
Abstract
Background
Anti-leucine-rich glioma-inactivated 1 (LGI-1) autoimmune encephalitis (AE), characterized by rapid decline of memory, seizures, and neuropsychiatric abnormalities, is a rare but devastating disorder. Early diagnosis and treatment are essential to prevent long-term sequelae. In this report, we provide a detailed description of clinical characteristics, laboratory test results, imaging, and electroencephalography (EEG) findings, as well as treatment responses of eight patients with anti-LGI-1 AE treated at our center.
Case presentation
At the onset, all eight patients presented with confusion/memory deterioration, seizures (including faciobrachial dystonic seizures or other types of seizure), and behavioral changes such as hallucination, paranoia, and anxiety. Four patients were found with severe hyponatremia. Anti-LGI1 antibodies were detected in the cerebrospinal fluid and/or serum of all patients. For patients with faciobrachial dystonic seizures, no discernible scalp EEG change was detected, while EEG recording of patients experiencing other types of seizure showed focal slowing, focal epileptiform discharges, and focal onset seizures. All patients showed abnormal brain magnetic resonance imaging signals, mainly involving the mesial temporal lobe and the hippocampus. In addition, one patient also experienced fulminant cerebral edema during the acute phase of the illness. All patients received immunotherapy and anti-seizure medications and achieved good seizure control. Nevertheless, these patients continued to experience cognitive impairment during their long-term follow-ups.
Conclusions
The care of anti-LGI1 AE patients requires rapid evaluation, prompt initiation of immunotherapy, and long-term follow-up. The long-term presence of neurocognitive complications observed in these patients underline the importance of develo** reliable biomarkers that can distinguish between different subtypes of this disease with heterogeneous clinico-electrographico-radiological features. Further research is needed to understand the molecular mechanisms underlying the heterogeneity, in order to facilitate development of more effective treatments for anti-LGI1 AE.
Similar content being viewed by others


Background
It has been over two decades since the discovery of voltage-gated potassium channel (VGKC) complex-related autoimmunity. The VGKC-complex antibodies do not directly interact with the VGKC complexes themselves; instead, they bind to two closely associated proteins: leucine-rich glioma-inactivated 1 (LGI1) and contactin-associated protein-like 2 (Caspr2) [1]. Since the pioneering work by Dalmau's group in 2010, anti-LGI1 autoimmune encephalitis (AE) has been recognized as the most common cause of limbic encephalitis and the second most common cause of AE [2, 3]. Diagnosis of anti-LGI1 AE is based on the detection of anti-LGI1 antibodies in the serum and/or the cerebrospinal fluid (CSF). Anti-LGI1 AE is a heterogeneous condition, as patients typically experience limbic symptoms such as rapidly declining memory and neuropsychiatric/behavioral problems; faciobrachial dystonic seizures (FBDS) or other types of seizure; and metabolic derangements such as hyponatremia [4]. The occurrence of anti-LGI1 AE may vary depending on age and sex, with males over the age of 60 being the most affected group. In some patients, the anti-LGI1 AE may be caused by an underlying tumor or other medical conditions that may trigger the autoimmune response, while others may have no identifiable causes. The heterogeneity of this condition highlights the importance of individualized examination. Timely detection of anti-LGI-1 antibody in either CSF and/or the serum and prompt use of immunotherapy are crucial for a favorable treatment outcome. The clinical course of anti-LGI1 AE is complex, but patients typically respond well to immunotherapy [5].
In this report, we present eight patients diagnosed with anti-LGI1 AE, in the aim to help clinicians better recognize, diagnose, and treat this disease.
Case presentation
We retrospectively analyzed the clinical data, electroencephalography (EEG) and imaging findings, treatment, and outcomes of eight patients diagnosed with anti-LGI1 AE at the Neurology Department of Kaiser Permanente Sacramento Medical Center between 2015 and 2022. The patients included five females and three males who met the following inclusion criteria: (1) rapid onset (within 3 months) of symptoms of memory impairment and/or mental and behavioral abnormalities; (2) occurrence of seizures during the acute or subacute phase of the disease; (3) detection of anti-LGI1 autoantibody in the CSF and/or the serum.
