
Abstract
Background
The unifying hypothesis of competing endogenous RNA (ceRNA) wherein crosstalk between coding (mRNAs) and long non-coding RNAs (lncRNAs) via microRNA (miRNA) response elements, creates a pervasive regulatory network across the transcriptome, has been implicated in complex disorders including schizophrenia. Even with a wide range of high-throughput data, the etiology of schizophrenia remains elusive, necessitating a more holistic understanding of the altered genetic landscape, shifting focus from solely candidate gene studies and protein-coding variants.
Objective
We developed lncRNA-associated ceRNA networks to elucidate global molecular/regulatory signatures underlying schizophrenia using diverse data in the public domain.
Methods
Microarray dataset associated with peripheral blood mononuclear cells (PBMCs) of schizophrenia and control patients was used to identify differentially expressed mRNAs. Weighted gene co-expression network analysis (WGCNA) was used to identify highly correlated hubs, and genes from these overlap** Kyoto Encyclopedia of Genes and Genomes (KEGG) and gene ontology (GO) term genesets were considered key mRNA players. StarBase, Human MicroRNA Disease Database, and miRWalk were used to derive mRNA-miRNA and miRNA-lncRNA relationships. Finally, the key mRNAs, interacting lncRNAs and miRNAs were chosen to reconstruct sub-ceRNA networks based on network centrality scores.
Results
Bioinformatics analysis revealed the involvement of three differentially expressed mRNAs, namely ADRA1A, HAP1 and HOMER3 in the schizophrenia ceRNA networks with lncRNAs NEAT1, XIST, and KCNQ1OT1 modulating their activity by a suggestive sequestering of miR-3163, miR-214-3p and miR-2467-3p, respectively.
Conclusions
Furthermore, based on contextual evidence, we propose how ceRNAs could orchestrate crosstalk between neurostructural dynamics and immune/inflammatory processes and enable unifying these disparate models of schizophrenia etiology.
Similar content being viewed by others


Introduction
Schizophrenia presents itself as a severe psychiatric disorder in early to late adolescence with a complex manifestation of positive symptoms (e.g., hallucinations and delusions), negative symptoms (e.g., social withdrawal, blunted affect) and pervasive cognitive deficits associated with functional decline [1, 2]. Though not as common as other psychiatric disorders with a lifetime prevalence of \(0.32\%\) [3] affected worldwide, it executes a high toll on society via multifaceted indices such as economic liabilities, human rights violations and suicides [4, 5]. Its pathophysiology is perplexing as uncovered through advanced genomic [6], developmental neurobiology [7], and systems biology investigations [8, 9] with irregularities in cellular, molecular, neuroanatomical, and neurophysiological domains [10] being implicated. This diverse milieu of factors and their enigmatic interplay contributes to its elusive etiology.
As far as the genetics of schizophrenia are concerned the strong links between schizophrenia and over \(100\) susceptibility loci, along with identified CNVs and SNVs, show promise. Thousands of common alleles with small effects collectively contribute substantially to schizophrenia risk. These findings may lead to new therapeutic insights. However, we must remember: (i) associations between genetic variants and schizophrenia do not necessarily imply causal pathways; (ii) many associations extend beyond schizophrenia to other mental disorders. The specifics of schizophrenia's origins and genotype-environment interactions are largely unknown, warranting caution in assessing the various genetic contributors to its development [11]. The evolution of pharmacological interventions to alleviate the patient's condition has been extremely slow since the advent of the first antipsychotics [12]. Efforts toward biomarker discovery for early identification of individuals at risk, improving diagnostic accuracy and precision, predicting treatment response, and to obtain new druggable targets are notable [13]. However, the prevalent body of work regarding this has mainly focused on the coding part of the genome but the mosaic manifestation of schizophrenia warrants a far more holistic understanding of the underlying pathophysiology.
Toward this end, integrating existing knowledge of schizophrenia etiology with the recent shift in our understanding of the ncRNAs as junk/black matter of the genome to being vital regulatory molecules is of prime importance [14,15,16]. By virtue of their ability to silence/alter expressions of multiple targets simultaneously, ncRNAs can affect entire signaling pathways [17]. Not surprisingly, understanding the role of ncRNAs in physiological and pathological conditions has significantly enriched our recognition and understanding of alternate/additional molecular pathways involved. More than a hundred miRNAs and though in its infancy \(\sim 30\) lncRNAs have been found dysregulated across the pre-frontal cortex, superior temporal gyrus, parietal cortex, amygdala, serum, and peripheral blood in schizophrenia [18]. SNPs in both miRNAs and lncRNAs have also been shown to have significant associations with the schizophrenia phenotype [19]. Collectively, substantial evidence has been generated as to the dysregulation of ncRNAs in schizophrenia and that the disruption of the networks they regulate is critical to schizophrenia as they are enriched with target genes pertaining to various neurophysiological processes [20, 21]. Considering all these, a new paradigm wherein the clinical utility of ncRNAs in the form of a next-generation is now being seriously assessed [22].
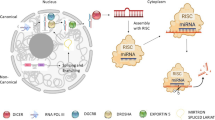
Nevertheless, making sense of the huge amount of data on ncRNAs in the public domain for targeted therapeutics is a herculean task [23,24,25,26]. Integrating the raw sequencing data from multiple sources and fitting them under logical models is one way of understanding how lncRNAs collectively interact with the coding genome. A possible route is the ceRNA theory, wherein ncRNAs such as lncRNAs that share common MREs with mRNAs can act as siphons and competitively sequester miRNAs, thus forming a complex regulatory network [27]. Any differential expression in these RNAs harboring common MREs could thereby lead to imbalances in the regulatory network and disease development [28,29,30]. ceRNA networks developed for diseases such as sarcopenia have identified lncRNAs, mRNAs and miRNAs which add to disease risk with very high accuracy [31]. Several lines of evidence have already shown a close association of ceRNA networks with several forms of cancer but very little is known for schizophrenia [\({\text{log}}_{2}(\text{fold change})\) values of all genes between schizophrenia and normal patients via limma package in R [41]. All p-values were also corrected via BH method in R. The genes were considered as differentially expressed corresponding to a \(\text{BH}-p-\text{value}<0.01\) and \(\left|{\text{log}}_{2}(\text{fold change})\right|>0.5\) [42]. DEGs having \({\text{log}}_{2}\left(\text{fold change}\right)>0.5\) and \({\text{log}}_{2}\left(\text{fold change}\right)<-0.5\) were designated as upregulated and downregulated, respectively. PCA method was utilized to assess the sample aggregation degree. It is an unsupervised method that can be used to understand the difference between two or more sample groups [43, 44]. Unsupervised PCA/dimensionality reduction was performed via R software based on the DEGs expression with respect to samples [3B–D). As observed, mRNA expression levels of all three hub genes were significant across control and schizophrenia samples.

