
Abstract
Phenolic compounds are ubiquitous plant secondary metabolites that possess various biological activities and are known to interact with proteins, altering their structure and properties. Therefore, interactions between these compounds and proteins has gained increasing attention due to their potential benefits to human health and for exploitation by the food industry. Phenolic compounds and proteins can form complexes via covalent linkages and/or non-covalent interactions through hydrophobic, electrostatic, van der Waals forces and hydrogen bonding. This review describes possible mechanisms of phenol-protein complex formation, their physiological action and activities that are important in the food industry, and possible outcomes in the terms of molecular docking and simulation analysis. The conformational changes of the protein upon binding with polyphenols can lead to the folding or unfolding of the protein molecules, forming insoluble or soluble complexes. The concentration of polyphenols, their molecular weight and structure, ions/cofactors and conditions of the system determine the precipitation or solubilization of the complex, affecting their nutritional and functional properties as well as their bioactivities. In this regard, molecular docking and simulation studies of phenolic-protein interactions allows comprehensive virtual screening of competitive/non-competitive and site-specific/non-specific conjugation of phenolics with different protein targets and facilitates understanding the observed effects. The docking analysis of flavonoids with enzymes and milk proteins has indicated their potential application in producing nutraceuticals and functional foods. Thus, combining molecular docking and simulation studies with experimental techniques is vital for better understanding the reactions that take place during digestion to engineer and manufacture novel food ingredients with desirable pharmacological properties and as potential food additives.
Graphical Abstract

Similar content being viewed by others

Introduction
Food is a complex and heterogenous system containing different major and minor constituents. The major macromolecular components include proteins, polysaccharides, and lipids while minor food components are organic acids, pigments, aroma compounds, vitamins, and minerals, among others, which also provide the essential nutrients (Samant et al. 1993). These constituents individually or as complexes play critical functional roles in foods and in different bioactivities. Most scientific literature has documented the nutrient composition of foods, identification of novel compounds and their functional properties, and bioactivities. In the past decades, most studies had focused on determining the content of bioactive compounds and bioactivity of vegetables, legumes, grains, fruits, herbs, and seafood. At present, the attention has been drawn to the fundamental understanding of the behavior of food macromolecules to interpret bioactivities at the molecular level.
In this contribution, an overview of the main interactions between phenolics and proteins in foods including their effects on biological activities and food properties are presented. In addition, particular emphasis is given to understanding and predicting these interactions and outcomes in terms of molecular docking simulations with model systems containing selected phenolic compounds and proteins. The insights obtained at the molecular level will provide a fundamental framework for in-vitro studies from nutritional, functional and pharmaceutical perspectives.
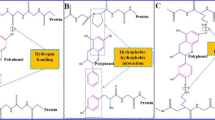
Proteins are one of the primary macronutrients and functional components of the diet that determine food’s textural, sensorial, and nutritional properties. According to the chemical structure, proteins are made up of 20 different amino acids that are linked together by peptide bonds in different combinations (Małecki et al. 2021). The linear sequence of amino acids within a protein is considered as the primary structure of the protein which drives the formation of the secondary structure through stable folding patterns such as alpha helices and beta sheets. The folding is further driven by non-specific hydrophobic interactions that ultimately determine the protein’s unique three-dimensional shape or tertiary structure and its function. The quaternary structure is a result of association of several protein chains into a specific spatial arrangement. Proteins undergo a wide range of structural and conformational changes through various complex interactions with other food components which provides numerous beneficial effects. Some of proteins have specific binding sites for other molecules that result in stable conformations upon binding and modulate the functioning of the system (Chen et al. 2021). In relation to the interactions between protein and phenolic compounds in food, proteins can form complexes with them, leading to changes in their structural, functional and nutritional properties which can broaden the range of functionalities achieved (Alu’datt et al. 2020; Ozdal et al. 2013).
Phenolic compounds are a major class of secondary metabolites in plants that are mainly derived from phenylalanine and tyrosine (Shahidi & Chandrasekara 2017). In this category, phenolic acids, namely hydroxybenzoic acid and hydroxycinnamic acid derivatives may be noted that are regarded as simple phenols consisting of a benzene ring substituted with hydroxyl groups. Meanwhile, polyphenols contain multiple rings with more than one phenolic group. Polyphenols can be classified as flavonoids and non-flavonoids according to their structures. The flavonoids include flavanones, flavanonols, flavones, flavonols, isoflavones, flavanols and tannins (hydrolysable and condensed) that are the most abundant and widespread dietary polyphenols (Fig. 1). The chemical structures of phenolic acids are given in Figs. 2 and 3. The different flavonoids differ greatly in their molecular structure based on the degree and pattern of glycosylation, hydroxylation, methoxylation and/or prenylation as shown in Figs. 4 and 5 (Guan et al. 2021). Most of them possess antioxidant activity, and hence they are frequently used as additives in functional dietary products (Li, He, et al. 2020). The nature of interactions and formation of thermodynamically favorable conformation were studied by docking and MD simulations of ferulic acid with the dimer and monomer forms of βLG. The results showed that the preferred binding site in the dimer form lies at the interface of the two monomers whereas it lies within the calyx shaped β-barrel of monomer form of βLG which exhibits the highest binding affinity. In two cases, the complexes were stabilized by hydrogen bonding and hydrophobic interactions. The binding of ferulic acid in monomer form reflects that the ligand entering at the calyx region of βLG following non-covalent interactions with the surrounding residues is essential in stabilizing the ligand within the receptor. This kind of information could be highly relevant to the food industry to enhance associative interactions for the development of novel bioactive compounds (Abdollahi et al. 2020).
The in-silico elucidation of the antidiabetic mechanism of EGCG with α-glucosidase indicated the binding of EGCG at the site close to the active site pocket of α-glucosidase and forms a stable complex with five intermolecular hydrogen bonds, which are the main forces between EGCG and α-glucosidase. It was identified that the binding site between α-glucosidase and EGCG was different from acarbose, a competitive inhibitor against α-glucosidase and there was no interaction between EGCG and the catalytic residues of Glu-277 and Asp-352. The docking studies illustrated that EGCG is a non-competitive inhibitor for α-glucosidase, providing the sense that EGCG might act as a promising antidiabetic agent (Xu et al. 2019).