Patients
The patients’ mean age at symptom onset was 64 ± 11 years (median 63.5 years). The latency from the onset of clinical symptoms to confirmed diagnosis ranged from 0.6 to 5 months (mean 2.4 ± 1.6 months, median 2 months), and the follow-up period ranged from 10 to 72 months. Table 1 summarizes the patients' clinical data.
Clinical features
The primary symptoms at onset were seizures and cognitive disorders. In addition to the most obvious cognitive disorders (memory impairment and intermittent confusion with acute or subacute onset), the patients also presented with other neurobehavioral/psychiatric symptoms, including paranoia and anxiety (n = 5 patients), personality changes (n = 1), hallucinations (n = 1), and hypersomnia (n = 1). These neurocognitive/neuropsychiatric manifestations led to earlier suspicion of autoimmune etiology in five patients, and AE panel testing was requested. Serum studies revealed severe hyponatremia in four patients. The CSF profile was less impressive, with only samples from two patients showing signs of pleocytosis. Five patients showed elevated CSF protein levels. Abnormal oligoclonal bands were seen in three of five patients tested (Table 1). Other autoantibodies were also detected in sera of 6 patients (Table 1). Tumor/cancer screening led to the discovery of thymoma in one patient (case #1) who was tested positive for anti-acetylcholine receptor antibodies and striated muscle antibodies in the serum. Case #1 did not have clinical symptoms of myasthenia gravis, nor did the neurodiagnostic testing with nerve conduction studies and electromyography reveal any evidence for myasthenia gravis.
Seizures were manifested as FBDS in four patients and as focal-onset seizures with impaired awareness (focal-onset dyscognitive seizures or complex partial seizures) in five patients. One patient did not exhibit clinical seizures at onset, but EEG monitoring confirmed non-convulsive status epilepticus. Three patients (cases #2, #3, and #4) presented with FBDS at onset. They were misrecognized as focal motor seizures or myoclonus seizures initially, and FBDS were not suspected until the patients were referred to a tertiary epilepsy center and underwent video EEG monitoring (vEEG). Thus, the failure to recognize FBDS caused a delay in making the accurate diagnosis. Once vEEG monitoring confirmed the diagnosis of FBDS, further tests were prompted for these patients, and the test results confirmed the diagnosis; therefore immunotherapies were initiated. In our series of cases, we initiated immunotherapies after the positive antibody diagnosis was confirmed for case #2, case #3, and case #6. Immunotherapies were initiated while waiting for confirmation of diagnosis for case #4 (Table 1).
EEG findings
Notably, FBDS were not accompanied by discernible cerebral EEG pattern changes during clinical events as recorded by vEEG monitoring (Fig. 1a). The most common EEG findings in our patients were (1) interictal focal slowing in the temporal regions, and (2) focal-onset seizures arising from the temporal regions when ictal patterns were recorded (Table 1). Lateralized rhythmic discharges (LRDs) in the left fronto-temporal regions (Fig. 1b) were observed in a patient who later developed non-convulsive status, presenting with asynchronized bilateral rhythmic triphasic-morphology sharp discharges with discernible evolutions (Fig. 1b).