A Overlap** hub genes between significant GO-BP, GO-MF, GO-CC, and pathway genesets. The red, blue, yellow, and green-colored areas signify KEGG, GO-BP, GO-MF, GO-CC genesets, respectively. Box-and-whisker plots showing expression intensity distribution of B HAP1, C HOMER3, D ADRA1A across control and schizophrenia patient samples. The top and bottom of the boxes signify 75th and 25th percentile of distribution. Horizontal lines within the boxes represent the median values while minimum and maximum values label the axes endpoints. P-values shown at the top of boxplots represent significance levels between sample groups for each hub gene
Schizophrenia-associated 3-node ceRNA network construction and topological analysis
The schizophrenia-associated 3-node ceRNA network comprised \(767\) nodes and \(3030\) edges as shown in Fig. 4. The breakup of nodes and edge pairs is summarized in Table S6. Tables S7−S8 shows top \(3\) lncRNAs and miRNAs within ceRNA network ranked based on betweenness, degree, and closeness centralities. Within this network, degree of lncRNAs, miRNAs and mRNAs ranged from \(1\) to \(117\), \(2\) to \(50\), and \(24\) to \(90\),respectively. Average degrees of lncRNAs, miRNAs, and mRNAs were \(4.64\), \(21.18\), \(47.66\), respectively. As observed from these centralities, ADRA1A hub gene was repressed and regulated by maximum miRNAs while lncRNA NEAT1 interacted with the highest number of miRNAs. Numerous miRNAs, lncRNAs, and mRNAs participating in higher-order subnetwork motifs were observed and the top three higher-order subnetwork motifs based on highest centrality scores of betweenness, degree, and closeness have been reported. The first-ranked subnetwork motif comprised one miRNA (miR-3163), one lncRNA (NEAT1), and one hub gene (ADRA1A). The second-ranked subnetwork motif comprised one miRNA (miR-214-3p), one lncRNA (XIST), and one hub gene (HOMER3). And, the third-ranked subnetwork motif comprised one miRNA (miR-2467-3p), one lncRNA (KCNQ1OT1), and one hub gene (ADRA1A) (Fig. 5A–C). Figure S6 shows centrality distributions like betweenness, closeness, ND, TC, NC, and ASPL of \(3\)-node ceRNA network.

Schizophrenia-associated 3-node ceRNA network comprising 767 nodes and 3030 edges. Magenta-colored diamond nodes represent the lncRNAs, red circular nodes represents the miRNAs, and green-colored octagonal nodes represents the hub genes

A Top higher-order subnetwork motif based on betweenness, degree, and closeness comprising one miRNA (miR-3163), one lncRNA (NEAT1), and one hub gene (ADRA1A). B The second higher-order subnetwork motif comprising one miRNA (miR-214-3p), one lncRNA (XIST), and one hub gene (HOMER3). C Third higher-order subnetwork motif comprising one miRNA (miR-2467-3p), one lncRNA (KCNQ1OT1), and one hub gene (ADRA1A). Magenta-colored diamond nodes represent the lncRNAs, red circular nodes represents the miRNAs, and green-colored octagonal nodes represents the hub genes
Discussion
Rare/ultra-rare protein-coding variants de novo or captured through WES of familial forms of schizophrenia have provided insights into a few genes from dopaminergic and neurodevelopmental pathways in schizophrenia [69,70,71,72] but heritability and etiology remain unexplained. Regulatory variants such as ncRNAs are emerging to be essential players in our understanding of the biology/etiology of common conditions such as schizophrenia [73, 74]. As one of the most common types of ncRNAs, lncRNAs are believed to play a pivotal role in the ceRNA machinery and elicit a significant effect in both physiological and pathological mechanisms. Multiple lines of evidence have implicated them in various psychiatric disorders [75,76,77]. Their differential expression in tissue, cell types, and developmental levels indicates that lncRNA expression is tightly regulated [78,79,80]. These give further credence to the idea that lncRNA-associated ceRNA networks may play a crucial role in schizophrenia etiology. However, the dire lack of studies on lncRNAs in the public domain has made it difficult for bioinformatic analyses to annotate their role in disease biology sufficiently. This is exacerbated further by the dearth of HTSeq studies in schizophrenia, including lncRNAs and the lack of postmortem data. To account for these, we employed an approach to shortlist coding genes and their interacting miRNAs and then extract the lncRNA-miRNA interactions reported in the public domain, thus making the mRNA-lncRNA-disease interaction hypothesis-free.
Furthermore, recent studies have indicated the link between brain and periphery via the circulatory system, which contains secreted regulatory molecules and hormones produced in the diffused NES that impact the peripheral markers' gene expression pattern [81,82,83,84]. These findings confirm that schizophrenia is a systemic disorder and support the notion that biomarkers in peripheral samples such as WB, PBMCs, lymphoblasts and olfactory epithelium may be insightful. Another line of evidence that dictated the choice of blood expression profiles for the analysis was based on the current evidence wherein immune/inflammatory processes are located in the disorder [33,34,35] and the strong connections established between altered immunity and lncRNAs [36, 85]. Understanding the networks at play in the peripheral system might help generate a holistic view of the underlying connection.
In the enrichment analysis using the highest-order WGCNA module, three genes were found overlap** in all the significant pathway and GO term libraries tested.
ADRA1A was enriched for terms such as neuroactive ligand-receptor interaction, cytosolic \({\text{Ca}}^{2+}\) ion transport, positive regulation of GABAergic synaptic transmission. HOMER3 was enriched for glutamatergic synapse and G protein-coupled glutamate receptor binding and was a cellular component of dendrites. HAP1 was associated with pathways involved in neurodegeneration, neurogenesis, neurotrophin binding and similar to HOMER3 was found located in the dendrites. All of the pathways have either been directly reported in schizophrenia etiology before or are of substantial importance in the processes involved in schizophrenia pathophysiology [86,87,88,89,90,91]. Thereby, any perturbation in their expression could potentially disturb these pathways and initiate or maintain the disease phenotype. With this hypothesis, we assessed the lncRNAs and miRNAs that could putatively dysregulate these key mRNAs leading to disease manifestation.
Among the three lncRNAs, NEAT1 has recently been reported to be upregulated [\({\text{log}}_{2}(\text{fold change})\) \(>2\)] in Brodmann area 46, hippocampus and striatum. NEAT1 is highly enriched in the mammalian brain and is an indispensable structural component of paraspeckles which are membrane-less cellular bodies involved in several cellular processes such as splicing and transcriptional modulation through chromatin structure modifications with emerging evidence suggesting their altered abundance with several innate immune activating responses stimuli such as sequestering to IFNGR1 [92]. NEAT1 itself has been cited as lncRNA-type immunoregulator (i) affecting monocyte-macrophage functions and T cell differentiation [93], (ii) assembly of inflammasomes by recruitment, maturation, and stabilization of CASP1 in activated macrophages [94], (iii) elicits pro-proliferative and anti-apoptotic roles and migration, invasion, and inflammatory cytokines secretion [95], (iv) exhibits innate immunity responses against viral infections [96]. Furthermore, multiple studies have shown miRNA sequestering tendencies of NEAT1, thereby attenuating target gene activity [97,98,99,100,101]. Based on our bioinformatic analyses, we propose that NEAT1 could be sequestering miR-3163 because of its higher number of transcripts in the disease state, thereby elevating the repression of ADRA1A which was downregulated in cases in the DEA (Fig. 3B).