Docking validation of potential human protein targets of apple polyphenols and possible mechanisms of chemoprevention in colorectal cancer revealed that some selected antioxidants can form stable complexes at cavities different from the active site. For instance, the binding of chlorogenic acid (CGA) to GTPase H-ras occurs at the allosteric site of the enzyme instead of the canonical binding site. In the presence and absence of ions/cofactors, direct docking analysis has been carried out to understand if the ligand is bound to a functionally active molecule and identify potential and stable protein-ligand interactions with selected targets. In this study, the lowest binding energy or the most stable complex corresponds to the interaction between quercitrin and hypoxanthine-guanine phosphoribosyltransferase enzyme. In addition, it was found that KDM1A gene coding lysine demethylase 1A and hyperin prefers binding whereas gene GAMT coding guanidinoacetate N-methyltransferase is the preferred target for hyperin, isoquercitrin, phloridzin and rutin present in apple. These observations clearly support the hypothesis that polyphenolic compounds act synergistically with proteins, providing cumulative effects on nucleotide metabolism and methyltransferase enzymes similar to the action of anti-cancer drugs (Scafuri et al. 2016).
Youn and Jun (2019) investigated the potential inhibitory action of cardamonin, pinocembrin, and pinostrobin on beta-site amyloid precursor protein cleaving enzyme1 (BACE1) as natural products-based therapy for Alzheimer’s disease (AD). Molecular docking analysis illustrated a non-competitive inhibitory activity for all these three compounds with reference to resveratrol used as the positive control. According to the binding energies, the most stable conjugates among proposed complexes were cadamonin-BACE1 which showed the strongest and effective inhibition. However, it was suggested that hydrophobic interaction between this compound involving the stabilization as cardamonin does not form any hydrogen bonds with BACE1. Stable conformations formed between P-glycoprotein and the flavonoids were analyzed through docking simulations to predict their possibility to pass the blood brain barrier. The computational analysis indicated that these flavonoids from B. rotunda may be promising AD preventative agents, and extensive examination needs to be carried out through in-vitro assays to support these predictions.
Identification of flavonoids that can regulate sirtuin 6 (SIRT6) activities is considered promising therapeutics for age-related diseases including cancer, diabetes, neurodegenerative diseases and metabolic disorders. Therefore, molecular docking has been carried out to discover their binding sites on SIRT6, and to identify principle interactions occurring on the enzyme active site with inhibitors and activators. The results have shown that the cleft present in enzyme forms the pocket for acetylated sirtuin substrate and for NAD+ cofactor, which requires the activation of the enzyme. Furthermore, they reported that activator compounds bind to a site outside of the cleft by forming interactions with a loop near the acetylated peptide substrate binding site. The activators may induce conformational changes in the loop upon binding to the putative activator site, improving acetylated substrates’ binding. Contrarily, the binding site of the majority of inhibitors was situated close to the binding site of known sirtuin inhibitors which can restrict NAD+ binding. Among different flavonoids, catechins with galloyl moiety exhibit a greater inhibitory activity by partially occupying the acetylated sirtuin substrate’s binding site and allowing more interactions with the binding pocket. It was also identified that kaempferol can act as a potent dual modulator due to its ability to bind multiple sites and form similar interactions with the same amino acid residues where the known activators and inhibitors interact with. An in-silico based mutation analysis was performed to examine the impact of activator site’s residues Gly-156, Asp-185, Trp-186, Glu-187 and Asp-188 on the action and/ structure of SIRT6. Extensive docking analysis revealed that different flavonoids could alter SIRT6 activity in a structure-dependent manner (Guan et al. 2021; Rahnasto-rilla et al. 2018).
Troxerutin (TX), a bioflavonoid, has been shown to exhibit anti-neoplastic and anti-cancer activities. Assessing the modulatory action of TX on transcription factors such as IKKβ, Nrf2 and Keap-1 through docking studies illustrated that TX interacts with the active sites of proteins by forming hydrogen bonds and π-cation interaction. TX with IKKβ conjugate has shown the most stable complex with the lowest binding energy compared to binding with other transcription factors, implying that high binding affinities are accompanied with hydrogen bond interactions. Based on the combination of docking analysis with immunoblotting and immunocytochemistry analysis, it was predicted that the formation of stable conjugates might result in a conformation change of transcription factors, followed by inactivating the expression of oncogenes and hence can be deemed as a potent drug in anti-cancer therapy (Thomas et al. 2018).
The structure-based virtual screening of flavonoids indicated that most flavonoids can bind to the acetylated lysine (KAc) binding site of BD1 of Brd4 receptor molecule and can act as novel natural bromodomain inhibitors. Flavonoids were found to occupy the active site, forming hydrogen bonds between the acetyl carbonyl oxygen and the amino group of the receptor’s conserved Asn-140 amino acid residue. Quercetin binds in a similar manner and shows the highest binding affinity while illustrating that these ligands may prevent the binding of Brd4 to acetylated lysines on nucleosome histones and inhibit RNA Polymerase II mediated transcription elongation. Moreover, the blocking of KAc site may inhibit the binding to the Myc gene resulting in a low level expression of the Myc gene, leading to low proliferation of cancer cells (Raj et al. 2017).
Investigation of inhibitory effects of anthraquinones on tyrosinase shows that they enter the active site of tyrosinase in the form of one molecule and competitively inhibit the activity of tyrosinase. The majority of interactions were electrostatic forces and hydrophobic interactions compared to hydrogen bonds due to the presence of aromatic rings in the structure of anthraquinones. Further analysis revealed that the inhibitory effect on tyrosinase activity was accomplished by acting on histidine residues bound to copper ions rather than chelating them. It was suggested that the binding of anthraquinones at the active site of tyrosinase results in conformational changes of secondary structure and prevented the entry of substrates, inhibiting the tyrosinase activity and in turn regulating the melanogenesis (Zeng et al. 2020). Similar molecular docking analyses were carried out to evaluate the synergetic effect of quercetin, cinnamic acid and ferulic acid on tyrosinase enzyme. Quercetin located at the hydrophobic pocket of the enzyme formed strong hydrophobic interactions and showed slightly higher binding affinity compared to binding with cinnamic and ferulic acids. Furthermore, computational docking simulations showed that quercetin, cinnamic acid and ferulic acid bind with different sites on tyrosinase in a non-competitive manner, exhibiting their ability to express synergistic action in inhibiting the tyrosinase activity (Yu et al. 2019).