a During faciobrachial dystonic seizure (FBDS), EEG recording did not show discernible changes when the patient displayed mouth tonic twitching (as indicated by the red arrow) (case #2). b Lateralized rhythmic discharges (LRD, prominently seen in the left hemisphere) were seen in the case #5 with non-convulsive status epilepticus (NCSE, ictal patterns recognized as asynchronized bilateral rhythmic triphasic-morphology sharp discharges with discernible evolutions with frequency and voltage amplitude) and fulminant cerebral edema
Imaging results
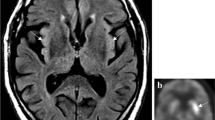
All patients showed abnormal signals in serial brain magnetic resonance imaging (MRI) examinations. Signal changes in the mesial temporal lobe were observed in all patients (Table 1 and Fig. 2a). In one patient (case #5), brain MRI showed diffuse cortical edema that progressed from supratentorial to subtentorial regions within 10 days, representing fulminant cerebral edema (Fig. 2b). Brain FDG-PET (fluorodeoxyglucose positron emission tomography) was performed in one patient with significant FBDS (case #2), revealing hypermetabolism in bilateral basal ganglia (Fig. 2c). In another patient (case #8), brain FDG-PET at 3 months after disease onset showed significant bilateral hypometabolism in the frontal lobes, temporal lobes, and posterior cingulate gyri (Fig. 2d).

a The case #6 developed asymmetric hippocampal atrophy as seen in serial brain MRI images. b Fulminant cerebral edema seen in case #5, more in the left hemisphere. c Brain PET revealed hypermetabolism in the basal ganglion in case #2 with FBDS. d The case #8 showed bilateral hypometabolism in the frontal and temporal lobes with rapid neurocognitive decline (brain PDG-PET scan); biodistribution shows statistically significant hypometabolism with very low Z-scores (-3, shown as purple-blue pseudo-color) in the posterior cingulate gyrus, as well as frontal (left worse than right) and temporal lobes (right worse than left). Upper panel shows the spatial structural localization of the hypometabolism; lower panel shows the fusion of images with low structural resolution to highlight the affected brain regions
Treatment and outcomes
All patients received anti-seizure medications (ASMs) as the first-line treatment. Two patients were empirically treated with antiviral medications at the onset of symptoms, and three patients received prompt immunotherapies while the autoimmune panel results were still pending. The average delay of immunotherapy initiation was 2.4 ± 1.6 months (median, 2 months) after symptom onset. Intravenous infusion of methylprednisolone and intravenous immune globulin were administered to all patients, and one patient also received plasma exchange therapy. Three patients received second-line immunotherapies with rituximab (Table 1). The patients experienced notable improvement of FBDS symptoms and other types of seizure after initiation of the immunotherapies. Only one patient suffered refractory focal dyscognitive seizures and required escalation of ASMs. None of the other patients experienced any recurrent seizures during the follow-up, and three of them successfully achieved ASM weaning.
During the 10–72 months of follow-up, our patients showed notable improvement of seizures and neurobehavioral/psychiatric symptoms. However, all patients continued to experience cognitive difficulties ranging from mild executive functioning, poor short-term memory to more severe neurocognitive disorders (Table 1), which were likely to affect their functional status measured by modified Rankin Scale (mRS). Six patients experienced at least one relapse with neurocognitive symptoms such as confusion or changes in the consciousness level, requiring hospitalization and escalated immunotherapies. The patient with fulminant cerebral edema continued to suffer from intermittent confusion and remained bed-bound during 3–6 months of follow-up. Maximal assistance was provided to this patient for daily activities such as feeding, ambulation, and transfer from bed to commode. At 12 months of follow-up, the patient showed some improvement in physical activity but remained with cognitive difficulty (Table 1).
Discussion
With the discovery of autoimmune encephalitis and the identification of autoantibodies targeting cell surface or synaptic proteins, the number of reported anti-LGI1 AE cases has increased exponentially. However, it remains challenging for healthcare providers to recognize and promptly treat patients with this type of AE due to the heterogeneity of clinical manifestations and the dependence on detecting neuronal autoantibodies [6]. Anti-LGI1 AE now accounts for 11.2% of all autoimmune encephalitis cases, the latter of which is also the most common form of limbic encephalitis. The annual incidence of anti-LGI1 encephalitis is estimated to be 0.83–2 per million persons [7]. Although the exact number of reported cases of LGI1 encephalitis is unclear, a recent systemic review has identified approximately 500 cases reported in literature [8]. Seizures, cognitive impairments, hyponatremia, and abnormal brain MRI T2/FLAIR signals are among the mostly observed clinical manifestations in patients with anti-LGI1 AE [4]. Unlike anti-NMDA AE, which presents more characteristic chronological clinical phases, patients with anti-LGI1 AE present more heterogeneous clinical features as observed in our cases [9]. Such heterogeneity in clinical features can make early and precise diagnosis of anti-LGI1 AE even more challenging.