The second lncRNA, XIST has also been previously associated with multiple mental disorders [102,103,104]. XIST is involved in the inactivation of the X chromosome, which has a long-standing reputation for harboring genes important for brain development and function [105]. Outside of its silencing roles, XIST i) stimulates proliferation and differentiation of naive \({\text{CD}4}^{+}\) T cells [106], (ii) is delocalized in B cells of female-biased autoimmunity [107], (iii) in-part promotes \({\text{CD}11\text{c}}^{+}\) atypical B cell formation [107], and (iv) has been shown to perturb PDL1 levels by probable competitive binding of miR-34a-5p [108]. Even though the expression profile of HOMER3 in schizophrenia is unknown, HOMER1 (member of the three-member HOMER family) has been shown to be up and downregulated in schizophrenia depending on the tissue type with variants in both found to be associated with schizophrenia [109]. Overexpression of XIST has been reported in bipolar disorder and major depressive disorder (phenotypes closely associated with schizophrenia) as well but is highly tissue-specific [102]. Therefore, in a similar fashion as NEAT1, XIST could be competitively sequestering to miR-214-3p, a miRNA already known to target the Qki [110], thereby leading to the altered HOMER3 levels.
The third lncRNA, KCNQ1OT1, targeting KCNQ1, though actively involved in epigenetic phenomenon via chromatin modifications, HMT G9a, and PRC2 [111], has no direct association with schizophrenia yet. However, it does seem to sponge miR-15a, leading to immune evasion and malignant progression of prostate cancer via upregulating PDL1, an essential immune checkpoint [112]. This might explain its expression correlation with \({\text{CD}4}^{+}\), \({\text{CD}8}^{+}\), and cytotoxic T cell levels and several other immune cell subsets in another ceRNA reported in colorectal cancer [85]. Furthermore, it might be indirectly associated with increased sudden cardiac arrest in schizophrenia patients [113].The KCNQ1 protein forms functional potassium channels [114]. Multiple lines of evidence, structural variants and mice knockouts, have shown KCNQ1 to be associated with LQT1, a condition synonymous with increased adverse cardiac events [115,116,117]. It is established that all atypical antipsychotics affect the cardiac potassium pump and that about \(\sim 6-10\text{\%}\) of schizophrenia patients show a longer QT interval under treatment [118, 119]. We propose that the expression of KCNQ1 as dictated by altered KCNQ1OT1 levels could be a putative cause of these adverse drug reactions.
Further investigations into this aspect could potentially lead to pre-emptive treatment strategies. Though the levels of KCNQ1OT1 in schizophrenia are not known, we can extrapolate from available knowledge that rs8234 [120] leads to lower expression of KCNQ1 and is also associated with reduced processing speed, reduced white matter FA and higher risk for schizophrenia [121], thereby implying that lower levels of KCNQ1 are associated with these impaired phenotypes. Elevated KCNQ1OT1 levels could also propagate this scenario. Considering these derived associations, studies establishing KCNQ1OT1 levels in schizophrenia could be informative.
We could thereby imply that elevated KCNQ1OT1 transcripts could be competitively sequestering to miR-2467-3p and inhibiting the expression of ADRA1A, thereby leading to its downregulated state in the DEA. Interestingly, ADRA1A is also associated with several cardiac conditions [122,123,124]. This gives a glimpse into the intricate mechanism in which ceRNAs could be acting in the pathophysiology of schizophrenia.
In conclusion, this study identified lncRNAs NEAT1, XIST and KCNQ1OT1-associated ceRNA networks which could be potentially relevant to schizophrenia by interacting with schizophrenia-relevant genes, ADRA1A and HOMER3. Furthermore, the affinity of the mRNAs to neurodevelopmental processes and that of the lncRNAs to immune/inflammatory processes might indicate a mechanism to unite the two most significant models proposed in schizophrenia etiology. Of note, though the current analyses is based on data specific to schizophrenia, neuroinflammation and its effect on neurodynamics is a well-established phenomenon in a variety of psychiatric illnesses such as depression, bipolar depression and obsessive–compulsive disorder [125]. ceRNAs established through this study and new ones discovered by using similar methods have the potential of uncovering further such pathways. Further refinements in such prediction strategies have the potential of unveiling additional interactions in schizophrenia biology, which, eventually, systems biology approaches coupled with artificial intelligence and machine learning technologies can integrate into a holistic picture. However, it is also important to note that the prediction strategy deployed in this study does not take into account the miRNA and potential ceRNA expression levels. This is important as it is well-established that both miRNAs and ceRNAs have temporal, spatial, and disease-specific expression patterns. Furthermore, studies have shown that ceRNAs and miRNAs with concentrations within a particular range are capable of eliciting such crosstalks. Even though we have provided evidence to give strong credence to the highlighted ceRNA axes, these must still be validated by qRT-PCR, luciferase reporter systems and co-IP assays. Furthermore, we have discussed in favor of the standalone components of the ceRNA networks. We believe that additional investigations into their roles in the diseased state would be valuable in assessing their role as important biomarkers for schizophrenia. Further wet lab experimentations would be an asset in proving the efficacy and accuracy of the predicted biomarkers. Also, design of lead compounds as potential drugs post successful clinical trials could be helpful for the treatment of schizophrenia in near future.
Data availability
The data underlying this article is available in NCBI-GEO at https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE66890 and can be accessed with GSE66890.