The preferred binding sites of phenolics on scallop gonad protein isolates (SGPIs) were identified and visualized by molecular docking simulation studies. Vitellogenin and β-actin, as the main crystal structures of SGPIs have been analyzed with EGCG to evaluate their ability to form stable complexes. It was found that EGCG inserts into the hydrophobic central cavity of vitellogenin or β-actin and stabilizes the conjugate via hydrogen bonds, van der Waals and hydrophobic interactions. Besides, interaction of these proteins with EGC and ECG resulted in decreasing order of SGPIs-binding capacity of phenolics as EGCG > ECG > EGC, indicating that binding affinities mainly rely on the number of –OH groups present in the ligand molecule. In the process of interactions, hydrogen bond and van der Waals forces dominated between SGPIs and EGCG while hydrophobic interaction forces were dominant in SGPIs-ECG complex. Therefore, a comprehensive theoretical understanding of the induced effect of these phenolic compounds on the structure and function of SGPIs could be used to accomplish desired practical aspects (Han et al. 2021).
Investigation on binding sites and binding affinities of catechin derivatives for bovine serum albumin (BSA) showed that ester catechins (EGCG and ECG) possess high binding affinities than non-ester catechins (EGC and EC) as they can form more hydrogen bonds (Yu et al. 2020). Further, interaction of EGCG with BSA by molecular operating environment (MOE) suite docking simulation program has indicated that EGCG interact with both Trp − 134 residue; at drug-binding site I by π–π stacking and Trp-213 residue; on the molecular surface of BSA (Ikeda et al. 2017). These results suggest that substitution at the C-3 position or galloyl moiety of catechins determines their binding affinities against serum albumin. Together, these simulation studies support the idea of formation of tea cream and controlling the performance of tea beverage products by introducing tea polyphenols (Yu et al. 2020). In studying the molecular nature of interactions between β-casein and p-coumaric acid, computational docking has been done to identify the location of their specific binding site and to obtain the thermodynamically stable conformation of β-casein with p-coumaric acid. Modeling outcomes have shown that the ligand molecule interacts with amino residues within the core of β-casein receptor molecule forming a hydrogen bond between hydroxyl group of p-coumaric acid and the carbonyl group of the peptide backbone of Ile-27 (Kaur et al. 2018).
Several studies have shown that docking algorithms are capable of identifying putative phenolic-protein binding sites of novel conjugates with their binding affinities, analyzing interactions and conformations that can be used to recognize and interpret their potential activities from practical perspectives. Moreover, it demonstrated that polyphenols with galloyl moiety bind most strongly to extended proteins with a high proline content to form thermodynamically favorable complexes. The degree of hydroxylation, methoxylation and steric hindrance of the polyphenol mainly determine its binding affinity for the protein molecule and structural conformation of the compound.
Conclusion
Complexation of phenolic compounds with proteins via covalent and/or non-covalent bonding entails changes in proteins that could result in favorable or unfavorable properties. The phenolic-protein interactions affect the compound’s activity and exert synergistic or antagonistic effects depending on the type and structure of compounds, molecular weight, concentration, pH, temperature, cofactors, method and food processing conditions, and physiological status. These conjugates are able to improve biological activities such as antioxidant, anti-inflammatory, anti-allergic and anti-cancer activities and bioavailability of polyphenols compared to individual components. In addition, they can also act as food preservation agents against microorganisms and lipid oxidation, emulsions for delivery of nutraceuticals, edible films for food packaging and drug releasing modulators and may also act as natural food color, flavor and texture modifier. It has been demonstrated that the underlying mechanism of these actions and interactions at the molecular level can be extensively investigated, specifically using molecular docking approaches with various analytical techniques. It can be concluded that polyphenols bind most strongly to extended proteins with a high proline content and polyphenols with galloyl moiety to form thermodynamically favorable complexes, by modulating their activities. As shown in earlier studies, molecular docking programs are able to successfully predict the binding modes between protein and polyphenols in a large scale. Therefore, the docking studies provide scientific basis to determine putative binding sites, stable conformations of conjugates and identifying promising compounds that can be developed further, to accomplish desired functional and health aspects. However, more in-depth studies are needed for a comprehensive understanding of the interaction between phenolic compounds and proteins and factors that affect their binding affinities for enhancing and expanding the application potential of different phenolic compounds in the food and pharmaceutical industries.
Availability of data and materials
All data generated are included in the references of this article.
Abbreviations
- AD:
-
Alzheimer’s disease
- BACE1:
-
Beta-site amyloid precursor protein cleaving enzyme 1
- BSA:
-
Bovine serum albumin
- CA:
-
(+)-Catechin
- CGA:
-
Chlorogenic acid
- CMC:
-
Critical micelle concentration
- EC:
-
(−) -epicatechin
- ECG:
-
(−)-epicatechin gallate
- EGC:
-
(−)-epigallocatechin
- EGCG:
-
(−)- epigallocatechin gallate
- EWP:
-
Egg white protein
- HPP:
-
Human plasma proteins
- MD:
-
Molecular dynamics
- MDA:
-
Malondialdehyde
- MOE:
-
Molecular operating environment
- PRP:
-
Proline-rich proteins
- SEM:
-
Sunflower extraction meal
- SGPIs:
-
Scallop gonad protein isolates
- SPI:
-
Soy protein isolate
- TBARS:
-
Thiobarbituric acid reactive substances
- TPP:
-
Type 2 diabetes plasma proteins
- UHT:
-
Ultrahigh-temperature
- α-LA:
-
α-lactalbumin
- βLG:
-
β-lactoglobulin
References
Abdollahi, K., Ince, C., Condict, L., Hung, A., & Kasapis, S. (2020). Combined spectroscopic and molecular docking study on the pH dependence of molecular interactions between β-lactoglobulin and ferulic acid. Food Hydrocolloids, 101, 105461. https://doi.org/10.1016/j.foodhyd.2019.105461.
Aewsiri, T., Benjakul, S., Visessanguan, W., Eun, J. B., Wierenga, P. A., & Gruppen, H. (2009). Antioxidative activity and emulsifying properties of cuttlefish skin gelatin modified by oxidised phenolic compounds. Food Chemistry, 117(1), 160–168. https://doi.org/10.1016/j.foodchem.2009.03.092.