Heterogeneity of seizure presentation
Patients with anti-LGI1 AE can present different types of seizure in addition to FBDS. In a recent study, patients were subgrouped based on seizure semiology: FBDS alone (FBDS), epileptic seizures without FBDS (non-FBDS), and coexistence of FBDS and other seizures (FBDS +) [10]. FBDS is the most common characteristic semiology, involving ipsilateral (less frequently, bilateral) dystonia-like seizures involving the face and/or the limb. It is estimated that 20-40% of the patients with anti-LGI1 AE have FBDS. However, FBDS is often misrecognized as focal motor or focal myoclonic seizures in general neurology practice due to its relative rarity. In addition, scalp EEG is often negative during FBDS, and the lack of ictal EEG pattern changes makes diagnosis challenging, leading to treatment delays [11]. In our case series, FBDS was not recognized in three patients until their referral to specialized epilepsy programs, representing challenges in diagnosis that often lead to treatment delays. One possible explanation for why scalp EEG fails to detect ictal pattern changes during FBDS is that FBDS is correlated with basal ganglia pathology, of which the electrophysiology changes are often difficult to be detected by scalp EEG [11]. In our patients, focal dyscognitive seizures (focal onset seizures with impaired awareness) and FBDS were two main types of seizure, which may correspond to hippocampus (mesial temporal types of seizures) and/or basal ganglia, two main targets of anti-LGI1 autoantibodies [11]. During treatments for seizure control, FBDS usually show a robust response to immunotherapies compared to other clinical symptoms. The presence of different types of seizures in addition to FBDS may indicate the involvement of a multi-foci epilepsy network, which may account for different seizure semiology and imply a poor prognosis [11]. Most of our patients showed promising seizure control outcome, and three patients even successfully achieved ASM weaning. Only one patient suffered from drug-resistant epilepsy, which might be correlated to the delay in diagnosis and initiation of immunotherapies.
Heterogeneity of brain imaging findings
At the acute phase of anti-LGI1 AE, cerebral involvement is rare. In this report, one of our patients had fulminant cerebral edema, a severe and rapidly progressing form of cerebral pathology often observed in patients with infectious encephalitis but rarely in patients with AE. Other etiologies of the fulminant cerebral edema such as infectious meningoencephalitis, cerebral angiitis, intoxication, and primary central nervous system (CNS) malignancy were ruled out (see Additional file 1: supplemental data). Significant elevation of CSF proteins indicated severe CNS inflammation secondary to the immune response induced by anti-LGI1 autoantibodies, which may potentially interrupt the blood–brain barrier integrity and lead to extensive cerebral inflammation. To our knowledge, this is the first report of fulminant cerebral edema in anti-LGI1 AE. For this patient, recognizing a possible autoimmune etiology prompted medical intervention, which prevented potentially life-threatening detrimental complications.