Abbreviations
- ncRNA:
-
Non-coding RNA
- lncRNA:
-
Long non-coding RNA
- SNP:
-
Single nucleotide polymorphism
- miRNA:
-
MicroRNA
- mRNA:
-
Messenger RNA
- ceRNA:
-
Competing endogenous RNA
- MRE:
-
MiRNA response element
- GEO:
-
Gene expression omnibus
- NCBI:
-
National Center for Biotechnology Information
- DEA:
-
Differential expression analysis
- QC:
-
Quality check
- HGNC:
-
HUGO Gene Nomenclature Committee
- BH:
-
Benjamini-Hochberg
- DEGs:
-
Differentially expressed genes
- WGCN:
-
Weighted gene co-expression network
- WGCNA:
-
Weighted gene co-expression network analysis
- ME:
-
Module eigengene
- MEdiss:
-
MEdissimilarity
- k.in:
-
Intramodular connectivity
- MM:
-
Module membership
- PPIN:
-
Protein–protein interaction network
- STRING:
-
Search tool for the retrieval of interacting genes/proteins
- GO:
-
Gene ontology
- BP:
-
Biological process
- MF:
-
Molecular function
- CC:
-
Cellular compartment
- KEGG:
-
Kyoto encyclopedia of genes and genomes
- ENCORI:
-
The encyclopedia of RNA interactomes
- TF:
-
Transcription factor
- CLIP:
-
Cross-linking and immunoprecipitation
- PBMC:
-
Peripheral blood mononuclear cell
- PCA:
-
Principal component analysis
- PC:
-
Principal component
- SFT:
-
Scale-free topology
- TOM:
-
Topological Overlap Matrix
- HTSeq:
-
High-throughput sequencing
- WB:
-
Whole blood
- ADRA1A:
-
Adrenoceptor Alpha 1A
- HOMER3:
-
Homer scaffold protein 3
- HAP1:
-
Huntingtin associated protein 1
- IFNGR1:
-
Interferon gamma receptor 1
- PDL1:
-
Programmed cell death receptor ligand 1
- Qki:
-
Schizophrenia-associated gene quaking
- PRC2:
-
Polycomb repressive complex 2
- HMT:
-
Histone methyltransferase
- LQT1:
-
Long QT syndrome type 1
- FA:
-
Fractional anisotropy
- qRT-PCR:
-
Quantitative real-time reverse-transcription polymerase chain reaction
- co-IP:
-
co-immunoprecipitation
- LV:
-
Low-variance
- DTC:
-
Dynamic tree cut
- CNV:
-
Copy number variation
- SNV:
-
Single nucleotide variant
- \({\text{Ca}}^{2+}\) :
-
Calcium
- NGS:
-
Next-generation sequencing
- CCL22:
-
C–C motif chemokine ligand 22
- TFF1:
-
Trefoil factor 1
- TNFRSF:
-
Tumor necrosis factor receptor superfamily
- PROK2:
-
Prokineticin 2
- DUSP4:
-
Dual specificity phosphatase 4
- PRICKLE2:
-
Prickle planar cell polarity protein 2
- MDGA1:
-
MAM domain containing glycosylphosphatidylinositol anchor 1
- PGK1:
-
Phosphoglycerate kinase 1
- NEAT1:
-
Nuclear paraspeckle assembly transcript 1
- XIST:
-
X inactive specific transcript
- KCNQ1:
-
Potassium voltage-gated channel subfamily Q member 1
- WES:
-
Whole exome sequencing
- NES:
-
Neuroendocrine system
- CASP1:
-
Caspase 1
References
Patel KR, Cherian J, Gohil K, Atkinson D (2014) Schizophrenia: overview and treatment options. P T 39:638–645
Rahman T, Lauriello J (2016) Schizophrenia: an overview. FOC 14:300–307. https://doi.org/10.1176/appi.focus.20160006
Perälä J, Suvisaari J, Saarni SI, Kuoppasalmi K, Isometsä E, Pirkola S et al (2007) Lifetime prevalence of psychotic and bipolar I disorders in a general population. Arch Gen Psychiatry 64:19. https://doi.org/10.1001/archpsyc.64.1.19
Poreddi V, Reddemma K, Math S (2013) People with mental illness and human rights: a develo** countries perspective. Indian J Psychiatry 55:117. https://doi.org/10.4103/0019-5545.111447
Hor K, Taylor M (2010) Review: Suicide and schizophrenia: a systematic review of rates and risk factors. J Psychopharmacol 24:81–90. https://doi.org/10.1177/1359786810385490
Giusti-Rodríguez P, Sullivan PF (2013) The genomics of schizophrenia: update and implications. J Clin Invest 123:4557–4563. https://doi.org/10.1172/JCI66031
Luvsannyam E, Jain MS, Pormento MKL, Siddiqui H, Balagtas ARA, Emuze BO et al (2022) Neurobiology of schizophrenia: a comprehensive review. Cureus 14:e23959. https://doi.org/10.7759/cureus.23959
Liu J, Li M, Luo X-J, Su B (2018) Systems-level analysis of risk genes reveals the modular nature of schizophrenia. Schizophr Res 201:261–269. https://doi.org/10.1016/j.schres.2018.05.015
Huang K-C, Tsao TT-H, Wang T-Y, Lee S-A (2016) Transcriptome analysis of systems biology for schizophrenia. In: Shen Y-C, editor. Schizophrenia treatment - the new facets, InTech; https://doi.org/10.5772/66864.
Kasai K, Iwanami A, Yamasue H, Kuroki N, Nakagome K, Fukuda M (2002) Neuroanatomy and neurophysiology in schizophrenia. Neurosci Res 43:93–110. https://doi.org/10.1016/S0168-0102(02)00023-8
Henriksen MG, Nordgaard J, Jansson LB (2017) Genetics of schizophrenia: overview of methods. Find Limit Front Hum Neurosci 11:322. https://doi.org/10.3389/fnhum.2017.00322
Hunter R, Barry S, Gaughan T (2013) 1835—antipsychotics for schizophrenia: too little progress after 50 years? Eur Psychiatry 28:1. https://doi.org/10.1016/S0924-9338(13)76799-3
Patel S, Sharma D, Uniyal A, Akhilesh GA, Tiwari V (2022) Recent advancements in biomarker research in schizophrenia: map** the road from bench to bedside. Metab Brain Dis 37:2197–2211. https://doi.org/10.1007/s11011-022-00926-5
Richard BC (2017) Non-coding RNA: it’s not junk. Dig Dis Sci 62:1107–1109. https://doi.org/10.1007/s10620-017-4506-1
Palazzo AF, Lee ES (2015) Non-coding RNA: what is functional and what is junk? Front Genet. https://doi.org/10.3389/fgene.2015.00002
Palazzo AF, Koonin EV (2020) Functional long non-coding RNAs evolve from junk transcripts. Cell 183:1151–1161. https://doi.org/10.1016/j.cell.2020.09.047
Statello L, Guo C-J, Chen L-L, Huarte M (2021) Gene regulation by long non-coding RNAs and its biological functions. Nat Rev Mol Cell Biol 22:96–118. https://doi.org/10.1038/s41580-020-00315-9
Esteller M (2011) Non-coding RNAs in human disease. Nat Rev Genet 12:861–874. https://doi.org/10.1038/nrg3074
Gibbons A, Udawela M, Dean B (2018) Non-coding RNA as novel players in the pathophysiology of schizophrenia. Noncoding RNA 4:11. https://doi.org/10.3390/ncrna4020011
Merelo V, Durand D, Lescallette AR, Vrana KE, Hong LE, Faghihi MA et al (2015) Associating schizophrenia, long non-coding RNAs and neurostructural dynamics. Front Mol Neurosci. https://doi.org/10.3389/fnmol.2015.00057
