
Abstract
Background
In the ice-free area of maritime Antarctica, fungi are the essential functioning group in terrestrial and marine ecosystems. Until now, no study has been conducted to comprehensively assess fungal communities in various habitats in Antarctica. We aimed to characterize fungal communities in the eleven habitats (i.e., soil, seawater, vascular plant, dung, moss, marine alga, lichen, green alga, freshwater, feather) in the Fildes Region (maritime Antarctica) using next-generation sequencing.
Results
A total of 12 known phyla, 37 known classes, 85 known orders, 164 known families, 313 known genera, and 320 known species were detected. Habitat specificity rather than habitat overlap determined the composition of fungal communities, suggesting that, although fungal communities were connected by dispersal at the local scale, the environmental filter is a key factor driving fungal assemblages in the ice-free Antarctica. Furthermore, 20 fungal guilds and 6 growth forms were detected. Many significant differences in the functional guild (e.g., lichenized, algal parasite, litter saprotroph) and growth form (e.g., yeast, filamentous mycelium, thallus photosynthetic) existed among different habitat types.
Conclusion
The present study reveals the high diversity of fungal communities in the eleven ice-free Antarctic habitats and elucidates the ecological traits of fungal communities in this unique ice-free area of maritime Antarctica. The findings will help advance our understanding of fungal diversity and their ecological roles with respect to habitats on a neighbourhood scale in the ice-free area of maritime Antarctica.
Similar content being viewed by others

Background
As a result of climate change, water availability, CO2, temperature, and UV levels have been changing in Antarctica [1]. Notably, ice-free areas currently cover less than one percent of Antarctica [2] and could expand by close to 25 percent by 2100 [3]. Many ice-free areas have emerged from the retreating ice in marine Antarctica, and glacial erosion is the dominant land-forming factor. As ice-free areas are home to Antarctic biodiversity (e.g., microbes, vascular plants, lichens, mosses, algae), the increase in ice-free areas could drastically change the availability and connectivity of biodiversity habitats (e.g., uncover potential new habitats for species [3]). Some non-native species have been introduced to Antarctic ice-free environments by natural dispersal or human activities (e.g., marine invertebrates, inserts, and plants) [4]. In addition, climate warming could influence the composition of microbial communities in Antarctica. Previous studies have shown that warming leads to significant changes in soil fungal abundance in Antarctica [5, 6].
To date, over 1000 non-lichenized fungal species [7] and 500 lichenized fungal species [8] have been recorded by collection or isolation from Antarctica. In recent years, many studies have used the next-generation sequencing (NGS) technique to reveal fungal diversity in various habitats of Antarctica, such as soil [6, 9,10,11,12,13,14], sediment [15], vegetation [14, 16], air [17], and freshwater [14, 18]. These molecular surveys suggest that the true fungal diversity may be far greater than that has been recorded. Besides, there are no studies that consider various habitats (e.g., soil, lichen, vascular plant, moss, freshwater, seawater, dung, air, feather, green alga, and marine alga) as a whole. In terms of microbes, it was believed that "everything is everywhere, but the environment selects" (the Baas-Becking’s hypothesis [19]). This hypothesis states the joint effect of dispersal capabilities (i.e., spores that aid in dispersal and propagation) and environmental selection. It is still unclear to what extent habitat specificity and habitat overlap determine fungal assemblages in a local ice-free area.
In both terrestrial and marine ecosystems, fungi typically live in highly diverse communities and serve a variety of ecological functions, such as saprotrophs (living on dung, leaf, plant, soil, wood), symbiotrophs (participating in mutualistic symbioses: ectomycorrhizal, ericoid mycorrhizal, endophyte, lichenized), and pathogens of plants and animals [20]. However, there is only fragmentary information on fungal ecological traits in the soil habitat of maritime Antarctica [13, 21,22,23]. Until now, the knowledge gap between fungal diversity and their ecological roles is still significant in other habitats of maritime Antarctica.
The present study aims to use the next-generation sequencing (NGS) approach to reveal: (1) the taxonomic diversity of fungal communities in an ice-free area of maritime Antarctica; (2) the extent of habitat specificity and habitat overlap in determining fungal assemblages in various habitat types, including soil, lichen, vascular plant, moss, freshwater, seawater, dung, air, feather, green alga, and marine alga; (3) the ecological traits of fungal communities in various habitats of maritime Antarctica. We hypothesized that environmental selection determined the taxonomic and functional compositions of fungal communities in various habitats from an ice-free area of maritime Antarctica. The findings will improve our understanding of the fungal diversity with respect to environments on a neighbourhood scale, and aid further analysis of fungal ecological roles in this unique ice-free area of maritime Antarctica.