Allahdad, Z., Varidi, M., Zadmard, R., Saboury, A. A., & Haertlé, T. (2019). Binding of β-carotene to whey proteins: Multi-spectroscopic techniques and docking studies. Food Chemistry, 277, 96–106. https://doi.org/10.1016/j.foodchem.2018.10.057.
Al-Shabib, N. A., Khan, J. M., Malik, A., Tabish Rehman, M., AlAjmi, M. F., Husain, F. M., … Altwaijry, N. (2020). Molecular interaction of tea catechin with bovine β-lactoglobulin: A spectroscopic and in silico studies. Saudi Pharmaceutical Journal, 28(3), 238–245. https://doi.org/10.1016/j.jsps.2020.01.002.
Alu’datt, M. H., Al-U’datt, D. G., Tranchant, C. C., Alhamad, M. N., Rababah, T., Gammoh, S., … Alli, I. (2020). Phenolic and protein contents of differently prepared protein co-precipitates from flaxseed and soybean and antioxidant activity and angiotensin inhibitory activity of their phenolic fractions. NFS Journal, 21, 65–72. https://doi.org/10.1016/j.nfs.2020.11.001.
Araghi, M., Moslehi, Z., Nafchi, A. M., Mostahsan, A., Salamat, N., & Garmakhany, A. D. (2015). Cold water fish gelatin modification by a natural phenolic cross-linker (ferulic acid and caffeic acid). Food Science and Nutrition, 3(5), 370–375. https://doi.org/10.1002/fsn3.230.
Asano, K., Shinagawa, K., & Hashimoto, N. (1982). Characterization of haze-forming proteins of beer and their roles in chill haze formation. American Society of Brewing Chemists, 40(4), 147–154. https://doi.org/10.1094/ASBCJ-40-0147.
Bandyopadhyay, P., Ghosh, A. K., & Ghosh, C. (2012). Recent developments on polyphenol-protein interactions: Effects on tea and coffee taste, antioxidant properties and the digestive system. Food and Function, 3(6), 592–605. https://doi.org/10.1039/c2fo00006g.
Banerjee, S., Ji, C., Mayfield, J. E., Goel, A., **ao, J., Dixon, J. E., & Guo, X. (2018). Ancient drug curcumin impedes 26S proteasome activity by direct inhibition of dual-specificity. Proceedings of the National Academy of Sciences, 1–6. https://doi.org/10.1073/pnas.1806797115.
Betz, M., Steiner, B., Schantz, M., Oidtmann, J., Mäder, K., Richling, E., & Kulozik, U. (2012). Antioxidant capacity of bilberry extract microencapsulated in whey protein hydrogels. Food Research International, 47(1), 51–57. https://doi.org/10.1016/j.foodres.2012.01.010.
Bohn, T. (2014). Dietary factors affecting polyphenol bioavailability. Nutrition Reviews, 72(7), 429–452. https://doi.org/10.1111/nure.12114.
Bongartz, V., Brandt, L., Gehrmann, M. L., Zimmermann, B. F., Schulze-kaysers, N., & Schieber, A. (2016). Evidence for the formation of benzacridine derivatives in alkaline-treated sunflower meal and model solutions. Molecules, 21(1), 1–9. https://doi.org/10.3390/molecules21010091.
Bouayed, J., Hoffmann, L., & Bohn, T. (2011). Total phenolics , flavonoids , anthocyanins and antioxidant activity following simulated gastro-intestinal digestion and dialysis of apple varieties: Bioaccessibility and potential uptake. Food Chemistry, 128(1), 14–21. https://doi.org/10.1016/j.foodchem.2011.02.052.
Buitimea-Cantúa, N. E., Gutiérrez-Uribe, J. A., & Serna-Saldívar, S. O. (2018). Phenolic-protein interactions: Effects on food properties and health benefits. Journal of Medicinal Food, 21(2), 188–198. https://doi.org/10.1089/jmf.2017.0057.
Canon, F., Ballivian, R., Chirot, F., Antoine, R., Sarni-manchado, P., Dugourd, P., & Lyon, D. (2011). Folding of a salivary intrinsically disordered protein upon binding to tannins. Journal of American Chemicals Society, 133(20), 7847–7852. https://doi.org/10.1021/ja200534f.
Chanphai, P., Bourassa, P., Kanakis, C. D., Tarantilis, P. A., Polissiou, M. G., & Tajmir-riahi, H. A. (2017). Review on the loading efficacy of dietary tea polyphenols with milk proteins. Food Hydrocolloids, 77, 322–328. https://doi.org/10.1016/j.foodhyd.2017.10.008.
Charlton, A. J., Baxter, N. J., Khan, M. L., Moir, A. J. G., Haslam, E., Davies, A. P., & Williamson, M. P. (2002). Polyphenol/peptide binding and precipitation. Journal of Agricultural and Food Chemistry, 50(6), 1593–1601. https://doi.org/10.1021/jf010897z.
Chen, W., Chao, C., Yu, J., Copeland, L., Wang, S., & Wang, S. (2021). Effect of protein-fatty acid interactions on the formation of starch-lipid-protein complexes. Food Chemistry, 364. https://doi.org/10.1016/j.foodchem.2021.130390.
Chen, Y. Z., & Zhi, D. G. (2001). Ligand - protein inverse docking and its potential use in the computer search of protein targets of a small molecule. Proteins, 43(2), 217–226. https://doi.org/10.1002/1097-0134(20010501)43:2<217::aid-prot1032>3.0.co;2-g. PMID: 11276090.
Colahan-Sederstrom, P. M., & Peterson, D. G. (2005). Inhibition of key aroma compound generated during ultrahigh-temperature processing of bovine milk via epicatechin addition. Journal of Agricultural and Food Chemistry, 53(2), 398–402. https://doi.org/10.1021/jf0487248.
Czubinski, J., & Dwiecki, K. (2017). A review of methods used for investigation of protein–phenolic compound interactions. International Journal of Food Science and Technology, 52(3), 573–585. https://doi.org/10.1111/ijfs.13339.
Fan, Y., Zhang, Y., Yokoyama, W., & Yi, J. (2017). β-Lactoglobulin-chlorogenic acid conjugate-based nanoparticles for delivery of (−)-epigallocatechin-3-gallate. RSC Advances, 7(35), 21366–21374. https://doi.org/10.1039/c6ra28462k.