Wang Z, Tong Q, Liao H, Rao S, Huang X (2018) Long non-coding RNAs in schizophrenia. Neurol Psychiatry Brain Res 30:132–136. https://doi.org/10.1016/j.npbr.2018.09.003
Borga C, Meeran SM, Fassan M (2019) Non-coding RNAs, a real next-gen class of biomarkers? Noncoding RNA Res 4:80–81. https://doi.org/10.1016/j.ncrna.2019.10.001
Winkle M, El-Daly SM, Fabbri M, Calin GA (2021) Noncoding RNA therapeutics—challenges and potential solutions. Nat Rev Drug Discov 20:629–651. https://doi.org/10.1038/s41573-021-00219-z
Leone S, Santoro R (2016) Challenges in the analysis of long noncoding RNA functionality. FEBS Lett 590:2342–2353. https://doi.org/10.1002/1873-3468.12308
Williams GT, Pickard MR (2016) Long non-coding RNAs: new opportunities and old challenges in cancer therapy. Transl Cancer Res 5:S564–S566. https://doi.org/10.2103/tcr.2016.09.04
Sacco LD, Baldassarre A, Masotti A (2011) Bioinformatics tools and novel challenges in long non-coding RNAs (lncRNAs) functional analysis. IJMS 13:97–114. https://doi.org/10.3390/ijms13010097
Salmena L, Poliseno L, Tay Y, Kats L, Pandolfi PP (2011) A ceRNA hypothesis: the rosetta stone of a hidden RNA language? Cell 146:353–358. https://doi.org/10.1016/j.cell.2011.07.014
Bai Z, Sun H, Li X, Wu J, Yuan H, Zhang G et al (2021) Time-ordered dysregulated ceRNA networks reveal disease progression and diagnostic biomarkers in ischemic and dilated cardiomyopathy. Cell Death Discov 7:296. https://doi.org/10.1038/s41420-021-00687-7
Song C, Zhang J, Qi H, Feng C, Chen Y, Cao Y et al (2017) The global view of mRNA-related ceRNA cross-talks across cardiovascular diseases. Sci Rep 7:10185. https://doi.org/10.1038/s41598-017-10547-z
Zhang X, Feng S, Fan Y, Luo Y, ** L, Li S (2020) Identifying a comprehensive ceRNA Network to reveal novel targets for the pathogenesis of parkinson’s disease. Front Neurol 11:810. https://doi.org/10.3389/fneur.2020.00810
Wang Y, Zhao Z-J, Kang X-R, Bian T, Shen Z-M, Jiang Y et al (2020) lncRNA DLEU2 acts as a miR-181a sponge to regulate SEPP1 and inhibit skeletal muscle differentiation and regeneration. Aging 12:24033–24056. https://doi.org/10.18632/aging.104095
Qi X, Zhang D-H, Wu N, **ao J-H, Wang X, Ma W (2015) ceRNA in cancer: possible functions and clinical implications. J Med Genet 52:710–718. https://doi.org/10.1136/jmedgenet-2015-103334
Debnath M (2015) Adaptive immunity in schizophrenia: functional implications of T cells in the etiology, course and treatment. J Neuroimmune Pharmacol 10:610–619. https://doi.org/10.1007/s11481-015-9626-9
Debnath M, Berk M, Maes M (2020) Changing dynamics of psychoneuroimmunology during the COVID-19 pandemic. Brain Behav Immun–Health 5:100096. https://doi.org/10.1016/j.bbih.2020.100096
Ma H, Cheng N, Zhang C (2022) Schizophrenia and alarmins. Medicina 58:694. https://doi.org/10.3390/medicina58060694
Mukhopadhyay A, Deshpande SN, Bhatia T, Thelma BK (2023) Significance of an altered lncRNA landscape in schizophrenia and cognition: clues from a case–control association study. Eur Arch Psychiatry Clin Neurosci 273:1677–1691. https://doi.org/10.1007/s00406-023-01596-9
Clough E, Barrett T (2016) The gene expression omnibus database. Methods Mol Biol 1418:93–110. https://doi.org/10.1007/978-1-4939-3578-9_5
Tarazona S, Furió-Tarí P, Turrà D, Pietro AD, Nueda MJ, Ferrer A et al (2015) Data quality aware analysis of differential expression in RNA-seq with NOISeq R/Bioc package. Nucleic Acids Res 43:e140. https://doi.org/10.1093/nar/gkv711
Tarazona S, García-Alcalde F, Dopazo J, Ferrer A, Conesa A (2011) Differential expression in RNA-seq: a matter of depth. Genome Res 21:2213–2223. https://doi.org/10.1101/gr.124321.111
Parkinson H, Kapushesky M, Shojatalab M, Abeygunawardena N, Coulson R, Farne A et al (2007) ArrayExpress–a public database of microarray experiments and gene expression profiles. Nucleic Acids Res 35:D747–D750. https://doi.org/10.1093/nar/gkl995
Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W et al (2015) limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res 43:e47–e47. https://doi.org/10.1093/nar/gkv007
Chang A, Loy CJ, Lenz JS, Steadman A, Andama A, Nhung NV et al (2023) Circulating cell-free RNA in blood as a host response biomarker for the detection of tuberculosis. Infect Dis (Except HIV/AIDS). https://doi.org/10.1101/2023.01.11.23284433
Zhu M, Gong Z, Wu Q, Shi X, Su Q, Zhang Y (2020) Sanguinarine suppresses migration and metastasis in colorectal carcinoma associated with the inversion of EMT through the Wnt/β-catenin signaling. Clin Transl Med 10:1–12. https://doi.org/10.1002/ctm2.1
Zhao Z, Li T, Dong X, Wang X, Zhang Z, Zhao C et al (2021) Untargeted metabolomic profiling of cuprizone-induced demyelination in mouse corpus callosum by UPLC-Orbitrap/MS reveals potential metabolic biomarkers of CNS demyelination disorders. Oxid Med Cell Longev 2021:7093844. https://doi.org/10.1155/2021/7093844
**ng J, Cai H, Lin Z, Zhao L, Xu H, Song Y et al (2023) Examining the function of macrophage oxidative stress response and immune system in glioblastoma multiforme through analysis of single-cell transcriptomics. Front Immunol 14:1288137. https://doi.org/10.3389/fimmu.2023.1288137
Zhao Z, Zheng R, Wang X, Li T, Dong X, Zhao C et al (2022) Integrating lipidomics and transcriptomics reveals the crosstalk between oxidative stress and neuroinflammation in central nervous system demyelination. Front Aging Neurosci 14:870957. https://doi.org/10.3389/fnagi.2022.870957
Langfelder P, Horvath S (2008) WGCNA: an R package for weighted correlation network analysis. BMC Bioinform 9:559. https://doi.org/10.1186/1471-2105-9-559
Singh P, Rai A, Dohare R, Arora S, Ali S, Parveen S et al (2020) Network-based identification of signature genes KLF6 and SPOCK1 associated with oral submucous fibrosis. Mol Clin Oncol 12:299–310. https://doi.org/10.3892/mco.2020.1991
Szklarczyk D, Gable AL, Lyon D, Junge A, Wyder S, Huerta-Cepas J et al (2019) STRING v11: protein–protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res 47:D607–D613
Shannon P (2003) Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res 13:2498–2504. https://doi.org/10.1101/gr.1239303
