
Abstract
Human herpes virus 6 (HHV-6) is a ubiquitous and most common pathogen that affects humans. Human herpes virus 6B (HHV-6B) is a wide spread human herpesvirus that infects most people when they are children, establishes latent infections in the central nervous system (CNS), especially in the hippocampus and amygdala, and induces neurologic diseases. HHV-6 can establish a latent infection and be reactivated by various stimuli. Recently, viral genomic DNA of HHV-6B has been detected in surgically removed brain tissues of intractable epilepsy patients, suggesting the involvement of HHV-6B in the pathogenesis of epilepsy. Temporal lobe epilepsy (TLE) has been shown to be closely related with HHV-6B. TLE patients with HHV-6B in their brains suffer from reiterative attacks of febrile seizures and hippocampal sclerosis. However, the mechanisms underlying the contribution of this virus to the development of TLE remains unknown. The direct damage and immune activation caused by the virus are involved in the process of neuron damage, abnormal neural circuit formation and glial cell proliferation. In addition, some cytokines like interleukin-17A (IL-17A), nuclear factor-kappa B (NF-κb), transforming growth factor-β (TGF-β), mitogen-activated protein kinase (MAPK) and phospholipase A2 are up-regulated and involved in the pathological process of TLE. More studies are needed to clarify the mechanisms underlying the link between HHV-6B and epilepsy, and identify biomarkers to recognize different patient groups for anti-inflammatory or immunomodulatory therapies.
Similar content being viewed by others


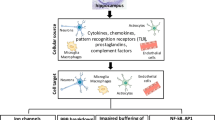
Background
The role of HHV-6B in the development of central nervous system (CNS) diseases is unclear, but the CNS has been suggested as the site of latent and persistent human herpes virus (HHV) infection [1]. Recent studies have suggested that the HHV-6B infection is one of the causal factors for febrile seizure (FS), febrile status epilepticus (FSE), and mesial temporal lobe epilepsy (MTLE) [2]. Inflammation is involved in epilepsy. Cytokines and inflammatory mediators produced by various injuries and infections may lower the seizure threshold, and the occurrence of seizures, in turn, induce up-regulation of inflammatory mediators in the brain [3]. The persistent infection and activation of HHV6B may be related to the chronic inflammatory response in the CNS, thus leading to seizure attacks. In this review, we particularly focus on the associations between HHV-6B infection and different epilepsy syndromes, and discuss the potential role of HHV-6B in the development of chronic epilepsy, which may offer insights into the development of therapeutic methods that target the viral or inflammatory mechanisms at early stage and prevent the progression of epilepsy.
History and structure of HHV-6 virus
In 1986, a novel HHV was first isolated from blood lymphocytes of patients with lymphoproliferative diseases [4], and finally designated as HHV-6. HHV-6 is a herpes simplex virus that has an envelope structure and linear double-stranded DNA. Electron microscopy shows that the virus particle has a diameter of 200 nm, encapsulated by an icosahedral symmetrical capsid surrounded by a crust. HHV-6A and HHV-6B are two variants of HHV-6 [5]. In 2012, the International Committee on Taxonomy of Viruses (ICTV) officially classified HHV-6A and HHV-6B as distinct viruses. HHV-6A and HHV-6B are both neurotropic, but differ in nucleotide sequence, cellular tropism, antigenicity, and etiology [6]. HHV-6B is a wide spread β-herpesvirus that can infect a variety of cell types. Acquired in early childhood, HHV-6B has been etiologically linked to exanthem subitum, a benign or self-limited disease [7, 8].
Some studies have supported that HHV-6 invades the brain by the route of olfactory pathway, which is connected to the limbic lobe [9]. The virus enters host cells through viral envelope proteins that attach to and react on host cell membrane. Approximately 1% of the general population carries HHV-6 DNA chromosomally integrated into every cell of their body [10]. Studies have shown that CD46 is a cellular receptor for HHV-6 [11], and that CD134 is a specific entry receptor for HHV-6B [12]. A life-long subclinical status or incubation period for infection is then established. The virus can remain latent for a long time in the CNS and peripheral blood mononuclear cells. It has been proposed that amygdala and adenoids are two other sites for viral incubation [13]. The virus can be reactivated under various stimuli, especially in the case of immunodeficiency [14].