Materials and methods
Sample collection
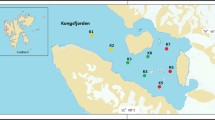
The sampling site is the Fildes Region (King George Island, maritime Antarctica), which is consisted of Fildes Peninsula, Ardley Island, and the northern part of Nelson Island. It is one of the largest ice-free areas in maritime Antarctica and has a relatively high level of biodiversity. The mean annual temperature in this region is − 2.2 °C [24] and has increased on average by 0.7 °C between 1969 and 2013 [25]. In January 2017, a total of 213 samples were collected during the 33rd Chinese National Antarctic Research Expedition (CHINARE-Antarctic) (Fig. 1, Additional file 1: Table S1), including 136 samples in this study (i.e., soil, lichen, vascular plant, freshwater, seawater, dung, air, feather, green alga, and marine alga) and 77 samples in the previous studies (i.e., soil, freshwater, moss, vascular plant) [12, 14].

The location of the sampling site and views of eleven habitats (i.e., soil, freshwater, seawater, air, lichen, vascular plant, moss, green alga, marine alga, dung, and feather) in the Fildes Region (maritime Antarctica)
DNA extraction and sequencing
(1) For samples of soil and dung, DNA extraction was conducted from about 0.25 g aliquot using MoBio PowerSoil DNA isolation kits (MoBio Laboratories Inc., USA). (2) For samples of freshwater and seawater, 1000 ml water was filtered through 0.2 μm-pore-size membranes (Pall Corporation, USA). Total DNA in membranes was extracted using the PowerWater DNA Isolation Kit (MO BIO Laboratories Inc., USA). (3) For samples of vascular plant, lichen, green alga, and marine alga, before DNA extraction, tissues were surface sterilized and crushed as described by Zhang et al. [26]. DNA extraction was performed using a PowerSoil DNA Isolation Kit (MO BIO Laboratories Inc., USA). (4) For bird feather, the feather was cut into small segments with sterilized scissors and then used a SuperFastPrep-1 Instrument (MP Biomedicals Co., USA) to crush segments. DNA extraction was conducted from segments using a PowerSoil DNA Isolation Kit (MO BIO Laboratories Inc., USA). (5) For air samples, total suspended particles of air were collected using portable ambient air samplers (Air Metrics, USA) as described by Yan et al. [27]. DNA extraction was conducted from the membranes of air samples using a PowerSoil DNA Isolation Kit (MO BIO Laboratories Inc., USA).
The obtained DNA was used for subsequent PCR and sequencing. The fungal nuclear ribosomal internal transcribed spacer 1 (ITS1, approximately 285 bp) was amplified using primer sets that were added a 10-nucleotide barcode to ITS1F [28] and ITS2 [29]. PCR was performed as previously described [12]. The PCR products of the ITS region were extracted from 2% agarose gels and purified using an AxyPrep DNA Gel Extraction Kit (Axygen Biosciences, USA) according to the manufacturer’s instructions and were quantified using QuantiFluor-ST (Promega, USA). Equimolar volumes of purified amplicons were pooled and were paired-end sequenced (2 × 300 bp) on an Illumina MiSeq platform at Majorbio Company (Shanghai, China). Both DNA extraction and PCR were applied to negative control samples. These negative controls did not undergo any more analysis because no quantifiable DNA was found in them. Sequencing was conducted using an Illumina MiSeq platform at Majorbio Company (Shanghai, China).
Sequencing data treatment
Paired-end reads were merged using FLASH software [30] and assigned to each sample according to the unique barcodes. The raw demultiplexed sequences were processed in QIIME 2 v2022.01 [31]. Paired-end reads were denoised, dereplicated, and filtered for chimeras using the DADA2 plugin [32], as implemented in QIIME 2. Raw reads were trimmed to include only bases with quality scores > 35. The first 26 and 26 nucleotides of the 5′ end of the forward and reverse sequences, respectively, also were trimmed. The 3′ ends of the forward and reverse sequences were truncated at positions 230 and 220, respectively. The number of sequences used to train the error model was set to 100,000. De novo clustering using a threshold of 100% of similarity was performed using VSEARCH [33], as implemented in QIIME 2. Taxonomic assignments were determined for amplicon sequence variants (ASVs) using classify-sklearn with a naïve Bayes classifer [34] against UNITE Fungi v8.3 reference database [35] pre-trained to ITS1. ASVs with an abundance of less than 5 sequences were removed. Samples were then subsampled to 20,180 sequences per sample. In addition, MAFFT [36] was used to align with representative sequences of ASVs (Additional file 2: Table S2) (FFT-NS-1 method), and a phylogenetic tree was constructed using Average linkage (UPGMA) method.