Ding Y, Zhao Z, Cai H, Zhou Y, Chen H, Bai Y et al (2023) Single-cell sequencing analysis related to sphingolipid metabolism guides immunotherapy and prognosis of skin cutaneous melanoma. Front Immunol 14:1304466. https://doi.org/10.3389/fimmu.2023.1304466
Lin Z, Sui X, Jiao W, Wang Y, Zhao J (2022) Exploring the mechanism and experimental verification of puerarin in the treatment of endometrial carcinoma based on network pharmacology and bioinformatics analysis. BMC Comp Med Ther 22:150. https://doi.org/10.1186/s12906-022-03623-z
Lin Z, Sui X, Jiao W, Chen C, Zhang X, Zhao J (2022) Mechanism investigation and experiment validation of capsaicin on uterine corpus endometrial carcinoma. Front Pharmacol 13:953874. https://doi.org/10.3389/fphar.2022.953874
Kanehisa M, Goto S (2000) KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res 28:27–30. https://doi.org/10.1093/nar/28.1.27
Gene Ontology Consortium (2004) The gene ontology (GO) database and informatics resource. Nucleic Acids Res 32:258D – 261. https://doi.org/10.1093/nar/gkh036
Chen EY, Tan CM, Kou Y, Duan Q, Wang Z, Meirelles GV et al (2013) Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinform 14:128. https://doi.org/10.1186/1471-2105-14-128
Kuleshov MV, Jones MR, Rouillard AD, Fernandez NF, Duan Q, Wang Z et al (2016) Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res 44:W90-97. https://doi.org/10.1093/nar/gkw377
Sticht C, De La Torre C, Parveen A, Gretz N (2018) miRWalk: An online resource for prediction of microRNA binding sites. PLoS ONE 13:e0206239. https://doi.org/10.1371/journal.pone.0206239
Li J-H, Liu S, Zhou H, Qu L-H, Yang J-H (2014) starBase v2.0: decoding miRNA-ceRNA, miRNA-ncRNA and protein–RNA interaction networks from large-scale CLIP-Seq data. Nucl Acids Res 42:D2-7. https://doi.org/10.1093/nar/gkt1248
Kariuki D, Asam K, Aouizerat BE, Lewis KA, Florez JC, Flowers E (2023) Review of databases for experimentally validated human microRNA–mRNA interactions. Database 2023:baas014. https://doi.org/10.1093/database/baad014
Pujato M, Kieken F, Skiles AA, Tapinos N, Fiser A (2014) Prediction of DNA binding motifs from 3D models of transcription factors; identifying TLX3 regulated genes. Nucleic Acids Res 42:13500–13512. https://doi.org/10.1093/nar/gku1228
Hoseth EZ, Ueland T, Dieset I, Birnbaum R, Shin JH, Kleinman JE et al (2017) A study of TNF pathway activation in schizophrenia and bipolar disorder in plasma and brain tissue. SCHBUL. https://doi.org/10.1093/schbul/sbw183
Verhoeven WMA, Egger JIM, Hovens JE, Hoefsloot L (2013) Kallmann syndrome and paranoid schizophrenia: a rare combination. Case Rep 2013:bcr2012007387–bcr2012007387. https://doi.org/10.1136/bcr-2012-007387
An N, Bassil K, Al Jowf GI, Steinbusch HWM, Rothermel M, De Nijs L et al (2021) Dual-specificity phosphatases in mental and neurological disorders. Prog Neurobiol 198:101906. https://doi.org/10.1016/j.pneurobio.2020.101906
Chen X, Long F, Cai B, Chen X, Chen G (2018) A novel relationship for schizophrenia, bipolar and major depressive disorder Part 3: Evidence from chromosome 3 high density association screen. J Comp Neurol 526:59–79. https://doi.org/10.1002/cne.24311
Bayat A, Iqbal S, Borredy K, Amiel J, Zweier C, Barcia G et al (2021) PRICKLE2 revisited—further evidence implicating PRICKLE2 in neurodevelopmental disorders. Eur J Hum Genet 29:1235–1244. https://doi.org/10.1038/s41431-021-00912-y
Li J, Liu J, Feng G, Li T, Zhao Q, Li Y et al (2011) The MDGA1 gene confers risk to schizophrenia and bipolar disorder. Schizophr Res 125:194–200. https://doi.org/10.1016/j.schres.2010.11.002
Hasler-Rapacz J, Ellegren H, Fridolfsson AK, Kirkpatrick B, Kirk S, Andersson L et al (1998) Identification of a mutation in the low density lipoprotein receptor gene associated with recessive familial hypercholesterolemia in swine. Am J Med Genet 76:379–386
John J, Sharma A, Kukshal P, Bhatia T, Nimgaonkar VL, Deshpande SN et al (2018) Rare variants in tissue inhibitor of metalloproteinase 2 as a risk factor for schizophrenia: evidence from familial and cohort analysis. Schizophr Bull. https://doi.org/10.1093/schbul/sbx196
John J, Kukshal P, Sharma A, Bhatia T, Nimgaonkar VL, Deshpande SN et al (2019) Rare variants in protein tyrosine phosphatase, receptor type A (PTPRA) in schizophrenia: evidence from a family based study. Schizophr Res 206:75–81. https://doi.org/10.1016/j.schres.2018.12.012
John J, Bhattacharyya U, Yadav N, Kukshal P, Bhatia T, Nimgaonkar VL et al (2020) Multiple rare inherited variants in a four generation schizophrenia family offer leads for complex mode of disease inheritance. Schizophr Res 216:288–294. https://doi.org/10.1016/j.schres.2019.11.041
John J, Kukshal P, Bhatia T, Chowdari KV, Nimgaonkar VL, Deshpande SN et al (2017) Possible role of rare variants in Trace amine associated receptor 1 in schizophrenia. Schizophr Res 189:190–195. https://doi.org/10.1016/j.schres.2017.02.020
Li S, Li J, Liu J, Wang J, Li X, Huo Y et al (2022) Regulatory variants at 2q33.1 confer schizophrenia risk by modulating distal gene TYW5 expression. Brain 145:770–786. https://doi.org/10.1093/brain/awab357
Ignatieva EV, Matrosova EA (2021) Disease-associated genetic variants in the regulatory regions of human genes: mechanisms of action on transcription and genomic resources for dissecting these mechanisms. Vavilovskii Zhurnal Genet Selektsii 25:18–29. https://doi.org/10.18699/VJ21.003
Lang Y, Zhang J, Yuan Z (2019) Construction and dissection of the ceRNA-ceRNA network reveals critical modules in depression. Mol Med Rep 19:3411–3420. https://doi.org/10.3892/mmr.2019.10009
He L, Zou P, Sun W, Fu Y, He W, Li J (2021) Identification of lncRNA NR_028138.1 as a biomarker and construction of a ceRNA network for bipolar disorder. Sci Rep 11:15653. https://doi.org/10.1038/s41598-021-94122-7
Li R, Wang Q, Qiu Y, Meng Y, Wei L, Wang H et al (2021) A potential autophagy-related competing endogenous RNA network and corresponding diagnostic efficacy in schizophrenia. Front Psychiatry 12:628361. https://doi.org/10.3389/fpsyt.2021.628361