HHV-6B may establish a life-long latency, and be reactivated to induce encephalitis when the host is at a state of immunodeficiency or hypoimmunity [15]. Almost all children have been infected with HHV-6B by the age of two, and the virus will persist throughout the life at a state of latency [16, 17]. In adults, latent HHV-6B infection may lead to some neurological diseases including multiple sclerosis (MS), MTLE and encephalitis. Patients frequently present seizures and severe neurological complications, such as meningitis or meningoencephalitis. HHV-6B is more associated with MTLE, while DNA and mRNA of HHV-6A are more commonly detected in patients with MS [18].
HHV-6B and FS
The HHV-6B virus infection may play an important role in the pathogenesis of FS, but the specific mechanism remains unclear [19]. Hall et al. have found that in infants and young children HHV-6 infection is a major cause of FS [20]. Suga et al. reported that children with primary HHV-6 infection have significantly higher frequency and older median age of FS than those without [21]. However, some studies have found no difference in the HHV-6 infection rate between patients with and without seizures [22, 23]. Recurrent FS is often associated with MTLE, possibly because that complex FS or a long history of FS may increase the risk of hippocampal sclerosis injury [24,25,26,27]. A large-scale analysis showed that the positive rate of HHV-6B DNA in TLE patients with a previous history of brain inflammation was higher than that in TLE patients without febrile convulsion or meningoencephalitis [26]. Evidence has shown that mesial temporal sclerosis (MTS) is a common cause of chronic mesial temporal epilepsy in adults, and is probably a result from severe febrile seizures at infancy. A FEBSTAT study suggested that FSE is a contributing factor to hippocampal injury [28]. A considerable number of patients remain at the seizure-free latent period after experiencing FS, and after several years develop chronic, progressive MTLE characterized by hippocampal sclerosis.
HHV-6B and TLE
TLE is the most common type of focal epilepsy with unclear pathological mechanism. MTLE patients often present resistance to anti-seizure medicine and need further surgical intervention. The pathologic findings in resected specimens from TLE patients mainly include hippocampal sclerosis, tumor, focal cortical dysplasias, post-infectious (most commonly after type 1 herpes simplex virus encephalitis), vascular malformation, ischemic lesion, old traumatic injuries, and inflammatory lesions [29], of which the hippocampal sclerosis is the most common etiology factor.
Recent studies have revealed that HHV-6B is associated with MTLE and hippocampal sclerosis. The detection of HHV-6B viral DNA were reported in resected temporal lobe and/or hippocampal tissues than in non-epileptogenic controls, with the frequency ranging from about 9 to 69% (Table 1) [2, 13, 25, 26, 30,31,32,33,34,35,36,37], suggest HHV-6B infection is one of the aetiological factor of TLE. In addition, the high levels of HHV-6B/HHV-7 expression have been detected only in patients with MTLE, but not in other focal epilepsies such as prefrontal epilepsy. A study in southwest China showed that the positive rate of HHV-6B DNA and HHV-7 DNA in patients with MTLE was 28 and 32%, respectively, and the mixed infection rate was 11% [34]. Esposito et al. detected the presence of HHV-6B DNA by PCR in 34 out of 346 (9.8%) TLE patients, and found that the positive rate of HHV-6B DNA was 15.0% in patients with clinical signs of previous brain inflammation, but only 6.3% in patients without febrile convulsion or meningoencephalitis [26]. These studies indicate that HHV-6B mainly inhabits in the temporal lobe, especially in the hippocampus, and co-localizes with astrocytes. Astrocyte are the most common host cells for HHV-6B infection. Donati et al. have detected a significant increase in HHV-6 levels in 4 out of 8 MTLE patients by viral-specific real-time polymerase chain reaction (PCR) methods, with highest concentrations in glial fibrillary acidic protein (GFAP, an astrocyte-specific cytoskeleton protein marker)-positive cells of the hippocampus [31]. The co-localization of HHV-6 with GFAP-positive cells confirms the localization of HHV-6 in astrocytes. It is widely known that astrocytes interact closely with neurons and play a key role in regulating synaptic transmission. Some studies have suggested an HHV-6-specific tropism for astrocytes [38]. Viral infection of astrocytes may interfere their function of maintaining the homeostasis of the excitatory neurotransmitter glutamate, finally leading to MTLE.