Feng, J., Cai, H., Wang, H., Li, C., & Liu, S. (2017). Improved oxidative stability of fish oil emulsion by grafted ovalbumin-catechin conjugates. Food Chemistry, 241, 60–69. https://doi.org/10.1016/j.foodchem.2017.08.055.
Fu, S., Wu, C., Wu, T., Yu, H., Yang, S., & Hu, Y. (2017). Preparation and characterisation of Chlorogenic acid-gelatin: A type of biologically active film for coating preservation. Food Chemistry, 221, 657–663. https://doi.org/10.1016/j.foodchem.2016.11.123.
Fujimoto, A., & Masuda, T. (2012). Chemical interaction between polyphenols and a cysteinyl thiol under radical oxidation conditions. Journal of Agricultural and Food Chemistry, 60(20), 5142–5151. https://doi.org/10.1021/jf3008822.
Ginsburg, I., Koren, E., Shalish, M., Kanner, J., & Kohen, R. (2012). Saliva increases the availability of lipophilic polyphenols as antioxidants and enhances their retention in the oral cavity. Archives of Oral Biology, 57(10), 1327–1334. https://doi.org/10.1016/j.archoralbio.2012.04.019.
Gorelik, S., Ligumsky, M., Kohen, R., & Kanner, J. (2008). A novel function of red wine polyphenols in humans: Prevention of absorption of cytotoxic lipid peroxidation products. The FASEB Journal, 22(1), 41–46. https://doi.org/10.1096/fj.07-9041com.
Grgić, J., Šelo, G., Planinić, M., Tišma, M., & Bucić-Kojić, A. (2020). Role of the encapsulation in bioavailability of phenolic compounds. Antioxidants (Basel), 9(10), 923. https://doi.org/10.3390/antiox9100923.
Gu, L., Su, Y., Zhang, M., Chang, C., Li, J., McClements, D. J., & Yang, Y. (2017). Protection of β-carotene from chemical degradation in emulsion-based delivery systems using antioxidant interfacial complexes: Catechin-egg white protein conjugates. Food Research International, 96, 84–93. https://doi.org/10.1016/j.foodres.2017.03.015.
Guan, H., Zhang, W., Sun-Waterhouse, D., Jiang, Y., Li, F., Waterhouse, G. I. N., & Li, D. (2021). Phenolic-protein interactions in foods and post ingestion: Switches empowering health outcomes. Trends in Food Science and Technology, 118, 71–86. https://doi.org/10.1016/j.tifs.2021.08.033.
Han, J., Du, Y., Yan, J., Jiang, X., Wu, H., & Zhu, B. (2021). Effect of non-covalent binding of phenolic derivatives with scallop (Patinopecten yessoensis) gonad protein isolates on protein structure and in vitro digestion characteristics. Food Chemistry, 357. https://doi.org/10.1016/j.foodchem.2021.129690.
Haratifar, S., Meckling, K. A., & Corredig, M. (2014). Antiproliferative activity of tea catechins associated with casein micelles, using HT29 colon cancer cells. Journal of Dairy Science, 97(2), 672–678. https://doi.org/10.3168/jds.2013-7263.
Hasni, I., Bourassa, P., Hamdani, S., Samson, G., & Carpentier, R. (2011). Interaction of milk a - and b -caseins with tea polyphenols. Food Chemistry, 126(2), 630–639. https://doi.org/10.1016/j.foodchem.2010.11.087.
Hassan, B., Chatha, S. A. S., Hussain, A. I., Zia, K. M., & Akhtar, N. (2018). Recent advances on polysaccharides, lipids and protein based edible films and coatings: A review. International Journal of Biological Macromolecules, 109, 1095–1107. https://doi.org/10.1016/j.ijbiomac.2017.11.097.
Hassan, N. M., Alhossary, A. A., Mu, Y., & Kwoh, C. K. (2017). Protein-ligand blind docking using quickVina-W with inter-process spatio-temporal integration. Scientific Reports, 7(1), 15451. https://doi.org/10.1038/s41598-017-15571-7.
Ikeda, M., Ueda-Wakagi, M., Hayashibara, K., Kitano, R., Kawase, M., Kaihatsu, K., … Ashida, H. (2017). Substitution at the C-3 position of catechins has an influence on the binding affinities against serum albumin. Molecules, 22(2), 1–12. https://doi.org/10.3390/molecules22020314.
Jiang, L., Liu, Y., Li, L., Qi, B., Ju, M., Xu, Y., … Sui, X. (2019). Covalent conjugates of anthocyanins to soy protein: Unravelling their structure features and in vitro gastrointestinal digestion fate. Food Research International, 120, 603–609. https://doi.org/10.1016/j.foodres.2018.11.011.
Kanakis, C. D., Hasni, I., Bourassa, P., Tarantilis, P. A., Polissiou, M. G., & Tajmir-riahi, H. (2011). Milk b -lactoglobulin complexes with tea polyphenols. Food Chemistry, 127(3), 1046–1055. https://doi.org/10.1016/j.foodchem.2011.01.079.
Kaur, J., Katopo, L., Hung, A., Ashton, J., & Kasapis, S. (2018). Combined spectroscopic, molecular docking and quantum mechanics study of β-casein and p-coumaric acid interactions following thermal treatment. Food Chemistry, 252, 163–170. https://doi.org/10.1016/j.foodchem.2018.01.091.
Keppler, J. K., Schwarz, K., & van der Goot, A. J. (2020). Covalent modification of food proteins by plant-based ingredients (polyphenols and organosulphur compounds): A commonplace reaction with novel utilization potential. Trends in Food Science and Technology, 101, 38–49. https://doi.org/10.1016/j.tifs.2020.04.023.
Lacroix, S., Klicic Badoux, J., Scott-Boyer, M.-P., Parolo, S., Matone, A., Priami, C., … Moco, S. (2018). A computationally driven analysis of the polyphenol-protein interactome. Scientific Reports, 8, 2232. https://doi.org/10.1038/s41598-018-20625-5.
Le Bourvellec, C., & Renard, C. M. G. C. (2012). Interactions between polyphenols and macromolecules: Quantification methods and mechanisms. Critical Reviews in Food Science and Nutrition, 52(3), 213–248. https://doi.org/10.1080/10408398.2010.499808.