Cabili MN, Trapnell C, Goff L, Koziol M, Tazon-Vega B, Regev A et al (2011) Integrative annotation of human large intergenic noncoding RNAs reveals global properties and specific subclasses. Genes Dev 25:1915–1927. https://doi.org/10.1101/gad.17446611
Ramos AD, Diaz A, Nellore A, Delgado RN, Park K-Y, Gonzales-Roybal G et al (2013) Integration of genome-wide approaches identifies lncRNAs of adult neural stem cells and their progeny in vivo. Cell Stem Cell 12:616–628. https://doi.org/10.1016/j.stem.2013.03.003
Hangauer MJ, Vaughn IW, McManus MT (2013) Pervasive transcription of the human genome produces thousands of previously unidentified long intergenic noncoding RNAs. PLoS Genet 9:e1003569. https://doi.org/10.1371/journal.pgen.1003569
Ikegame T, Bundo M, Sunaga F, Asai T, Nishimura F, Yoshikawa A et al (2013) DNA methylation analysis of BDNF gene promoters in peripheral blood cells of schizophrenia patients. Neurosci Res 77:208–214. https://doi.org/10.1016/j.neures.2013.08.004
Cheng J, Wang Y, Zhou K, Wang L, Li J, Zhuang Q et al (2014) Male-specific association between dopamine receptor D4 gene methylation and schizophrenia. PLoS ONE 9:e89128. https://doi.org/10.1371/journal.pone.0089128
Nour El Huda AR, Norsidah KZ, Nabil Fikri MR, Hanisah MN, Kartini A, Norlelawati AT (2018) DNA methylation of membrane-bound catechol-O-methyltransferase in Malaysian schizophrenia patients. Psychiatry Clin Neurosci 72:266–279. https://doi.org/10.1111/pcn.12622
Nabil Fikri RM, Norlelawati AT, Nour El-Huda AR, Hanisah MN, Kartini A, Norsidah K et al (2017) Reelin (RELN) DNA methylation in the peripheral blood of schizophrenia. J Psychiatr Res 88:28–37. https://doi.org/10.1016/j.jpsychires.2016.12.020
Liu J, Lv W, Li S, Deng J (2021) Regulation of long non-coding RNA KCNQ1OT1 network in colorectal cancer immunity. Front Genet 12:684002. https://doi.org/10.3389/fgene.2021.684002
Liu Y, Li Z, Zhang M, Deng Y, Yi Z, Shi T (2013) Exploring the pathogenetic association between schizophrenia and type 2 diabetes mellitus diseases based on pathway analysis. BMC Med Genomics 6:S17. https://doi.org/10.1186/1755-8794-6-S1-S17
De Jonge JC, Vinkers CH, Hulshoff Pol HE, Marsman A (2017) GABAergic mechanisms in schizophrenia: linking postmortem and in vivo studies. Front Psychiatry 8:118. https://doi.org/10.3389/fpsyt.2017.00118
Boczek T, Mackiewicz J, Sobolczyk M, Wawrzyniak J, Lisek M, Ferenc B et al (2021) The role of G protein-coupled receptors (GPCRs) and calcium signaling in schizophrenia. Focus on GPCRs activated by neurotransmitters and chemokines. Cells 10:1228. https://doi.org/10.3390/cells10051228
Coyle JT, Basu A, Benneyworth M, Balu D, Konopaske G (2012) Glutamatergic synaptic dysregulation in schizophrenia: therapeutic implications. Handb Exp Pharmacol. https://doi.org/10.1007/978-3-642-25758-2_10
Archer T (2010) Neurodegeneration in schizophrenia. Expert Rev Neurother 10:1131–1141. https://doi.org/10.1586/ern.09.152
Karageorgiou V, Milas GP, Michopoulos I (2019) Neutrophil-to-lymphocyte ratio in schizophrenia: a systematic review and meta-analysis. Schizophr Res 206:4–12. https://doi.org/10.1016/j.schres.2018.12.017
Zan J, Zhao X, Deng X, Ding H, Wang B, Lu M et al (2021) Paraspeckle promotes hepatocellular carcinoma immune escape by sequestering IFNGR1 mRNA. Cell Mol Gastroenterol Hepatol 12:465–487. https://doi.org/10.1016/j.jcmgh.2021.02.010
Gast M, Rauch BH, Haghikia A, Nakagawa S, Haas J, Stroux A et al (2019) Long noncoding RNA NEAT1 modulates immune cell functions and is suppressed in early onset myocardial infarction patients. Cardiovasc Res 115:1886–1906. https://doi.org/10.1093/cvr/cvz085
Zhang P, Cao L, Zhou R, Yang X, Wu M (2019) The lncRNA Neat1 promotes activation of inflammasomes in macrophages. Nat Commun 10:1495. https://doi.org/10.1038/s41467-019-09482-6
Wang Y, Hou L, Yuan X, Xu N, Zhao S, Yang L et al (2020) LncRNA NEAT1 targets fibroblast-Like synoviocytes in rheumatoid arthritis via the miR-410-3p/YY1 Axis. Front Immunol 11:1975. https://doi.org/10.3389/fimmu.2020.01975
Ma H, Han P, Ye W, Chen H, Zheng X, Cheng L et al (2017) The long noncoding RNA NEAT1 exerts antihantaviral effects by acting as positive feedback for RIG-I signaling. J Virol 91:e02250-e2316. https://doi.org/10.1128/JVI.02250-16
Zhang P, Lu B, Zhang Q, Xu F, Zhang R, Wang C et al (2020) LncRNA NEAT1 sponges MiRNA-148a-3p to suppress choroidal neovascularization and M2 macrophage polarization. Mol Immunol 127:212–222. https://doi.org/10.1016/j.molimm.2020.08.008
Gao M, Liu L, Zhang D, Yang Y, Chang Z (2020) Long non-coding RNA NEAT1 serves as sponge for miR-365a-3p to promote gastric cancer progression via regulating ABCC4. OTT 13:3977–3985. https://doi.org/10.2147/OTT.S245557
Guo Z, He C, Yang F, Qin L, Lu X, Wu J (2019) Long non-coding RNA-NEAT1 a sponge for miR-98-5p, promotes expression of oncogene HMGA2 in prostate cancer. Biosci Rep 39:BSR20190635. https://doi.org/10.1042/BSR20190635
**e Q, Lin S, Zheng M, Cai Q, Tu Y (2019) Long noncoding RNA NEAT1 promotes the growth of cervical cancer cells via sponging miR-9-5p. Biochem Cell Biol 97:100–108. https://doi.org/10.1139/bcb-2018-0111
Yan H, Liang H, Liu L, Chen D, Zhang Q (2019) Long noncoding RNA NEAT1 sponges miR-125a-5p to suppress cardiomyocyte apoptosis via BCL2L12. Mol Med Report. https://doi.org/10.3892/mmr.2019.10095
Ji B, Higa KK, Kelsoe JR, Zhou X (2015) Over-expression of XIST, the master gene for X chromosome inactivation, in females with major affective disorders. EBioMedicine 2:909–918. https://doi.org/10.1016/j.ebiom.2015.06.012
Yan X-W, Liu H-J, Hong Y-X, Meng T, Du J, Chang C (2022) lncRNA XIST induces Aβ accumulation and neuroinflammation by the epigenetic repression of NEP in Alzheimer’s disease. J Neurogenet 36:11–20. https://doi.org/10.1080/01677063.2022.2028784
Chanda K, Mukhopadhyay D (2020) LncRNA **st, X-chromosome instability and Alzheimer’s disease. CAR 17:499–507. https://doi.org/10.2174/1567205017666200807185624
Nguyen DK, Disteche CM (2006) High expression of the mammalian X chromosome in brain. Brain Res 1126:46–49. https://doi.org/10.1016/j.brainres.2006.08.053
