Based on the results, it can be concluded that the tailored simvastatin patch can be an alternative to treat hyperlipidemia.
Availability of data and materials
The data that support the findings of this study are available from the corresponding author, upon reasonable request.
Abbreviations
- SIM:
-
Simvastatin
- SIM-TP:
-
Simvastatin transdermal patch
- SEM:
-
Scanning electron microscopy
- HPMC:
-
Hydroxy methyl propyl cellulose K100
References
Jeong WY, Kwon M, Choi HE, Kim KS (2021) Recent advances in transdermal drug delivery systems: a review. Biomater Res 25:24. https://doi.org/10.1186/S40824-021-00226-6
Prajapati ST, Patel CG, Patel CN (2011) Formulation and evaluation of transdermal patch of repaglinide. ISRN Pharm 2011:1–9. https://doi.org/10.5402/2011/651909
Pastore MN, Kalia YN, Horstmann M, Roberts MS (2015) Transdermal patches: history, development, and pharmacology. Br J Pharmacol 172:2179–2209. https://doi.org/10.1111/BPH.13059
Zondek B (1942) Chemotherapeutical use of halogenized phenols as external disinfectants. Nature 149:334–335. https://doi.org/10.1038/149334a0
Pastore MN, Kalia YN, Horstmann M, Roberts MS (2015) Transdermal patches: history, development and pharmacology. Br J Pharmacol 172:2209. https://doi.org/10.1111/BPH.13059
Subedi RK, Oh SY, Chun MK, Choi HK (2010) Recent advances in transdermal drug delivery. Arch Pharm Res 33:339–351. https://doi.org/10.1007/S12272-010-0301-7/METRICS
Fong Yen W, Basri M, Ahmad M, Ismail M (2015) Formulation and evaluation of galantamine gel as drug reservoir in transdermal patch delivery system. Sci World J 2015:1–7. https://doi.org/10.1155/2015/495271
Tanner T, Marks R (2008) Delivering drugs by the transdermal route: review and comment. Skin Res Technol 14:249–260. https://doi.org/10.1111/J.1600-0846.2008.00316.X
Richhariya A, Rohit N, Singh Dangi Y (2022) brief review on transdermal patches: present & future prospective. Int J Mod Pharm Res 6:40
de Barros NR, Chagas PAM, Borges FA et al (2015) Diclofenac potassium transdermal patches using natural rubber latex biomembranes as carrier. J Mater 2015:1–7. https://doi.org/10.1155/2015/807948
Subramaniam S, Devi M, Rajesh C et al (2016) Preparation, evaluation, and optimization of atorvastatin nanosuspension incorporated transdermal patch. Asian J Pharm 10:S487–S491
Zidan AS, Hosny KM, Ahmed OAA, Fahmy UA (2016) Assessment of simvastatin niosomes for pediatric transdermal drug delivery. Drug Deliv 23:1536–1549. https://doi.org/10.3109/10717544.2014.980896
Newman A, Clutterbuck RD, Powles RL et al (2009) A comparison of the effect of the 3-hydroxy-3-methylglutaryl coenzyme a (hmg-coa) reductase inhibitors simvastatin, lovastatin, and pravastatin on leukaemic and normal bone marrow progenitors. Leuk Lymphoma 24:533–537. https://doi.org/10.3109/10428199709055590
Rezvanian M, Mohd Amin MCI, Ng SF (2016) Development and physicochemical characterization of alginate composite film loaded with simvastatin as a potential wound dressing. Carbohydr Polym 137:295–304. https://doi.org/10.1016/J.CARBPOL.2015.10.091
Bakhaidar RB, Naveen NR, Basim P et al (2022) Response Surface Methodology (RSM) powered formulation development, optimization, and evaluation of thiolated based mucoadhesive nanocrystals for local delivery of simvastatin. Polymers (Basel) 14:5184. https://doi.org/10.3390/POLYM14235184/S1
Tiwari R, Pathak K (2011) Nanostructured lipid carrier versus solid lipid nanoparticles of simvastatin: comparative analysis of characteristics, pharmacokinetics, and tissue uptake. Int J Pharm 415:232–243. https://doi.org/10.1016/J.IJPHARM.2011.05.044