Li, H., Guo, A., & Wang, H. (2008). Mechanisms of oxidative browning of wine. Food Chemistry, 108(1), 1–13. https://doi.org/10.1016/j.foodchem.2007.10.065.
Li, M., Ritzoulis, C., Du, Q., Liu, Y., Ding, Y., Liu, W., & Liu, J. (2021). Recent progress on protein-polyphenol complexes: Effect on stability and nutrients delivery of oil-in-water emulsion system. Frontiers in Nutrition, 8, 765589. https://doi.org/10.3389/fnut.2021.765589.
Li, X., Dai, T., Hu, P., Zhang, C., Chen, J., Liu, C., & Li, T. (2020). Characterization the non-covalent interactions between beta lactoglobulin and selected phenolic acids. Food Hydrocolloids, 105, 105761. https://doi.org/10.1016/j.foodhyd.2020.105761.
Li, Y., He, D., Li, B., Lund, M. N., **ng, Y., Wang, Y., … Li, L. (2021). Engineering polyphenols with biological functions via polyphenol-protein interactions as additives for functional foods. Trends in Food Science and Technology, 110, 470–482. https://doi.org/10.1016/j.tifs.2021.02.009.
Liu, F., Ma, C., Gao, Y., & McClements, D. J. (2017). Food-grade covalent complexes and their application as nutraceutical delivery systems: A review. Comprehensive Reviews in Food Science and Food Safety, 16(1), 76–95. https://doi.org/10.1111/1541-4337.12229.
Liu, F., Sun, C., Yang, W., Yuan, F., & Gao, Y. (2015). Structural characterization and functional evaluation of lactoferrin – Polyphenol conjugates formed by free-radical graft copolymerization. RSC Advances, 5, 15641–15651. https://doi.org/10.1039/c4ra10802g.
Liu, J., Yong, H., Yao, X., Hu, H., Yun, D., & **ao, L. (2019). Recent advances in phenolic-protein conjugates: Synthesis, characterization, biological activities and potential applications. RSC Advances, 9, 35825–35840. https://doi.org/10.1039/c9ra07808h.
Liu, L., Gao, K., Wang, Z., Lin, X., Huang, J., Wu, X., … Wu, H. (2018). Reducing the allergenic capacity of β -lactoglobulin by covalent conjugation with dietary polyphenols. Food Chemistry, 256, 427–434. https://doi.org/10.1016/j.foodchem.2018.02.158.
Liu, Z., & Hu, M. (2009). Natural polyphenol disposition via coupled metabolic pathways. Expert Opinion on Drug Metabolism & Toxicology, 3(3), 389–406. https://doi.org/10.1517/17425255.3.3.389.
Lou, Z., Wang, H., Zhu, S., Ma, C., & Wang, Z. (2011). Antibacterial activity and mechanism of action of chlorogenic acid. Journal of Food Science, 76(6), 398–403. https://doi.org/10.1111/j.1750-3841.2011.02213.x.
Lund, M. N., & Ray, C. A. (2017). Control of maillard reactions in foods: Strategies and chemical mechanisms. Journal of Agricultural and Food Chemistry, 65(23), 4537–4552. https://doi.org/10.1021/acs.jafc.7b00882.
Małecki, J., Muszyński, S., Sołowiej, B., & B.J.Sources (2021). Proteins in food systems— Bionanomaterials, conventional and unconventional sources, functional properties, and development opportunities. Polymers (Basel), 13(15), 2506. https://doi.org/10.3390/polym13152506.
Manach, C., Scalbert, A., Morand, C., Rémésy, C., & Jiménez, L. (2004). Polyphenols: Food sources and bioavailability. American Journal of Clinical Nutrition, 79(5), 727–747. https://doi.org/10.1093/ajcn/79.5.727.
Meng, X. Y., Zhang, H. X., Mezei, M., & Cui, M. (2012). Molecular docking: A powerful approach for structure-based drug discovery. Current Computer-Aided Drug Design, 7(2), 146–157. https://doi.org/10.2174/157340911795677602.
Minatel, I. O., Borges, C. V., Ferreira, M. I., Gomez, H. A. G., Chen, C.-Y. O., & Lima, G. P. P. (2017). Phenolic compounds: Functional properties, impact of processing and bioavailability. Phenolic Compounds - Biological Activity.https://doi.org/10.5772/66368.
Mohammadi, F., & Moeeni, M. (2015). Study on the interactions of trans-resveratrol and curcumin with bovine α-lactalbumin by spectroscopic analysis and molecular docking. Materials Science & Engineering C-Materials for Biological Applications, 50, 358–366. https://doi.org/10.1016/j.msec.2015.02.007.
Mori, T., Ishii, T., Akagawa, M., Nakamura, Y., & Nakayama, T. (2010). Covalent binding of tea catechins to protein thiols: The relationship between stability and electrophilic reactivity. Bioscience, Biotechnology and Biochemistry, 74(12), 2451–2456. https://doi.org/10.1271/bbb.100509.
Murray, N. J., Williamson, M. P., Lilley, T. H., & Haslam, E. (1994). Study of the interaction between salivary proline-rich proteins and a polyphenol by 1H-NMR spectroscopy. European Journal of Biochemistry, 219(3), 923–935. https://doi.org/10.1111/j.1432-1033.1994.tb18574.x.
Oh, H. I., Hoff, J. E., Armstrong, G. S., & Haff, L. A. (1980). Hydrophobic interaction in tannin-protein complexes. Journal of Agricultural and Food Chemistry, 28(2), 394–398. https://doi.org/10.1021/jf60228a020.
Ozdal, T., Capanoglu, E., & Altay, F. (2013). A review on protein-phenolic interactions and associated changes. Food Research International, 51(2), 954–970. https://doi.org/10.1016/j.foodres.2013.02.009.
Penalva, R., Esparza, I., Larraneta, E., Gamazo, C., & Irache, J. M. (2015). Zein-based nanoparticles improve the oral bioavailability of resveratrol and its anti-inflammatory effects in a mouse model of endotoxic shock. Journal of Agricultural and Food Chemistry, 63(23), 5603–5611. https://doi.org/10.1021/jf505694e.
Petsko, G. A., & Yates, J. R. (2011). Analyzing molecular interactions. Current Protocols in Bioinformatics.https://doi.org/10.1002/0471250953.bi0801s36.