Abnormal T2/FLAIR signals in the mesial temporal structures are commonly observed in anti-LGI1 AE patients and are believed to be associated with cognitive symptoms [12]. Neuroimaging studies have identified structural and functional differences between anti-LGI1 AE patients and healthy individuals, revealing that the hippocampus, amygdala, and other mesial temporal structures are the most affected brain structures [12]. In the acute phase, the most characteristic alterations are edema and T2/FLAIR hyperintensity in the hippocampus and the mesial temporal structures. Additionally, extratemporal lesions such as basal ganglia hyperintensities have been reported in patients with FBDS during the acute phase [13]. Beyond the subacute phase, the degree of hippocampal atrophy is associated with the severity of long-term cognitive impairment [14]. In our case series, we observed hippocampal atrophy in 4 patients and mesial temporal sclerosis in one patient. The mesial temporal and hippocampal atrophy persisted even after disease remission. In one patient, the brain FDG-PET showed hypometabolism patterns similar to those with fronto-temporal lobe dementia at as early as 3 months after disease onset. These findings indicate that with disease progression of anti-LGI 1 AE, critical structures essential for learning, memory, and executive function are rapidly damaged, leading to long-term or permanent hippocampal and global cerebral dysfunctions despite receiving immunotherapies [15,16,17]. In this report, most of the patients had positive outcomes during long-term follow-up after seizure treatment, yet persistent deficits in memory were observed. This highlights the critical importance of early and effective immunotherapy [16]. In our case series, three patients were only treated with ASMs when presenting with FBDS or other types of seizure. Two patients (cases #6 and #8) experienced relapse with profound neurocognitive impairment. After consultation with a specialized epilepsy program, immunotherapy was initiated, and the patients achieved good neurobehavioral recovery. Unfortunately, brain MRI showed notable hippocampal atrophy, which was likely associated with the lingering poor short-term memory of these patients (Fig. 2a). Other published case studies have shown that FBDS often precede cognitive decline. Therefore, recognizing FBDS before other clinical manifestations and treatment with immunotherapy may help reduce the long-term complications.
Molecular mechanism of the anti-LGI1 antibody-induced pathogenesis
LGI1 is a secreted neuronal protein that is primarily expressed in CA1 and CA3 of the hippocampus as well as the dentate gyrus. The protein has two domains: the N-terminal leucine-rich repeat (LRR) domain, which forms LGI1 dimerization, and the C-terminal epitempin-repeat (EPTP) domain, which mediates LGI1 interaction with a disintegrin and metalloproteinase 23/22 (ADAM23/22), a disintegrin, and metalloproteinase (Fig. 3a). The trans-synaptic LGI1-ADAM complex is vital for synaptic transmission and neuronal excitability [18]. At the presynaptic terminal, ADAM23 interacts with the Kv1 subunit of the voltage-gated potassium channel (VGKC), which is essential for the localization of the Kv1.1 and Kv1.2 subunit complexes to the synaptic terminals. At the postsynaptic terminal, ADAM22 interacts with α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptors (AMPARs) via postsynaptic density protein 95 (PSD95, Fig. 3b) [18]. The exact mechanisms of the effects of autoantibodies against LGI1 are unknown. Nevertheless, research shows that the interaction between the presynaptic LGI1-ADAM23 heterodimer and the postsynaptic LGI1-ADAM22 heterodimer is responsible for synaptic transmission and necessary for hippocampal long-term synaptic plasticity (Fig. 3b). The anti-LGI1 antibody derived from patients’ serum inhibits LGI1 binding to ADAM22 and ADAM23 in vitro, causing a significant reduction in Kv1.1 and synaptic density in the hippocampus of mice, and reversibly reducing synaptic AMPARs in vitro [19]. This provides a potential molecular mechanism for cognitive impairment seen in patients with anti-LGI1 AE. Fels et al. showed that neutralization of LGI1 by anti-LGI1 antibodies reduces AMPAR expression at the surface of both excitatory and inhibitory neurons [19]. They also showed evidence of increased hyperexcitability in mouse hippocampal slices treated with patient’s anti-LGI1 auto-antibodies. Such increased hyperexcitability of the neuronal network is independent from Kv1.1 channels, but rather due to a disturbance of the inhibitory network. Downregulation of the inhibitory network may be a mechanism for the generation of epileptic seizures in patients with anti-LGI1 AE [19].