She C, Yang Y, Zang B, Yao Y, Liu Q, Leung PSC et al (2022) Effect of LncRNA XIST on immune cells of primary biliary cholangitis. Front Immunol 13:816433. https://doi.org/10.3389/fimmu.2022.816433
Yu B, Qi Y, Li R, Shi Q, Satpathy AT, Chang HY (2021) B cell-specific XIST complex enforces X-inactivation and restrains atypical B cells. Cell 184:1790-1803.e17. https://doi.org/10.1016/j.cell.2021.02.015
Li J, Che L, Xu C, Lu D, Xu Y, Liu M et al (2022) XIST/miR-34a-5p/PDL1 axis regulated the development of lung cancer cells and the immune function of CD8 + T cells. J Recept Signal Transduct 42:469–478. https://doi.org/10.1080/10799893.2021.2019274
Mudge J, Miller NA, Khrebtukova I, Lindquist IE, May GD, Huntley JJ et al (2008) Genomic convergence analysis of schizophrenia: mRNA sequencing reveals altered synaptic vesicular transport in post-mortem cerebellum. PLoS ONE 3:e3625. https://doi.org/10.1371/journal.pone.0003625
Irie K, Tsujimura K, Nakashima H, Nakashima K (2016) MicroRNA-214 promotes dendritic development by targeting the schizophrenia-associated gene quaking (Qki). J Biol Chem 291:13891–13904. https://doi.org/10.1074/jbc.M115.705749
Pandey RR, Mondal T, Mohammad F, Enroth S, Redrup L, Komorowski J et al (2008) Kcnq1ot1 antisense noncoding RNA mediates lineage-specific transcriptional silencing through chromatin-level regulation. Mol Cell 32:232–246. https://doi.org/10.1016/j.molcel.2008.08.022
Chen Q-H, Li B, Liu D-G, Zhang B, Yang X, Tu Y-L (2020) LncRNA KCNQ1OT1 sponges miR-15a to promote immune evasion and malignant progression of prostate cancer via up-regulating PD-L1. Cancer Cell Int 20:394. https://doi.org/10.1186/s12935-020-01481-8
Vohra J (2020) Sudden cardiac death in schizophrenia: a review. Heart Lung Circ 29:1427–1432. https://doi.org/10.1016/j.hlc.2020.07.003
Wang Y, Eldstrom J, Fedida D (2020) Gating and regulation of KCNQ1 and KCNQ1 + KCNE1 channel complexes. Front Physiol 11:504. https://doi.org/10.3389/fphys.2020.00504
Huang H, Kuenze G, Smith JA, Taylor KC, Duran AM, Hadziselimovic A et al (2018) Mechanisms of KCNQ1 channel dysfunction in long QT syndrome involving voltage sensor domain mutations. Sci Adv 4:eaar631. https://doi.org/10.1126/sciadv.aar2631
Crotti L, Celano G, Dagradi F, Schwartz PJ (2008) Congenital long QT syndrome. Orphanet J Rare Dis 3:18. https://doi.org/10.1186/1750-1172-3-18
Moss AJ, Shimizu W, Wilde AAM, Towbin JA, Zareba W, Robinson JL et al (2007) Clinical aspects of type-1 long-QT syndrome by location, coding type, and biophysical function of mutations involving the KCNQ1 gene. Circulation 115:2481–2489. https://doi.org/10.1161/CIRCULATIONAHA.106.665406
Ramos-Ríos R, Arrojo-Romero M, Paz-Silva E, Carballal-Calvo F, Bouzón-Barreiro JL, Seoane-Prado J et al (2010) QTc interval in a sample of long-term schizophrenia inpatients. Schizophr Res 116:35–43. https://doi.org/10.1016/j.schres.2009.09.041
Cao H, Zhou Y, Li T, Yao C, Yang W, Kong S et al (2021) The prevalence, risk factors and clinical correlates of QTc prolongation in chinese hospitalized patients with chronic schizophrenia. Front Psychiatry 12:704045. https://doi.org/10.3389/fpsyt.2021.704045
Amin AS, Giudicessi JR, Tijsen AJ, Spanjaart AM, Reckman YJ, Klemens CA et al (2012) Variants in the 3′ untranslated region of the KCNQ1-encoded Kv7.1 potassium channel modify disease severity in patients with type 1 long QT syndrome in an allele-specific manner. Eur Heart J 33:714–723. https://doi.org/10.1093/eurheartj/ehr473
Bruce HA, Kochunov P, Paciga SA, Hyde CL, Chen X, **e Z et al (2017) Potassium channel gene associations with joint processing speed and white matter impairments in schizophrenia. Genes Brain Behav 16:515–521. https://doi.org/10.1111/gbb.12372
Matsunaga T, Yasuda K, Adachi T, Gu N, Yamamura T, Moritani T et al (2007) Alpha-adrenoceptor gene variants and autonomic nervous system function in a young healthy Japanese population. J Hum Genet 52:28–37. https://doi.org/10.1007/s10038-006-0076-3
Zhang J, Simpson PC, Jensen BC (2021) Cardiac α1A-adrenergic receptors: emerging protective roles in cardiovascular diseases. Am J Physiol-Heart Circ Physiol 320:H725–H733. https://doi.org/10.1152/ajpheart.00621.2020
Jensen BC, Swigart PM, De Marco T, Hoopes C, Simpson PC (2009) α1-adrenergic receptor subtypes in nonfailing and failing human myocardium. Circ Heart Fail 2:654–663. https://doi.org/10.1161/CIRCHEARTFAILURE.108.846212
Najjar S, Pearlman DM, Alper K, Najjar A, Devinsky O (2013) Neuroinflammation and psychiatric illness. J Neuroinflammation 10:816. https://doi.org/10.1186/1742-2094-10-43
Acknowledgements
The authors would like to thank the University of Delhi South Campus and Jamia Millia Islamia for providing infrastructure, journal access, and internet facilities. Prithvi Singh would like to thank the ICMR for awarding him Senior Research Fellowship [Grant Number: BMI/11(89)/2020]. This work is also partially supported by the Science & Engineering Research Board (SERB), Government of India [File Number: EEQ/2023/000980].
Funding
This research work did not receive any external funding.
Author information
Authors and Affiliations
Contributions
Anirban Mukhopadhyay involved in conceptualization, methodology, software, formal analysis, data curation, writing—original draft, and writing—review and editing. Prithvi Singh involved in methodology, software, formal analysis, data curation, writing—original draft, and writing—review and editing. Ravins Dohare involved in writing—review and editing, supervision, and project administration. BK Thelma involved in writing—review and editing, supervision, and project administration. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare that they have no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Mukhopadhyay, A., Singh, P., Dohare, R. et al. Deciphering the landscape of lncRNA-driven ceRNA network in schizophrenia etiology. Egypt J Med Hum Genet 25, 71 (2024). https://doi.org/10.1186/s43042-024-00542-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s43042-024-00542-1