Statistical analyses
Statistical analysis was carried out on the MicrobiomeAnalyst (marker data profiling, MDP) [37]. The fungal community compositions in the samples were ordinated using Principal Coordinates Analysis (PCoA) with the unweighted UniFrac distance method. Dendrogram analysis (clustering algorithm: Ward; distance measure: unweighted UniFrac distance) of fungal communities in the samples of eleven habitats was also performed to explore their relationships. Analysis of similarity (unweighted UniFrac distances; 999 permutations) was used to validate the dissimilarity of fungal communities among different habitat types. A Venn diagram showing the number of fungal ASVs in eleven habitats was constructed online (www.omicshare.com/tools/Home/Soft/venn). A correlation network analysis was conducted to explore interactions of fungal genera based on Spearman’s rank correlation test (Permutation: 100, p-value threshold: 0.01, correlation threshold: 0.75). A linear discriminant analysis effect size (LEfSe) analysis was used to explore the significantly different fungal taxonomic groups (i.e., phylum, class, order, family, and genus) among the eleven habitat types based on the factorial Kruskal–Wallis test. The ecological traits of fungal communities were determined using FungalTraits [38]. Potentially pathogenic fungal species of interest were determined according to the Atlas of Clinical Fungi [39].
Results
Characteristics of fungal community compositions in the eleven habitats
A total of 9,864,604 raw reads from 213 samples were obtained. The number of raw reads ranged from 30,400 to 139,071 per sample. After being denoised, dereplicated, and filtered for chimeras, the number of reads was reduced to 8,040,848 for a total number of 17,236 ASVs. The number of trimmed reads ranged from 7 to 125,173 per sample (Additional file 3: Table S3). The read number was reduced to 8,035,065 for 15,382 ASVs in 213 samples following the elimination of rare ASVs (less than five reads). Each sequence library was then rarefied to 20,812 reads which retained 4,204,024 reads in 202 samples and 14,814 ASVs. The ASV number detected in each sample was in the range of 2 to 502 (Additional file 4: Table S4). The ASVs could be identified at different taxonomic levels of precision: 9,892 ASVs were assigned to unknown phyla, whereas 4,922 ASVs were assigned to 12 known phyla, 37 known classes, 85 known orders, 164 known families, 313 known genera, and 320 known species.
The stacked bar plots showed that the fungal community compositions differed among the eleven habitats (i.e., air, soil, seawater, vascular plant, dung, moss, marine alga, lichen, green alga, freshwater, feather) (Fig. 2). For example, reads assigned to unknown phylum accounted for the largest proportion of reads in samples, especially in seawater and air. There were more phyla in freshwater than in other habitats (Fig. 2a). At the order level, Helotiales accounted for the largest proportion among the orders in vascular plant, while Thelebolales predominated in dung, Kriegeriales in feather, and Lecanorales in lichen (Fig. 2b).

Bar plots showing the abundance of (a) fungal phyla, (b) fungal orders in the 202 samples collected from eleven habitats in the Fildes Region (maritime Antarctica)
Differentiation between fungal assemblages across habitats
The PCoA diagram showed that the spatial patterns of the fungal communities were highly related to habitat types (Fig. 3a). ANOSIM tests also indicated the fungal community compositions were significantly different among the eleven different habitats (R = 0.5153, p < 0.001). Additionally, ANOSIM tests for the pairwise comparisons revealed the different degrees of similarity of fungal communities among different habitats (Table 1). For example, soil and air did not harbor significantly different fungal communities (R = 0.064393, p > 0.05), whereas fungal communities in air were significantly different from those in green alga (R = 0.91608, p < 0.001). A Venn diagram indicated none of ASVs were shared by all eleven habitats (Fig. 3b). In addition, Dendrogram analysis revealed the relationships of fungal communities among 202 samples collected from the eleven habitats, indicating that the samples clustered by habitat types (Additional file 1: Fig. S1). For example, with regard to fungal community composition, seawater samples clustered together and were separated from the samples in other habitats.

a Principal Coordinates Analysis (PCoA) ordination plot showing spatial pattern of fungal communities in the 202 samples from the eleven habitats; b Venn diagram showing the number of ASVs in the eleven habitats.
The co-occurrence patterns showed that the correlations between the fungal genera were highly related to habitats (Fig. 4). For example, five fungal genera (i.e., Tremella, Trichothecium, Wickerhamomyces, Podospora, and Flavocetraria) were in one module with more connections and occurred in the soil habitat (Fig. 4a), whereas two fungal genera (i.e., Humicola and Debaryomyces) were in another module and occurred in the marine alga habitat (Fig. 4b).