LGI1 protein structure (a) and LGI1 interaction with other synaptic proteins (b). LGI1: leucin-rich glioma-inactivated 1; LRR: leucin-rich repeat; EPTP: epitempin-repeat; ADAM: a disintegrin and metalloproteinase; Glu: glutamate; Kv: voltage-gated potassium channel; AMPA-R: α-amino-3-hydroxy-5-menthyl-4-isoxazolepropionic acid receptor; PSD-95: postsynaptic density protein 95
Clinicians and researchers have observed a variability in the clinical features of anti-LGI1 AE and the lingering sequelae after treatment. Studies have investigated whether epitope specificity and the antibody’s ability to crosslink LGI1 play a crucial role in the pathogenic mechanisms [20,21,22]. Ramberger et al. found that the administration of the antibody that specifically binds to the LRR domain impairs recognition memory in mice and substantially abrogates long-term potentiation (LTP) induction at CA3-CA1 synapses [20]. Similarly, Extremet et al. discovered that the treatment of neurons with LRR domain-specific antibody increases the intrinsic excitability and reduces the sensitivity to a selective Kv1.1-channel blocker, while the EPTP domain-specific antibody does not [23]. In vitro studies have also found that antibodies binding to the N-terminal LRR domain tend to generate more severe clinical presentations, including FBDS plus seizures, FBDS, and cognitive impairment, whereas those binding to the C-terminal EPTP domain tend to have milder clinical presentations, often with isolated FBDS and absence of cognitive impairment [21]. Ludewig applied purified patient antibodies to mouse hippocampal slices and found that incubation with anti-LGI1 antibodies derived from patients with non-FBDS seizures and cognitive impairment resulted in a significant decline in long-term potentiation or short-term plasticity at CA3-CA1 neurons, as well as decreased hippocampal synaptic density. In contrast, applying anti-LGI1 antibodies derived from FBDS-only patients without cognitive symptoms did not render the same effects [24]. These findings suggest that different epitopes targeted by autoantibodies may play a role in determining the severity of clinical symptoms in anti-LGI1 AE. Therefore, accumulating evidence for the heterogeneities in the cellular and molecular pathogenesis as well as the clinical features of anti-LGI1 AE, underscores the need for vigilance from clinicians and timely treatment with appropriate immunotherapies to avoid widespread disruption of cognitive networks and long-term neurocognitive impairments in verbal and visuospatial memory, executive function, and semantic and phonemic fluency.
Conclusions
Our findings demonstrate a wide spectrum of clinical, electrographic, and radiological features in anti-LGI1 AE, a disease that has varied disease severities and treatment responses. Anti-LGI1 AE should be considered as a disease with several distinct clinical syndromes. As anti-LGI1 AE has similar clinical presentations as viral encephalitis, Hashimoto's encephalopathy, other forms of autoimmune encephalitis, and rapidly progressive dementia (such as Creutzfeldt-Jakob disease), differential diagnosis is important. When patients present new-onset seizures and are resistant to ASM treatments, early referral to a specialized epilepsy program or center, vEEG monitoring, and serial brain imaging are helpful for further diagnosis clarification and treatment guidance. Multidisciplinary approaches may significantly improve the prognosis, potentially improving patients' quality of life.
Availability of data and materials
The datasets during and/or analyzed during the current study are available from the corresponding author on reasonable request.