In 2007, Fotheringham et al. reported detection of HHV-6 antigen in cultured primary astrocytes from epilepsy brain resections that were positive for HHV-6 DNA, and found a marked decrease in the expression of glutamate transporter excitatory amino acid transporter-2 (EAAT-2) after in vitro infection of astrocyte cultures with HHV-6 [2]. As EAAT-2 mediates most of the glutamate uptake in the CNS the decreased EAAT-2 expression indicates that glutamate intake in infected astrocytes is lower than that in uninfected astrocytes. Some studies have shown that the glutamate uptake is increased upon acute infection of astrocytes with HHV-6, and the glutamate uptake would be impaired during persistent HHV-6 infection [39]. In MTLE, glutamate is crucial in the generation and maintenance of seizures. The impaired glutamate reuptake leads imbalance of glutamate concentration inside and outside the cell. Excessive glutamate may have excitotoxicity effects and contribute to neuronal depolarization.
Inflammatory factors induced by HHV-6B
Persistent HHV-6B infection of glial cells can reduce the seizure threshold by triggering an inflammatory response of the CNS. Recent evidence suggests that the inflammatory cytokines play a key role in the pathogenesis of epilepsy [40]. Mao et al. found that the interictal increase of interleukin-17A (IL-17A) was highly correlated with seizure severity, and that IL-17A may promote inflammatory response by activating neutrophils and stimulating the production of proinflammatory cytokines, thus aggravating epilepsy [41]. In 2013, Vezzania summarized the role and mechanisms of inflammatory response in the development of epilepsy, and concluded that cytokines and risk signals induce the expression of inflammatory molecules to directly activate glial or neuronal signal pathways, or lead to blood-brain barrier rupture, lowering the threshold of seizures and causing a cell loss and gliosis, which would further promote inflammatory response, thus forming a vicious circle [42]. Some key inflammatory factors include NF-kB, transforming growth factor-β (TGF-β), mitogen-activated protein kinase (MAPK) and phospholipase A2. Some studies have shown that the levels of transforming growth factor-α (TGF-α) and interleukin-12(IL-12) are higher in patients with elevated HHV-6 load than those without [43]. HHV-6 viral infection may be involved in the development of seizure by promoting the activation and expression of inflammatory factors. In patients with HHV-6 encephalopathy, the level of interleukin-8 (IL-8) in cerebrospinal fluid is significantly higher than the serum level, suggesting that IL-8 plays a role in brain impairment [44]. In addition, high levels of interleukin-1β (IL-1β) and IL-8 have been detected in saliva of seizure patients, especially in patients with HHV-6 infection [23]. Moreover, both inflammation and HHV-6 infection can interrupt the glutamate homeostasis in astrocytes, which may play an important role in the pathogenesis of epilepsy. Inflammatory cytokines such as IL-1β can also suppress glutamate transporter-1 (GLT-1) expression, consequently inhibiting astrocytic reuptake of glutamate and eliciting excitotoxic destruction of neurons [45]. Kawamura et al. have detected elevated levels of GFAP and monocyte chemotactic protein-1(MCP-1) in amygdala samples from MTS patients with positive HHV-6B infection [13]. NF-kB is a well-known nuclear transcription factor involved in multiple physiological and pathological processes such as inflammation, immunity, cell transformation, apoptosis, and excitatory toxicity [46]. NF-kB can be activated at the active state of viral replication [47], and induce the expression of inflammatory factors such as tumor necrosis factor, IL-1, IL-6, P53, CD95 and Bcl-XS. However, overactivation of immune responses induced by HHV-6B was not reported. It showed long-term immunocompatibility of virus in the hippocampus and cortex, which is consistent with the chronic progression of TLE.