Pianet, I., André, Y., Ducasse, M. A., Tarascou, I., Lartigue, J. C., Pinaud, N., … Laguerre, M. (2008). Modeling procyanidin self-association processes and understanding their micellar organization: A study by diffusion NMR and molecular mechanics. Langmuir, 24(19), 11027–11035. https://doi.org/10.1021/la8015904.
Poklar Ulrih, N. (2017). Analytical techniques for the study of polyphenol–protein interactions. Critical Reviews in Food Science and Nutrition, 57(10), 2144–2161. https://doi.org/10.1080/10408398.2015.1052040.
Prigent, S. V. E., Gruppen, H., Visser, A. J. W. G., van Koningsveld, G. A., de Jong, G. A. H., & Voragen, A. G. J. (2003). Effects of non-covalent interactions with 5-O-caffeoylquinic acid (chlorogenic acid) on the heat denaturation and solubility of globular proteins. Journal of Agricultural and Food Chemistry, 51(17), 5088–5095. https://doi.org/10.1021/jf021229w.
Rahnasto-rilla, M., Tyni, J., Huovinen, M., Ja, E., & Kulikowicz, T. (2018). Natural polyphenols as sirtuin 6 modulators. Scientific Reports, 8, 4163. https://doi.org/10.1038/s41598-018-22388-5.
Rai, S., Kureel, A. K., Dutta, P. K., & Mehrotra, G. K. (2018). Phenolic compounds based conjugates from dextran aldehyde and BSA: Preparation, characterization and evaluation of their anti-cancer efficacy for therapeutic applications. International Journal of Biological Macromolecules, 110, 425–436. https://doi.org/10.1016/j.ijbiomac.2017.11.049.
Raj, U., Kumar, H., & Varadwaj, P. K. (2017). Molecular docking and dynamics simulation study of flavonoids as BET bromodomain inhibitors. Journal of Biomolecular Structure and Dynamics, 35(11), 2351–2362. https://doi.org/10.1080/07391102.2016.1217276.
Rawel, H. M., Huschek, G., Sagu, S. T., & Homann, T. (2019). Cocoa bean proteins-characterization, changes and modifications due to ripening and post-harvest processing. Nutrients, 11(2), 428. https://doi.org/10.3390/nu11020428.
Rawel, H. M., & Rohn, S. (2010). Nature of hydroxycinnamate-protein interactions. Phytochemistry Reviews, 9(1), 93–109. https://doi.org/10.1007/s11101-009-9154-4.
Rocasalbas, G., Francesko, A., Touriño, S., Fernández-Francos, X., Guebitz, G. M., & Tzanov, T. (2013). Laccase-assisted formation of bioactive chitosan/gelatin hydrogel stabilized with plant polyphenols. Carbohydrate Polymers, 92(2), 989–996. https://doi.org/10.1016/j.carbpol.2012.10.045.
Rocha, B. A. M., Teixeira, C. S., Silva-filho, J. C., Nóbrega, R. B., Alencar, D. B., Nascimento, K. S., … Delatorre, P. (2015). Structural basis of ConM binding with resveratrol, an anti-inflammatory and antioxidant polyphenol. International Journal of Biological Macromolecules, 72, 1136–1142. https://doi.org/10.1016/j.ijbiomac.2014.08.031.
Roche, D. B., Brackenridge, D. A., & McGuffin, L. J. (2015). Proteins and their interacting partners: An introduction to protein-ligand binding site prediction methods. International Journal of Molecular Sciences, 16(12), 29829–29842. https://doi.org/10.3390/ijms161226202.
Rohn, S., Rawel, H. M., & Kroll, J. (2002). Inhibitory effects of plant phenols on the activity of selected enzymes. Journal of Agricultural and Food Chemistry, 50(12), 3566–3571. https://doi.org/10.1021/jf011714b.
Rohn, S., Rawel, H. M., & Kroll, J. (2004). Antioxidant activity of protein-bound quercetin. Journal of Agricultural and Food Chemistry, 52(15), 4725–4729. https://doi.org/10.1021/jf0496797.
Sahihi, M., Heidari-Koholi, Z., & Bordbar, A. K. (2012). The interaction of polyphenol flavonoids with -lactoglobulin: Molecular docking and molecular dynamics simulation studies. Journal of Macromolecular Science, Part B: Physics, 51(12), 2311–2323. https://doi.org/10.1080/00222348.2012.672854.
Samant, S. K., Singhal, R. S., Kulkarni, P. R., & Rege, D. V. (1993). Protein-polysaccharide interactions: A new approach in food formulations. International Journal of Food Science and Technology, 28(6), 547–562. https://doi.org/10.1111/j.1365-2621.1993.tb01306.x.
Sang, S., Lambert, J. D., Hong, J., Tian, S., Lee, M. J., Stark, R. E., … Yang, C. S. (2005). Synthesis and structure identification of thiol conjugates of (−)-epigallocatechin gallate and their urinary levels in mice. Chemical Research in Toxicology, 18(11), 1762–1769. https://doi.org/10.1021/tx050151l.
Scafuri, B., Marabotti, A., Carbone, V., Minasi, P., Dotolo, S., & Facchiano, A. (2016). A theoretical study on predicted protein targets of apple polyphenols and possible mechanisms of chemoprevention in colorectal cancer. Scientific Reports, 6, 32516. https://doi.org/10.1038/srep32516.
Shahidi, F., & Ambigaipalan, P. (2015). Phenolics and polyphenolics in foods, beverages and spices: Antioxidant activity and health effects - A review. Journal of Functional Foods, Part B, 18, 820–897. https://doi.org/10.1016/j.jff.2015.06.018.
Shahidi, F., & Chandrasekara, A. (2017). Interaction of phenolics and their association with dietary fiber: From plant to gut. Dietary Fiber Functionality in Food and Nutraceuticals, 21–44. https://doi.org/10.1002/9781119138105.ch2.
Silva, C., Correia-branco, A., Andrade, N., Ferreira, A. C., Soares, L., Sonveaux, P., … Martel, F. (2019). Selective pro-apoptotic and antimigratory effects of polyphenol complex catechin:lysine 1:2 in breast, pancreatic and colorectal cancer cell lines. European Journal of Pharmacology, 859, 172533. https://doi.org/10.1016/j.ejphar.2019.172533.