Correlation network analyses showing the correlation of fungal genera among (a) six habitat types (i.e., air, dung, feather, freshwater, seawater, and soil), (b) five habitat types (i.e., green alga, lichen, marine alga, moss, and vascular plant). Each node represents a fungal genus and the size of a node is based on sequence number. The nodes are colored based on habitat types
Many taxonomic groups showed habitat specificity in LEfse analysis (Fig. 5, Additional file 1: Figs. S2, S3, and S4). For example, phylum Ascomycota dominated in the vascular plant and lichen habitats, phylum Basidiomycota in the feather and dung habitats, and phylum Chytridiomycota in the air and freshwater habitats (Additional file 1: Fig. S2). Order Lecanorales was dominated in the lichen and green alga habitats, and order Helotiales in the vascular plant habitat. Moreover, order Wallemiales dominated in the dung habitat, and order Kriegeriales in the feather habitat (Fig. 5a). Genus Phenoliferia and Goffeauzyma dominated in the feather habitat, and genus Simplicillium in the seawater habitat; genera Mastodia and Piskurozyma were more abundant in the green alga habitat; genus Himantormia was more abundant in the lichen habitat; genus Antarctomyces was more abundant in the dung habitat (Fig. 5b).

LEfSe analysis showing (a) the fungal orders and (b) genera that differ significantly among the eleven habitat types in the Fildes Region. Significant orders or genera are ranked by their LDA scores (x-axis). The right heatmap shows whether the relative abundances of orders or genera are higher (red) or lower (blue)
Ecological traits of fungal communities in various habitats
In terms of fungal ecological trait, 20 functional guilds and 6 growth forms were detected in this study. Using Fungaltratis, 2481 fungal ASVs identified at generic level were given guild assignments. A majority were assigned as saprophytic fungi (1899 ASVs), followed by plant pathogens (231 ASVs) and lichenized fungi (138 ASVs) (Additional file 4: Table S4).
By using LEfse analysis, many ecological traits were significantly distinguished among different habitats. With regards to growth form, yeast predominated in the feather and dung habitats, but had low contributions in the moss and vascular plant habitats; thallus photosynthetic predominated in the lichen and green alga habitats (Fig. 6a). With regard to functional guild, lichenized fungi dominated in the lichen habitat, while had low contributions in the marine alga and feather habitats; algal parasite fungi dominated in the green alga and marine alga habitats (Fig. 6b).

LEfSe analysis showing (a) the growth forms and (b) functional guilds that differ significantly among the eleven habitat types in the Fildes Region. Significant growth forms or lifestyles are ranked by their LDA scores (x-axis). The right heatmap shows whether the relative abundances of growth forms or functional guilds are higher (red) or lower (blue)
A total of 43 fungal species were detected as potential human pathogens (Additional file 1: Table S5). The common species were Pseudogymnoascus destructans (68 samples), Malassezia restricta (61 samples), Alternaria tenuissima (33 samples), Cladosporium cladosporioides (30 samples), Parengyodontium album (21 samples), Aspergillus sydowii (18 samples), Aspergillus penicillioides (16 samples), and Fusarium solani (11 samples). In contrast, several fungal species (e.g., Acremonium spinosum, Wickerhamomyces anomalus) were infrequently detected.
Discussion
Antarctic ice-free areas are a unique laboratory for understanding cold adaptation, the spread, and the colonization of microbes in extreme habitats. To date, the majority of studies on fungal communities have been conducted based on single habitat in the Antarctic ice-free areas and a comprehensive study involving various habitats remains scarce.
In this study, a total of 14,814 fungal ASVs, 313 known genera, and 320 known species were detected from the eleven habitats in the Fildes Region (maritime Antarctica). In previous studies that used next-generation sequencing, 87 genera and 123 species were detectable in the Antarctic soils [10]; Rosa et al. [17] identified 186 fungal amplicon sequence variants (ASVs) from air collected from King George Island (marine Antarctica); Rosa et al. [11] identified 346 fungal ASVs from soil samples in Deception Island (marine Antarctica). A total of 12 known phyla, including Ascomycota, Basidiomycota, Chytridiomycota, Mortierellomycota, Rozellomycota, Monoblepharomycota, Glomeromycota, Kickxellomycota, Zoopagomycota, Aphelidiomycota, Olpidiomycota, and Basidiobolomycota, were detected in this study. In a previous study, the phyla Ascomycota, Mortierellomycota, Basidiomycota, Chytridiomycota, Rozellomycota, Mucoromycota, Calcarisporiellomycota, and Zoopagomycota were detected in the soils from maritime Antarctica using DNA metabarcoding [13]. The phylum Ascomycota was dominant in Antarctic glacial ice fragments, followed by Basidiomycota and Mortierellomycota as revealed by amplicon-metagenome analysis [40]. de Souza et al. [18] found phyla Ascomycota, Basidiomycota, Mortierellomycota, Chytridiomycota, and Rozellomycota in two Antarctic lakes using amplicon-metagenome analysis. Overall, our data provided a considerably more comprehensive exploration of the fungal diversity in the Antarctic ice-free area.