Conclusion
In conclusion, accumulating evidence has highlighted the capacity of the HHV-6 virus to establish latent infection and the viral reactivation in the CNS may be related to the pathogenesis of epilepsy. Data on various tissue specimens from epileptic patients have suggested that HHV-6 induces complex inflammatory responses and selectively enhances neuronal excitability, thus triggering seizures. But how this virus contributes to disease pathology and the casual relationship of HHV-6 with epilepsy remain elusive. It is important to confirm the direct or indirect damage of activated virus in the development of TLE, and the role of up-regulated cytokines or inflammatory factors. Meanwhile, more studies are needed to clarify the mechanisms underlying the link between HHV-6 and TLE.
Availability of data and materials
Not applicable.
Abbreviations
- CNS:
-
Central nervous system
- EAAT-2:
-
Excitatory amino acid transporter-2
- FS:
-
Febrile seizures
- GFAP:
-
Glial fibrillary acidic protein
- GLT-1:
-
Glutamate transporters-1
- HHV-6:
-
Human herpes virus-6
- HS:
-
Hippocampal sclerosis
- ICTV:
-
International Committee on Taxonomy of Viruses
- IL-17A:
-
Interleukin-17A
- IL-1β:
-
Interleukin-1β
- IL-1:
-
Interleukin-1
- IL-6:
-
Interleukin-6
- IL-8:
-
Interleukin-8
- IL-12:
-
Interleukin-12
- MAPK:
-
Mitogen-activated protein kinase
- MCP-1:
-
Monocyte chemotactic protein-1
- MS:
-
Multiple sclerosis
- MTLE:
-
Mesial temporal lobe epilepsy
- MTS:
-
Mesial temporal sclerosis
- NF-κb:
-
Nuclear factor-kappa B
- PCR:
-
Polymerase chain reaction
- TGF-β:
-
Transforming growth factor-β
References
Torigoe S, Koide W, Yamada M, Miyashiro E, Tanaka-Taya K, Yamanishi K. Human herpesvirus 7 infection associated with central nervous system manifestations. J Pediatr. 1996;129(2):301–5. https://doi.org/10.1016/S0022-3476(96)70259-7.
Fotheringham J, Donati D, Akhyani N, Fogdell-Hahn A, Vortmeyer A, Heiss JD, et al. Association of human herpesvirus-6B with mesial temporal lobe epilepsy. PLoS Med. 2007;4(5):e180. https://doi.org/10.1371/journal.pmed.0040180.
Vezzani A. Epilepsy and inflammation in the brain: overview and pathophysiology. Epilepsy Curr. 2014;14(2_suppl):3–7. https://doi.org/10.5698/1535-7511-14.s2.3.
Salahuddin S, Ablashi D, Markham P, Josephs S, Sturzenegger S, Kaplan M, et al. Isolation of a new virus, HBLV, in patients with lymphoproliferative disorders. Science (New York, NY). 1986;234(4776):596–601.
Ablashi D, Agut H, Berneman Z, Campadelli-Fiume G, Carrigan D, Ceccerini-Nelli L, et al. Human herpesvirus-6 strain groups: a nomenclature. Arch Virol. 1993;129(1-4):363–6. https://doi.org/10.1007/BF01316913.
Ablashi D, Agut H, Alvarez-Lafuente R, Clark D, Dewhurst S, DiLuca D, et al. Classification of HHV-6A and HHV-6B as distinct viruses. Arch Virol. 2014;159(5):863–70. https://doi.org/10.1007/s00705-013-1902-5.
Yamanishi K, Okuno T, Shiraki K, Takahashi M, Kondo T, Asano Y, et al. Identification of human herpesvirus-6 as a causal agent for exanthem subitum. Lancet (London, England). 1988;1(8594):1065–7.
Campadelli-Fiume G, Mirandola P, Menotti L. Human herpesvirus 6: An emerging pathogen. Emerg Infect Dis. 1999;5(3):353–66. https://doi.org/10.3201/eid0503.990306.