Thomas, N. S., George, K., & Anand, A. A. (2018). Anticancer mechanism of troxerutin via targeting Nrf2 and NF-κB signalling pathways in hepatocarcinoma cell line. Toxicology In Vitro, 54, 317–329. https://doi.org/10.1016/j.tiv.2018.10.018.
Vardhan, P. V., & Shukla, L. I. (2017). Gamma irradiation of medicinally important plants and the enhancement of secondary metabolite production. International Journal of Radiation Biology, 93(9), 967–979. https://doi.org/10.1080/09553002.2017.1344788.
Von Staszewski, M., Jara, F. L., Ruiz, A. L. T. G., Jagus, R. J., Carvalho, J. E., & Pilosof, A. M. R. (2012). Nanocomplex formation between β-lactoglobulin or caseinomacropeptide and green tea polyphenols: Impact on protein gelation and polyphenols antiproliferative activity. Journal of Functional Foods, 4(4), 800–809. https://doi.org/10.1016/j.jff.2012.05.008.
Walle, T., Browning, A. M., Steed, L. L., Reed, S. G., & Walle, U. K. (2005). Flavonoid glucosides are hydrolyzed and thus activated in the oral cavity in humans. Journal of Nutrition, 135(1), 48–52. https://doi.org/10.1093/jn/135.1.48.
Wu, H., Zhuo, L., He, Q., Liao, X., & Shi, B. (2009). Heterogeneous hydrogenation of nitrobenzenes over recyclable Pd(0) nanoparticle catalysts stabilized by polyphenol-grafted collagen fibers. Applied Catalysis A: General, 366(1), 44–56. https://doi.org/10.1016/j.apcata.2009.06.024.
**e, Y., **ao, J., Kai, G., & Chen, X. (2012). Glycation of plasma proteins in type II diabetes lowers the non-covalent interaction affinities for dietary polyphenols. Integrative Biology, 4(5), 502–507. https://doi.org/10.1039/c2ib00185c.
Xu, L., Li, W., Chen, Z., Guo, Q., Wang, C., Santhanam, R. K., & Chen, H. (2019). Inhibitory effect of epigallocatechin-3-O-gallate on α-glucosidase and its hypoglycemic effect via targeting PI3K/AKT signaling pathway in L6 skeletal muscle cells. International Journal of Biological Macromolecules, 125, 605–611. https://doi.org/10.1016/j.ijbiomac.2018.12.064.
Yi, J., Zhang, Y., Liang, R., Zhong, F., & Ma, J. (2015). Beta-carotene chemical stability in nanoemulsions was improved by stabilized with beta-lactoglobulin-catechin conjugates through free radical method. Journal of Agricultural and Food Chemistry, 63(1), 297–303. https://doi.org/10.1021/jf5056024.
Yin, J., Hedegaard, R. V., Skibsted, L. H., & Andersen, M. L. (2014). Epicatechin and epigallocatechin gallate inhibit formation of intermediary radicals during heating of lysine and glucose. Food Chemistry, 146, 48–55. https://doi.org/10.1016/j.foodchem.2013.09.032.
You, J., Luo, Y., & Wu, J. (2014). Conjugation of ovotransferrin with catechin shows improved antioxidant activity. Journal of Agricultural and Food Chemistry, 62(12), 2581–2587. https://doi.org/10.1021/jf405635q.
Youn, K., & Jun, M. (2019). Biological evaluation and docking analysis of potent BACE1 inhibitors from Boesenbergia rotunda. Nutrients, 11(3), 662. https://doi.org/10.3390/nu11030662.
Yu, Q., Fan, L., & Duan, Z. (2019). Five individual polyphenols as tyrosinase inhibitors : Inhibitory activity, synergistic effect, action mechanism, and molecular docking. Food Chemistry. https://doi.org/10.1016/j.foodchem.2019.05.184.
Yu, X., Cai, X., Luo, L., Wang, J., Ma, M., Wang, M., & Zeng, L. (2020). Influence of tea polyphenol and bovine serum albumin on tea cream formation by multiple spectroscopy methods and molecular docking. Food Chemistry, 333, 127432. https://doi.org/10.1016/j.foodchem.2020.127432.
Zanchi, D., Vernhet, A., Poncet-Legrand, C., Cartalade, D., Tribet, C., Schweins, R., & Cabane, B. (2007). Colloidal dispersions of tannins in water-ethanol solutions. Langmuir, 23(20), 9949–9959. https://doi.org/10.1021/la700694b.
Zeng, H. J., Sun, D. Q., Chu, S. H., Zhang, J. J., Hu, G. Z., & Yang, R. (2020). Inhibitory effects of four anthraquinones on tyrosinase activity: Insight from spectroscopic analysis and molecular docking. International Journal of Biological Macromolecules, 160, 153–163. https://doi.org/10.1016/j.ijbiomac.2020.05.193.
Zhang, Y., & Zhong, Q. (2012). Binding between bixin and whey protein at pH 7.4 studied by spectroscopy and isothermal titration calorimetry. Journal of Agricultural and Food Chemistry, 60(7), 1880–1886. https://doi.org/10.1021/jf2050262.
Zhao, J., Fu, Y., Yasvoina, M., Shao, P., Hitt, B., O’Connor, T., … Vassar, R. (2007). β-site amyloid precursor protein cleaving enzyme 1 levels become elevated in neurons around amyloid plaques: Implications for Alzheimer’s disease pathogenesis. Journal of Neuroscience, 27(14), 3639–3649. https://doi.org/10.1523/JNEUROSCI.4396-06.2007.
Acknowledgements
Not applicable.
Funding
We are grateful to the National Science and Engineering Research Council (NSERC) of Canada for financial support.
Author information
Authors and Affiliations
Contributions
The study was conceptualized by FS and, CDS participated in literature survey, data collection and drafting the manuscript. The finalization of the draft with editing, providing suggestions and extensive revising was done by FS. Authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
Author Dr. Fereidoon Shahidi is editor-in-chief of Food Production, Processing and Nutrition and he was not involved in the journal's review of, or decisions related to this manuscript..
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Shahidi, F., Dissanayaka, C.S. Phenolic-protein interactions: insight from in-silico analyses – a review. Food Prod Process and Nutr 5, 2 (2023). https://doi.org/10.1186/s43014-022-00121-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s43014-022-00121-0