Antarctica has a number of important environmental pressures (i.e., temperature, solar radiation, salinity, soil parameters, pH), which may be an important factor affecting fungal community composition. In this study, we found that fungal communities were significantly different among the eleven habitats. At a local scale, high habitat specificity in fungal communities may be attributable to the differences in current habitat factors (e.g., physicochemical factors, nutrient contents, antagonistic factors) or historical factors (e.g., availability for fungal colonization). For example, vegetation micro-niche availability (e.g., green alga in the moist niches and lichens in the dry niches) and physiological attributes (i.e., chemical defenses, or nutrient contents) may directly affect fungal communities in different hosts (i.e., lichen, moss, vascular plant, green alga, and marine alga) [41, 42]. Solar radiation is an important environmental factor affecting the composition of Antarctic fungal communities [43] and different habitats in the Fildes Region (e.g., soil, air, lichen thallus, or plant tissue), have different levels of solar radiation. The environmental conditions of marine habitats (e.g., salinity) are very different from those of terrestrial habitats and thereby fungal communities in freshwater and seawater were significantly different.
In this study, many fungal guilds were significantly different among various habitats. The most prevalent fungal guild identified in this study was saprotroph (e.g., soil saprotroph, litter saprotroph, wood saprotroph) (Additional file 4: Table S4). In soils, saprotroph fungi play an important ecological role in organic matter decomposition and nutrient cycling through their secretion of extracellular enzyme activities [44]. In seawater, fungi as saprotrophs, interact with marine phytoplankton and can have a significant impact on primary production dynamics and carbon flux in the marine food chain [45]. We found lichenized (functional guild), thallus photosynthetic (growth form), and Lecanorales (fungal order consisting mainly of lichenized taxa) dominated in the lichen habitat (Figs. 5a and 6). In this ice-free area of maritime Antarctica, lichenized fungi, as dominant vegetation, are the main supports for primary production (capable of supporting photosynthesis) [46]. In addition, Kriegeriales (order consisting mainly of yeast taxa) and yeast (growth form) predominated in the feather habitat (Figs. 5a and 6a). In our unpublished data, the colonization rate of cultured yeasts was also higher in feather samples than in other sample types.
Temperature changes caused by global climate change are an important factor affecting the composition of fungal communities. Previous studies have shown that warming leads to significant changes in fungal abundance [5, 6]. In the soils of maritime Antarctica, the richness, relative abundance, and composition of fungal guilds and growth forms are influenced by air temperature and edaphic factors [21]. In addition, new ice-free areas and new climatically suitable habitats may facilitate the establishment of fungal species, either naturally (e.g., lichen-, plant-, moss-, green alga-, air-, and seawater-associated fungi) or by the accidental introduction from animals (e.g., penguin-, bird-, and human-associated fungi). It is estimated that climate warming in marine Antarctica may lead to changes of habitats in ice-free areas, and then affects whole fungal communities in the ice-free area.
Climate warming in the Antarctic regions may also increase the risk of fungal diseases. Schütte et al. [47] noted an increase in the relative abundance of potential fungal pathogens after the thawing of the permafrost in Alaska. According to the Atlas of Clinical Fungi [39], this study revealed the broad spectrum of potential fungal pathogens of humans (43 species) in an Antarctic ice-free area. Previous studies have revealed the occurrence of potential fungal pathogens in the Antarctic environments, including Aspergillus fumigatus, Byssochlamys spectabilis, Chrysosporium keratinophilum, Cryptococcus laurentii, Penicillium chrysogenum, Rhizopus oryzae, and Rhodotorula mucilaginosa, which were isolated from ornithogenic soils and displayed virulence capabilities [48]. Our results based on next-generation sequencing provide an indicator of the potential health risk and further analyses of fungal isolates are needed to assess their virulences which are crucial for pathogenicity.