Harberts E, Yao K, Wohler JE, Maric D, Ohayon J, Henkin R, et al. Human herpesvirus-6 entry into the central nervous system through the olfactory pathway. Proc Natl Acad Sci U S A. 2011;108(33):13734–9. https://doi.org/10.1073/pnas.1105143108.
Agut H, Bonnafous P, Gautheret-Dejean A. Laboratory and clinical aspects of human herpesvirus 6 infections. Clin Microbiol Rev. 2015;28(2):313–35. https://doi.org/10.1128/CMR.00122-14.
Santoro F, Kennedy P, Locatelli G, Malnati M, Berger E, Lusso P. CD46 is a cellular receptor for human herpesvirus 6. Cell. 1999;99(7):817–27. https://doi.org/10.1016/S0092-8674(00)81678-5.
Tang H, Serada S, Kawabata A, Ota M, Hayashi E, Naka T, et al. CD134 is a cellular receptor specific for human herpesvirus-6B entry. Proc Natl Acad Sci U S A. 2013;110(22):9096–9. https://doi.org/10.1073/pnas.1305187110.
Kawamura Y, Nakayama A, Kato T, Miura H, Ishihara N, Ihira M, et al. Pathogenic role of human herpesvirus 6B infection in mesial temporal lobe epilepsy. J Infect Dis. 2015;212(7):1014–21. https://doi.org/10.1093/infdis/jiv160.
Caserta M, Mock D, Dewhurst S. Human herpesvirus 6. Clin Infect Dis. 2001;33(6):829–33. https://doi.org/10.1086/322691.
Studahl M, Hagberg L, Rekabdar E, Bergström T. Herpesvirus DNA detection in cerebral spinal fluid: differences in clinical presentation between alpha-, beta-, and gamma-herpesviruses. Scand J Infect Dis. 2000;32(3):237–48. https://doi.org/10.1080/00365540050165857.
Okuno T, Takahashi K, Balachandra K, Shiraki K, Yamanishi K, Takahashi M, et al. Seroepidemiology of human herpesvirus 6 infection in normal children and adults. J Clin Microbiol. 1989;27(4):651–3. https://doi.org/10.1128/JCM.27.4.651-653.1989.
Chapenko S, Trociukas I, Donina S, Chistyakov M, Sultanova A, Gravelsina S, et al. Relationship between beta-herpesviruses reactivation and development of complications after autologous peripheral blood stem cell transplantation. J Med Virol. 2012;84(12):1953–60. https://doi.org/10.1002/jmv.23412.
Alvarez-Lafuente R, De las Heras V, Bartolomé M, Picazo J, Arroyo R. Relapsing-remitting multiple sclerosis and human herpesvirus 6 active infection. Arch Neurol. 2004;61(10):1523–7. https://doi.org/10.1001/archneur.61.10.1523.
Barone S, Kaplan M, Krilov L. Human herpesvirus-6 infection in children with first febrile seizures. J Pediatr. 1995;127(1):95–7. https://doi.org/10.1016/S0022-3476(95)70263-6.
Hall C, Long C, Schnabel K, Caserta M, McIntyre K, Costanzo M, et al. Human herpesvirus-6 infection in children. A prospective study of complications and reactivation. N Engl J Med. 1994;331(7):432–8. https://doi.org/10.1056/NEJM199408183310703.
Suga S, Suzuki K, Ihira M, Yoshikawa T, Kajita Y, Ozaki T, et al. Clinical characteristics of febrile convulsions during primary HHV-6 infection. Arch Dis Child. 2000;82(1):62–6. https://doi.org/10.1136/adc.82.1.62.
Bertolani M, Portolani M, Marotti F, Sabbattini A, Chiossi C, Bandieri M, et al. A study of childhood febrile convulsions with particular reference to HHV-6 infection: pathogenic considerations. Child’s Nerv Syst. 1996;12(9):534–9. https://doi.org/10.1007/BF00261607.
Bartolini L, Piras E, Sullivan K, Gillen S, Bumbut A, Lin C, et al. Detection of HHV-6 and EBV and cytokine levels in saliva from children with seizures: results of a multi-center cross-sectional study. Front Neurol. 2018;9:834. https://doi.org/10.3389/fneur.2018.00834.