Abbreviations
- AchR:
-
Acetylcholine receptor
- ADAM:
-
A disintegrin and metalloproteinase
- AE:
-
Autoimmune encephalitis
- AMPA-R:
-
α-amino-3-hydroxy-5-menthyl-4-isoxazolepropionic acid receptor
- ASMs:
-
Anti-seizure medications
- CLQT:
-
Cognitive linguistic quick test
- CNS:
-
Central nervous system
- CSF:
-
Cerebrospinal fluid
- EEG:
-
Electroencephalography
- EPTP:
-
Epitempin-repeat
- FBDS:
-
Faciobrachial dystonic seizures
- FDG-PET:
-
Fluorodeoxyglucose positron emission tomography
- GAD65:
-
Glutamic acid decarboxylase 65-kilodalton isoform
- IVIG:
-
Intravenous immune globulin
- LGI-1:
-
Leucine-rich glioma-inactivated 1
- LRDs:
-
Lateralized rhythmic discharges
- LRR:
-
leucine-rich repeat
- LTP:
-
Long-term potentiation
- MRI:
-
Magnetic resonance imaging
- mRS:
-
Modified rankin scale
- PLEX:
-
Plasmapheresis
- PSD-95:
-
Postsynaptic density protein 95
- TPO:
-
Thyroid peroxidase
- vEEG:
-
Video electroencephalography
- VGKC:
-
Voltage-gated potassium channel
References
Binks SNM, Klein CJ, Waters P, Pittock SJ, Irani SR. LGI1, CASPR2 and related antibodies: a molecular evolution of the phenotypes. J Neurol Neurosurg Psychiatry. 2018;89:526–34.
Lai M, Huijbers MG, Lancaster E, Graus F, Bataller L, Balice-Gordon R, et al. Investigation of LGI1 as the antigen in limbic encephalitis previously attributed to potassium channels: a case series. The Lancet Neurology. 2010;9:776–85.
Zuliani L, Marangoni S, De Gaspari P, Rosellini I, Nosadini M, Fleming JM, et al. Epidemiology of neuronal surface antibody-mediated autoimmune encephalitis and antibody-based diagnostics. J Neuroimmunology. 2021;357:577598.
van Sonderen A, Thijs RD, Coenders EC, Jiskoot LC, Sanchez E, de Bruijn MA, et al. Anti-LGI1 encephalitis: clinical syndrome and long-term follow-up. Neurology. 2016;87(14):1449–56.
van Sonderen A, Schreurs MW, Wirtz PW, Sillevis Smitt PA, Titulaer MJ. From VGKC to LGI1 and Caspr2 encephalitis: the evolution of a disease entity over time. Autoimmun Rev. 2016;15(10):970–4.
Abboud H, Probasco JC, Irani S, Ances B, Benavides DR, Bradshaw M, et al. Autoimmune encephalitis: proposed best practice recommendations for diagnosis and acute management. J Neurol Neurosurg Psychiatry. 2021;92:757–68.
Dubey D, Pittock SJ, Kelly CR, McKeon A, Lopez-Chiriboga AS, Lennon VA, et al. Autoimmune encephalitis epidemiology and a comparison to infectious encephalitis: autoimmune encephalitis. Ann Neurol. 2018;83:166–77.
Teng Y, Li T, Yang Z, Su M, Ni J, Wei M, et al. Clinical Features and Therapeutic Effects of Anti-leucine-rich Glioma Inactivated 1 Encephalitis: A Systematic Review. Front Neurol. 2022;12:791014.
Uy CE, Binks S, Irani SR. Autoimmune encephalitis: clinical spectrum and management. Pract Neurol. 2021;21:412–23.
Chen C, Wang X, Zhang C, Cui T, Shi W-X, Guan H-Z, et al. Seizure semiology in leucine-rich glioma-inactivated protein 1 antibody-associated limbic encephalitis. Epilepsy Behav. 2017;77:90–5.
Roberto KT, Espiritu AI, Fernandez MLL, Gutierrez JC. Electroencephalographic findings in antileucine-rich glioma-inactivated 1 (LGI1) autoimmune encephalitis: A systematic review. Epilepsy Behav. 2020;112:107462.
Rissanen E, Carter K, Cicero S, Ficke J, Kijewski M, Park M-A, et al. Cortical and subcortical dysmetabolism are dynamic markers of clinical disability and course in anti-LGI1 encephalitis. Neurol Neuroimmunol Neuroinflamm. 2022;9(2):e1136.