Anod HV, Gupta NV, Gowda DV, Manohar M (2018) Preparation and evaluation of simvastatin transdermal film. Int J Appl Pharm 10:235–238. https://doi.org/10.22159/IJAP.2018V10I5.26657
Singh A, Bali A (2016) Formulation and characterization of transdermal patches for controlled delivery of duloxetine hydrochloride. J Anal Sci Technol 7:1–13. https://doi.org/10.1186/s40543-016-0105-6
Humama F, Shalini S (2015) Development and evaluation of transdermal therapeutic system of an antihypertensive drug. Int Res J Pharm 6:213–218. https://doi.org/10.7897/2230-8407.06347
Rehman A, Iqbal M, Khan BA et al (2022) Fabrication, in vitro, and in vivo assessment of eucalyptol-loaded nanoemulgel as a novel paradigm for wound healing. Pharmaceutics. https://doi.org/10.3390/pharmaceutics14091971
Nayak BS, Ellaiah P, Pattanayak D, Das S (2011) Formulation design preparation and in vitro characterization of nebivolol transdermal patches. Asian J Pharm. https://doi.org/10.22377/AJP.V5I3.104
Serrano Castañeda P, Juan Escobar-Chávez J, Aguado AT et al (2017) Design and evaluation of a transdermal patch with atorvastatin. Farmacia 65:908–916
Madan RJ, Argade SN, Dua K (2015) Formulation and evaluation of transdermal patches of donepezil. Recent Pat Drug Deliv Formul 9:95–103. https://doi.org/10.2174/1872211308666141028213615
Akhlaq M, Arshad MS, Mudassir AM et al (2016) Formulation and evaluation of anti-rheumatic dexibuprofen transdermal patches: a quality-by-design approach. J Drug Target 24:603–612. https://doi.org/10.3109/1061186X.2015.1116538
Shehata T, Mohafez O, Hanieh H (2018) Pharmaceutical formulation and biochemical evaluation of atorvastatin transdermal patches. Indian J Pharm Educ Res 52:54–61. https://doi.org/10.5530/ijper.52.1.6
Brito Raj S, Chandrasekhar KB, Reddy KB (2019) Formulation, in-vitro and in-vivo pharmacokinetic evaluation of simvastatin nanostructured lipid carrier loaded transdermal drug delivery system. Futur J Pharm Sci 5:1–14. https://doi.org/10.1186/s43094-019-0008-7
Patel J, Jani R (2016) Enhancing effect of natural oils as permeation enhancer for transdermal delivery of diltiazem hydrochloride through Wistar rat skin. Int J Pharm Sci Rev Res 36:9–16
Jain S, Goswami M, Bhandari A, Arora V (2011) Skin irritation study of intradermal patch of chitosan containing trazodone-HCl on rat skin. Int J Res Pharmaceut Biomed Sci 2:1082–1084
Funding
This research received no funding.
Author information
Authors and Affiliations
Contributions
All authors contributed to this work. N: Research Scholar, Collection of data, manuscript writing. AAand: Concept and designing, manuscript writing. DP: in-vivo activity and manuscript writing. Thank you very much for considering our manuscript for publication.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Animal study was endorsed by the Institutional Animal Ethics Committee (IAEC) Hygia Institute of Pharmaceutical Education & Research, Lucknow, Uttar Pradesh under IAEC Approved no. HIPER/IAEC/95/02/2022.
Consent for publication
The authors declare no conflict of interest.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Neetu, Anand, A. & Panjwani, D. Optimization of simvastatin transdermal patch for hyperlipidemia treatment in rat model. Futur J Pharm Sci 10, 31 (2024). https://doi.org/10.1186/s43094-024-00606-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s43094-024-00606-4