Kothur K, Bandodkar S, Wienholt L, Chu S, Pope A, Gill D, et al. Etiology is the key determinant of neuroinflammation in epilepsy: elevation of cerebrospinal fluid cytokines and chemokines in febrile infection-related epilepsy syndrome and febrile status epilepticus. Epilepsia. 2019;60(8):1678–88. https://doi.org/10.1111/epi.16275.
Karatas H, Gurer G, Pinar A, Soylemezoglu F, Tezel G, Hascelik G, et al. Investigation of HSV-1, HSV-2, CMV, HHV-6 and HHV-8 DNA by real-time PCR in surgical resection materials of epilepsy patients with mesial temporal lobe sclerosis. J Neurol Sci. 2008;264(1-2):151–6. https://doi.org/10.1016/j.jns.2007.08.010.
Esposito L, Drexler JF, Braganza O, Doberentz E, Grote A, Widman G, et al. Large-scale analysis of viral nucleic acid spectrum in temporal lobe epilepsy biopsies. Epilepsia. 2015;56(2):234–43. https://doi.org/10.1111/epi.12890.
Kälviäinen R, Salmenperä T. Do recurrent seizures cause neuronal damage? A series of studies with MRI volumetry in adults with partial epilepsy. Prog Brain Res. 2002;135:279–95. https://doi.org/10.1016/S0079-6123(02)35026-X.
Lewis D, Shinnar S, Hesdorffer D, Bagiella E, Bello J, Chan S, et al. Hippocampal sclerosis after febrile status epilepticus: the FEBSTAT study. Ann Neurol. 2014;75(2):178–85. https://doi.org/10.1002/ana.24081.
Al Sufiani F, Ang L. Neuropathology of temporal lobe epilepsy. Epilepsy Res Treat. 2012;2012:624519.
Uesugi H, Shimizu H, Maehara T, Arai N, Nakayama H. Presence of human herpesvirus 6 and herpes simplex virus detected by polymerase chain reaction in surgical tissue from temporal lobe epileptic patients. Psychiatry Clin Neurosci. 2000;54(5):589–93. https://doi.org/10.1046/j.1440-1819.2000.00758.x.
Donati D, Akhyani N, Fogdell-Hahn A, Cermelli C, Cassiani-Ingoni R, Vortmeyer A, et al. Detection of human herpesvirus-6 in mesial temporal lobe epilepsy surgical brain resections. Neurology. 2003;61(10):1405–11. https://doi.org/10.1212/01.WNL.0000094357.10782.F9.
Eeg-Olofsson O, Bergström T, Andermann F, Andermann E, Olivier A, Rydenhag B. Herpesviral DNA in brain tissue from patients with temporal lobe epilepsy. Acta Neurol Scand. 2004;109(3):169–74. https://doi.org/10.1046/j.1600-0404.2003.00238.x.
Niehusmann P, Mittelstaedt T, Bien CG, Drexler JF, Grote A, Schoch S, et al. Presence of human herpes virus 6 DNA exclusively in temporal lobe epilepsy brain tissue of patients with history of encephalitis. Epilepsia. 2010;51(12):2478–83. https://doi.org/10.1111/j.1528-1167.2010.02741.x.
Li J, Lei D, Peng F, Zeng Y, Li L, **a Z, et al. Detection of human herpes virus 6B in patients with mesial temporal lobe epilepsy in West China and the possible association with elevated NF-κB expression. Epilepsy Res. 2011;94(1-2):1–9. https://doi.org/10.1016/j.eplepsyres.2010.11.001.
Huang C, Yan B, Lei D, Si Y, Li H, Chen M, et al. Apolipoprotein 4 may increase viral load and seizure frequency in mesial temporal lobe epilepsy patients with positive human herpes virus 6B. Neurosci Lett. 2015;593:29–34. https://doi.org/10.1016/j.neulet.2014.12.063.