Conclusion
The present study reveals the high diversity of fungal communities in the eleven different habitats and elucidates the ecological traits of fungal communities in an Antarctica ice-free area. We thereby conclude that habitat specificity rather than habitat overlap determined the distribution of fungal communities, suggesting that although fungal communities were connected by dispersal at the local scale, the environmental filter is a key factor driving fungal assemblages in this Antarctic ice-free area.
Availability of data and materials
The datasets generated during and/or analysed during the current study are available in the National Center for Biotechnology Information under the BioProject ID PRJNA509411.
References
Turner J, Colwell SR, Marshall GJ, Lachlan-Cope TA, Carleton AM, Jones PD, et al. Antarctic climate change during the last 50 years. Int J Climatol. 2005;25(3):279–94.
Burton-Johnson A, Black M, Fretwell PT, Kaluza-Gilbert J. An automated methodology for differentiating rock from snow, clouds and sea in Antarctica from Landsat 8 imagery: a new rock outcrop map and area estimation for the entire Antarctic continent. Cryosphere. 2016;10(4):1665–77.
Lee JR, Raymond B, Bracegirdle TJ, Chades I, Fuller RA, Shaw JD, Terauds A. Climate change drives expansion of Antarctic ice-free habitat. Nature. 2017;547:49–54.
Hughes KA, Pescott OL, Peyton J, Adriaens T, Cottier-Cook EJ, Key G, et al. Invasive non-native species likely to threaten biodiversity and ecosystems in the Antarctic Peninsula region. Global Change Biol. 2020;26(4):2702–16.
Yergeau E, Bokhorst S, Kang S, Zhou J, Greer CW, Aerts R, et al. Shifts in soil microorganisms in response to warming are consistent across a range of Antarctic environments. ISME J. 2012;6:692–702.
Newsham KK, Davey ML, Hopkins DW, Dennis PG. Regional diversity of maritime antarctic soil fungi and predicted responses of guilds and growth forms to climate change. Front Microbiol. 2020;11: 615659.
Bridge PD, Spooner BM. Non-lichenized Antarctic fungi: transient visitors or members of a cryptic ecosystem? Fungal Ecol. 2012;5:381–94.
Øvstedal DO, Schaefer CEGR. A new species from Heritage Range, Ellsworth mountains, Antarctica. Hoehnea. 2013;40:361–4.
Newsham KK, Hopkins DW, Carvalhais LC, Fretwell PT, Rushton SP, O’Donnell AG, et al. Relationship between soil fungal diversity and temperature in the maritime Antarctic. Nat Clim Change. 2016;6:182.
Baeza M, Barahona S, Alcaino J, Cifuentes V. Amplicon-metagenomic analysis of fungi from Antarctic terrestrial habitats. Front Microbiol. 2017;8:2235.
Rosa LH, da Silva TH, Ogaki MB, Pinto OHB, Stech M, Convey P, et al. DNA metabarcoding uncovers fungal diversity in soils of protected and non-protected areas on Deception Island, Antarctica. Sci Rep. 2020;10:21986.
Zhang T, Wang NF, Yu LY. Soil fungal community composition differs significantly among the Antarctic, Arctic, and Tibetan Plateau. Extremophiles. 2020;24:821–9.
da Silva TH, Câmara PE, Pinto OHB, Carvalho-Silva M, Oliveira FS, Convey P, et al. Diversity of fungi present in permafrost in the South Shetland Islands, maritime Antarctic. Microb Ecol. 2022;83:58–67.
Zhang T, Wang NF, Yu LY. Geographic distance and habitat type influence fungal communities in the Arctic and Antarctic sites. Microb Ecol. 2021;82:224–32.
Ogaki MB, Camara P, Pinto OHB, Lirio JM, Coria SH, Vieira R, et al. Diversity of fungal DNA in lake sediments on Vega Island, north-east Antarctic Peninsula assessed using DNA metabarcoding. Extremophiles. 2021;25:257–65.
Park CH, Kim KM, Elvebakk A, Kim OS, Jeong G, Hong SG. Algal and fungal diversity in Antarctic lichens. J Eukaryot Microbiol. 2015;62:196–205.
Rosa LH, Pinto OHB, Convey P, Carvalho-Silva M, Rosa CA, Camara P. DNA Metabarcoding to assess the diversity of airborne fungi present over Keller Peninsula, King George Island, Antarctica. Microb Ecol. 2021;82:165–72.
de Souza LM, Ogaki MB, Câmara PE, Pinto OH, Convey P, Carvalho-Silva M, et al. Assessment of fungal diversity present in lakes of Maritime Antarctica using DNA metabarcoding: a temporal microcosm experiment. Extremophiles. 2021;25:77–84.