Qiao J, Zhao X, Wang S, Li A, Wang Z, Cao C, et al. Functional and structural brain alterations in encephalitis with LGI1 antibodies. Front Neurosci. 2020;14:304.
Szots M, Blaabjerg M, Orsi G, Iversen P, Kondziella D, Madsen CG, et al. Global brain atrophy and metabolic dysfunction in LGI1 encephalitis: a prospective multimodal MRI study. J Neurol Sci. 2017;376:159–65.
Guery D, Cousyn L, Navarro V, Picard G, Rogemond V, Bani-Sadr A, et al. Long-term evolution and prognostic factors of epilepsy in limbic encephalitis with LGI1 antibodies. J Neurol. 2022;269:5061–9.
Day GS. Rethinking outcomes in leucine-rich, glioma-inactivated 1 protein encephalitis: “Good” isn’t good enough. JAMA Neurol. 2017;74:19.
Finke C, Prüss H, Heine J, Reuter S, Kopp UA, Wegner F, et al. Evaluation of cognitive deficits and structural hippocampal damage in encephalitis with leucine-rich, glioma-inactivated 1 Antibodies. JAMA Neurol. 2017;74(1):50–9.
Kegel L, Aunin E, Meijer D, Bermingham J.R. LGI proteins in the nervous system. ASN Neuro. 2013;5(3).
Fels E, Muñiz-Castrillo S, Vogrig A, Joubert B, Honnorat J, Pascual O. Role of LGI1 protein in synaptic transmission: From physiology to pathology. Neurobiol Dis. 2021;160:105537.
Ramberger M, Berretta A, Tan JMM, Sun B, Michael S, Yeo T, et al. Distinctive binding properties of human monoclonal LGI1 autoantibodies determine pathogenic mechanisms. Brain. 2020;143:1731–45.
Ohkawa T, Fukata Y, Yamasaki M, Miyazaki T, Yokoi N, Takashima H, et al. Autoantibodies to epilepsy-related LGI1 in limbic encephalitis neutralize LGI1-ADAM22 interaction and reduce synaptic AMPA receptors. J Neurosci. 2013;33:18161–74.
Debanne D, El Far O. Pre- and postsynaptic effects of LGI1 autoantibodies in a murine model of limbic encephalitis. Brain. 2018;141:3092–5.
Extrémet J, El Far O, Ankri N, Irani SR, Debanne D, Russier M. An epitope-specific LGI1-autoantibody enhances neuronal excitability by modulating Kv1.1 Channel. Cells. 2022;11(17):2713.
Ludewig S, Salzburger L, Goihl A, Rohne J, Leypoldt F, Bittner D, Düzel E, et al. Antibody Properties Associate with Clinical Phenotype in LGI1 Encephalitis. Cells. 2023;12:282.
Acknowledgements
Not applicable.
Funding
The study was not supported by any funding.
Author information
Authors and Affiliations
Contributions
NZ collected the clinical information. EH, HG and NZ drafted the manuscript. EH and NZ reviewed the literature and extracted the data. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This study was approved by the Ethics Committee of Kaiser Permanente Medical Center. All patients or the patient’s next-of-kin provided written informed consent to participate if a patient could not sign due to disability. A copy of the written consent is available for review by the Editor of this journal.
Consent for publication
Written informed consent was obtained from the patients or the next-of-kin of patients for publication of this research and any accompanying images.
Competing interests
The authors declare that they have no competing interests.
Supplementary Information
Additional file 1.
Supplement Data for case #5 to explore other etiologies for the cerebral edema.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Huang, E.Y., Gao, H. & Zhong, N. Heterogeneity of clinical features, EEG and brain imaging findings in anti-leucine-rich glioma-inactivated protein 1 autoimmune encephalitis: a retrospective case series study and review of the literature. Acta Epileptologica 5, 21 (2023). https://doi.org/10.1186/s42494-023-00132-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s42494-023-00132-5