Leibovitch EC, Jacobson S. Human Herpesvirus 6 as a viral trigger in mesial temporal lobe epilepsy. J Infect Dis. 2015;212(7):1011–3. https://doi.org/10.1093/infdis/jiv162.
Wipfler P, Dunn N, Beiki O, Trinka E, Fogdell-Hahn A. The viral hypothesis of mesial temporal lobe epilepsy - is human herpes virus-6 the missing link? A systematic review and meta-analysis. Seizure. 2018;54:33–40. https://doi.org/10.1016/j.seizure.2017.11.015.
Wainwright M, Martin P, Morse R, Lacaze M, Provenzale J, Coleman R, et al. Human herpesvirus 6 limbic encephalitis after stem cell transplantation. Ann Neurol. 2001;50(5):612–9. https://doi.org/10.1002/ana.1251.
Fotheringham J, Williams EL, Akhyani N, Jacobson S. Human herpesvirus 6 (HHV-6) induces dysregulation of glutamate uptake and transporter expression in astrocytes. J NeuroImmune Pharmacol. 2008;3(2):105–16. https://doi.org/10.1007/s11481-007-9084-0.
Pitkänen A, Löscher W, Vezzani A, Becker A, Simonato M, Lukasiuk K, et al. Advances in the development of biomarkers for epilepsy. Lancet Neurol. 2016;15(8):843–56. https://doi.org/10.1016/S1474-4422(16)00112-5.
Mao L, Ding J, Peng W, Ma Y, Zhang Y, Fan W, et al. Interictal interleukin-17A levels are elevated and correlate with seizure severity of epilepsy patients. Epilepsia. 2013;54(9):e142–5. https://doi.org/10.1111/epi.12337.
Vezzani A, Aronica E, Mazarati A, Pittman Q. Epilepsy and brain inflammation. Exp Neurol. 2013;244:11–21. https://doi.org/10.1016/j.expneurol.2011.09.033.
Rasa-Dzelzkaleja S, Gravelsina S, Chapenko S, Krukle Z, Svirskis S, Suna N, et al. Persistent Roseoloviruses infection in adult patients with epilepsy. Brain Sci. 2020;10(5):287. https://doi.org/10.3390/brainsci10050287.
Kawabe S, Ito Y, Ohta R, Sofue A, Gotoh K, Morishima T, et al. Comparison of the levels of human herpesvirus 6 (HHV-6) DNA and cytokines in the cerebrospinal fluid and serum of children with HHV-6 encephalopathy. J Med Virol. 2010;82(8):1410–5. https://doi.org/10.1002/jmv.21808.
Prow N, Irani D. The inflammatory cytokine, interleukin-1 beta, mediates loss of astroglial glutamate transport and drives excitotoxic motor neuron injury in the spinal cord during acute viral encephalomyelitis. J Neurochem. 2008;105(4):1276–86. https://doi.org/10.1111/j.1471-4159.2008.05230.x.
Hayden M, Ghosh S. Shared principles in NF-kappaB signaling. Cell. 2008;132(3):344–62. https://doi.org/10.1016/j.cell.2008.01.020.
Rahman M, McFadden G. Modulation of NF-κB signalling by microbial pathogens. Nat Rev Microbiol. 2011;9(4):291–306. https://doi.org/10.1038/nrmicro2539.
Acknowledgements
We gratefully acknowledge Professor Dong Zhou from Neurology of Sichuan University for support, as well as **nyue Jiang, Le Zhang and Mintao Lin for help and suggestions.
Funding
This review was funded by the Science & Technology Department of Sichuan Province (No.2019YFH0145) and National Natural Science Foundation of China (No.81571272).
Author information
Authors and Affiliations
Contributions
The topic of this review and the management framework were proposed by JML. The manuscript was drafted by JQW. The construction of the framework and the manuscript were further improved by JML. The authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
All authors consented for the publication of this review.
Competing interests
The authors declare no conflicts of interest.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wang, J., Li, J. Temporal lobe epilepsy associated with human herpes virus 6. Acta Epileptologica 3, 10 (2021). https://doi.org/10.1186/s42494-021-00044-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s42494-021-00044-2