Baas Becking LGM. Geobiologie of inleiding tot de milieukunde.The Hague, the Netherlands: W.P. Van Stockum & Zoon. 1934.
Nguyen NH, Song Z, Bates ST, Branco S, Tedersoo L, Menke J, et al. FUNGuild: an open annotation tool for parsing fungal community datasets by ecological guild. Fungal Ecol. 2016;20:241–8.
Newsham KK, Davey ML, Hopkins DW, Dennis PG. Regional diversity of maritime Antarctic soil fungi and predicted responses of guilds and growth forms to climate change. Front Microb. 2021;11: 615659.
Canini F, Geml J, Buzzini P, Turchetti B, Onofri S, D’Acqui LP, et al. Growth forms and functional guilds distribution of soil fungi in coastal versus inland sites of Victoria Land, Antarctica. Biology. 2021;10(4):320.
Canini F, Geml J, D’Acqui LP, Buzzini P, Turchetti B, Onofri S, et al. Fungal diversity and functionality are driven by soil texture in Taylor Valley Antarctica. Fungal Ecol. 2021;50: 101041.
Michel RFM, Schaefer CEGR, Simas FMB, Francelino MR, Fernandes-Filho EI, Lyra GB, et al. Active-layer thermal monitoring on Fildes Peninsula, King George Island, maritime Antarctica. Solid Earth. 2014;5(2):1361–74.
Braun C, Esefeld J, Peter HU. Monitoring the consequences of local climate change on the natural resources of the ice-free regions of Maxwell Bay (King George Island, Antarctic). Umweltbundesamt Texte. 2017;26:1–177.
Zhang T, Wei XL, Zhang YQ, Liu HY, Yu LY. Diversity and distribution of lichen-associated fungi in the Ny-Ålesund Region (Svalbard, High Arctic) as revealed by 454 pyrosequencing. Sci Rep. 2015;5:14850.
Yan D, Zhang T, Su J, Zhao LL, Wang H, Fang XM, Zhang YQ, Liu HY, Yu LY. Diversity and composition of airborne fungal community associated with particulate matters in Bei**g during haze and non-haze days. Front Microb. 2016;7:487.
Gardes M, Bruns TD. ITS primers with enhanced specificity for basidiomycetes—application to identification of mycorhizae and rusts. Microb Ecol. 1993;2:113–8.
White TJ, Bruns T, Lee SJWT, Taylor JL. Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics. PCR Protoc Guide Methods Appl. 1990;18(1):315–22.
Magoč T, Salzberg SL. FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinformatics. 2011;27:2957–63.
Bolyen E, Rideout JR, Dillon MR, Bokulich NA, Abnet C, Al-Ghalith GA, et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat Biotechnol. 2019;3:852–7.
Callahan BJ, McMurdie PJ, Rosen MJ, Han AW, Johnson AJA, Holmes SP. DADA2: high-resolution sample inference from Illumina amplicon data. Nat Methods. 2016;13:581–3.
Rognes T, Flouri T, Nichols B, Quince C, Mahé F. VSEARCH: a versatile open source tool for metagenomics. PeerJ. 2016;4: e2584.
Bokulich NA, Kaehler BD, Rideout JR, Dillon M, Bolyen E, Knight R, et al. Optimizing taxonomic classification of marker-gene amplicon sequences with QIIME 2’s q2-feature-classifier plugin. Microbiome. 2018;6:90.
Abarenkov K, Zirk A, Piirmann T, Pöhönen R, Ivanov F, Nilsson RH, et al. UNITE QIIME release for Fungi 2. Version 10.05.2021. 2021;UNITE Community.
Katoh K, Rozewicki J, Yamada KD. MAFFT online service: multiple sequence alignment, interactive sequence choice and visualization. Brief Bioinform. 2019;20:1160–6.
Dhariwal A, Chong J, Habib S, King IL, Agellon LB, **a J. MicrobiomeAnalyst: a web-based tool for comprehensive statistical, visual and meta-analysis of microbiome data. Nucleic Acids Res. 2017;5:W180-8.
Põlme S, Abarenkov K, Henrik Nilsson R, Lindahl BD, Clemmensen KE, Kauserud H, et al. FungalTraits: a user-friendly traits database of fungi and fungus-like stramenopiles. Fungal Divers. 2020;105:1–16.
de Hoog GS, Guarro J, Gené J, Ahmed SA, Al-Hatmi AMS, Figueras MJ, et al. Atlas of clinical fungi: the ultimate benchtool for diagnostics, 4th edition. Utrecht/Reus.2020.
de Menezes GCA, Câmara PEAS, Pinto OHB, Convey P, Carvalho-Silva M, Simoes JC, et al. Fungi in the Antarctic cryosphere: Using DNA metabarcoding to reveal fungal diversity in glacial ice from the Antarctic Peninsula Region. Microb Ecol. 2022;83:647–57.
Roser DJ, Melick DR, Ling HU, Seppelt RD. Polyol and sugar content of terrestrial plants from continental Antarctica. Antarct Sci. 1992;4:413–20.
Davey ML, Nybakken L, Kauserud H, Ohlson M. Fungal biomass associated with the phyllosphere of bryophytes and vascular plants. Mycol Res. 2009;113:1254–60.
Ruisi S, Barreca D, Selbmann L, Zucconi L, Onofri S. Fungi in Antarctica. Rev Environ Sci Bio. 2006;6:127–41.
Talbot JM, Bruns TD, Smith DP, Branco S, Glassman SI, Erlandson S, et al. Independent roles of ectomycorrhizal and saprotrophic communities in soil organic matter decomposition. Soil Biol Biochem. 2013;57:282–91.
Hassett BT, Borrego EJ, Vonnahme TR, Rama T, Kolomiets MV, Gradinger R. Arctic marine fungi: biomass, functional genes, and putative ecological roles. ISME J. 2019;13:1484–96.
Lindsay DC. The role of lichens in Antarctic ecosystems. Bryologist. 1978;2:268–76.
Schütte UME, Henning JA, Ye Y, Bowling A, Ford J, Genet H, et al. Effect of permafrost thaw on plant and soil fungal community in a boreal forest: does fungal community change mediate plant productivity response? J Ecol. 2019;107:1737–52.
de Sousa JRP, Goncalves VN, de Holanda RA, Santos DA, Bueloni CFLG, Costa AO, et al. Pathogenic potential of environmental resident fungi from ornithogenic soils of Antarctica. Fungal Biol. 2017;121(12):991–1000.
Acknowledgements
No applicable.
Funding
This research was supported by Projects of the Chinese Arctic and Antarctic Administration, State Oceanic Administration; CAMS Innovation Fund for Medical Sciences (Grant No. 2021-I2M-1–055); National Microbial Resource Center (Grant No. NMRC-2020–3); Non-profit Central Research Institute Fund of CAMS (Grant No. 2021-PT350-001); the Chinese Polar Environmental Comprehensive Investigation & Assessment Programs (CHINARE-02-01).
Author information
Authors and Affiliations
Contributions
The study was designed by TZ. TZ collected samples, conducted lab work and data analysis, and wrote the manuscript; DY and XFC conducted parts of data analysis; ZQJ collected seawater samples; YLY contributed to planning the project. All authors reviewed the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1.
Table S1. Information on the 213 samples collected from the Fildes Region (maritime Antarctica). Table S5. An overview of potentially pathogenic fungi found in the eleven habitats from the Fildes Region (maritime Antarctica). Fig. S1. Dendrogram showing fungal communities in the 202 samples of eleven habitats from the Fildes Region (maritime Antarctica). Fig. S2. LEfSe analysis showing the fungal phyla that are significantly different among the eleven habitats in the Fildes Region (maritime Antarctica). Significant phyla are ranked by their LDA scores (x-axis). The right heatmap shows whether the relative abundances of phyla are higher (red) or lower (blue). Fig. S3. LEfSe analysis showing the fungal classes that are significantly different among the eleven habitats in the Fildes Region (maritime Antarctica). Significant classes are ranked by their LDA scores (x-axis). The right heatmap shows whether the relative abundances of classes are higher (red) or lower (blue). Fig. S4. LEfSe analysis showing the fungal families that are significantly different among the eleven habitats in the Fildes Region (maritime Antarctica). Significant families are ranked by their LDA scores (x-axis). The right heatmap shows whether the relative abundances of families are higher (red) or lower (blue).
Additional file 2.
Table S2. Representative sequence of fungal ASV detected in the 213 samples.
Additional file 3.
Table S3. Information on the 17,236 fungal ASVs in the 213 samples collected from the Fildes Region (maritime Antarctica).
Additional file 4.
Table S4. Information on the 14,814 fungal ASVs in the 202 samples collected from the Fildes Region (maritime Antarctica).
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Zhang, T., Yan, D., Ji, Z. et al. A comprehensive assessment of fungal communities in various habitats from an ice-free area of maritime Antarctica: diversity, distribution, and ecological trait. Environmental Microbiome 17, 54 (2022). https://doi.org/10.1186/s40793-022-00450-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40793-022-00450-0