
Abstract
Background
Fungi and bacteria coexist in a wide variety of environments, and their interactions are now recognized as the norm in most agroecosystems. These microbial communities harbor keystone taxa, which facilitate connectivity between fungal and bacterial communities, influencing their composition and functions. The roots of most plants are associated with arbuscular mycorrhizal (AM) fungi, which develop dense networks of hyphae in the soil. The surface of these hyphae (called the hyphosphere) is the region where multiple interactions with microbial communities can occur, e.g., exchanging or responding to each other’s metabolites. However, the presence and importance of keystone taxa in the AM fungal hyphosphere remain largely unknown.
Results
Here, we used in vitro and pot cultivation systems of AM fungi to investigate whether certain keystone bacteria were able to shape the microbial communities growing in the hyphosphere and potentially improved the fitness of the AM fungal host. Based on various AM fungi, soil leachates, and synthetic microbial communities, we found that under organic phosphorus (P) conditions, AM fungi could selectively recruit bacteria that enhanced their P nutrition and competed with less P-mobilizing bacteria. Specifically, we observed a privileged interaction between the isolate Streptomyces sp. D1 and AM fungi of the genus Rhizophagus, where (1) the carbon compounds exuded by the fungus were acquired by the bacterium which could mineralize organic P and (2) the in vitro culturable bacterial community residing on the surface of hyphae was in part regulated by Streptomyces sp. D1, primarily by inhibiting the bacteria with weak P-mineralizing ability, thereby enhancing AM fungi to acquire P.
Conclusions
This work highlights the multi-functionality of the keystone bacteria Streptomyces sp. D1 in fungal-bacteria and bacterial-bacterial interactions at the hyphal surface of AM fungi.
Video Abstract
Similar content being viewed by others

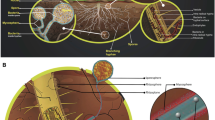
Background
Bacteria and fungi coexist in a wide variety of environments, and their interactions (so-called bacterial-fungal interactions (BFIs)) are crucial in the functioning of many agroecosystems, driving biogeochemical cycles and contributing to plant nutrition and health [1, 2]. Recently, co-occurrence analysis between bacteria and fungi revealed complex ecological processes such as cross-feeding, competition, and predation [3,4,5]. Interactions involving keystone species — defined as “highly interconnected species that drive community responses through microbe-microbe interactions” [1] — have been reported in the phyllosphere microbiome as well as in leaf litter, soils, and plants [3, 5,6,7,23,24,25,26,27,28]. The host plants were carrot, maize, soybean, cotton, and alfalfa, and the AM fungal species belonged to Glomus, Rhizophagus, Funneliformis, and Gigaspora. The t-test was used for comparing the significant difference between the two treatments. C Bacterial communities co-occurrence networks visualizing significant correlations (ρ > 0.65, P < 0.01; indicated with gray lines) in controls (− AMF) and on the surface of ERH (+ AMF). Circles indicate bacteria amplicon sequence variants (ASVs). The gray edges represent strong and significant correlation between two nodes. Keystone ASVs (black-bordered square nodes) were identified separately for the − AMF and + AMF and defined as those nodes within the top 5% of node degree (number of edges correlations to a node) values of each network. Keystone ASVs are represented with black-bordered squares. ASVs are colored by their genus classification. The number of keystone ASVs in different genera in two bar chart alongside of co-occurrence networks. D Mean relative abundance (%) of ASVs (in five soil types and four fungal species) that match the 20 bacterial isolates from the soil BJ inoculated in the control (− AMF) or ERH (+ AMF) treatments. E Relative abundance (%) of bacteria in two SynComs associated (+ AMF) or not (− AMF) to the ERH of Rhizophagus irregularis MUCL43194 (SynCom19: 19 bacterial isolates without Streptomyces sp. D1; SynCom20: 19 bacterial isolates plus Streptomyces sp. D1). Error bars represent the standard error of five independent replicates. ERH-enriched bacterial isolates in the absence of Streptomyces sp. D1, and depleted by ERH in the presence of Streptomyces sp. D1, are symbolized by stars. F Relative abundance (%) of the 20 isolates associated with the ERH of Rhizophagus irregularis MUCL 43194 or develo** in the absence of ERH as a SynComs (SynCom20) in the pot system
We then collected data from six publications [23,24,25,26,27,28] to explore whether the preference for the genus Streptomyces is a widespread phenomenon. We compared the relative abundance of this genus in bulk and hyphosphere soil. The results showed that the relative abundance of the genus Streptomyces increased in the presence of AM fungi (Fig. 1B) suggesting a preponderance of AM fungi to recruit bacteria belonging to this genus.
Two bacterial community co-occurrence network analyses were further constructed in the presence versus the absence of ERH. In the presence of ERH, 17 keystone amplicon sequence variants (ASVs) were identified among which 11 belonged to the genus Streptomyces (Fig. 1C). The keystone ASVs of this genus showed more than 48 degrees of co-occurrence network, suggesting that they might influence the presence and distribution of other ASVs in the network. In the absence of ERH, 31 keystone ASVs were identified that mainly belonged to the genera Variovorax, Allo-Neo-Para-Rhizobium, Pseudomonas, and Nocardioides. Ten other genera, also including Streptomyces, were represented by only one keystone ASV (Fig. 1C).
To study the mechanisms involved in AM fungi and bacteria interaction, 62 bacteria were isolated from the ERH surface of R. irregularis MUCL 43194 (from the soil BJ), and 20 were selected by clustering at 97% similarity in 16S rRNA gene sequences to eliminate redundancy. The relative abundance of the 20 isolates on the ERH surface ranged from < 0.01 to 10.01% (Fig. 1D and Table S1). Interestingly, the mean relative abundance of ASVs that matched Streptomyces sp. D1 increased (from 5.62 to 10.01%) between the control treatments and the ERH treatments (Fig. 1D).
Finally, two SynComs were constructed, composed of 20 bacterial isolates (see for composition Fig. 1E), thus including (SynCom20) or not (SynCom19) Streptomyces sp. D1. The two SynComs were spread on the surface of the hyphal compartment (HC) covered by ERH of R. irregularis MUCL 43194 or free of hyphae. In the absence of Streptomyces sp. D1 (i.e., SynCom19), 12 isolates were enriched at the surface of the ERH, while in the presence of this bacterium (i.e., SynCom20), only 6 isolates were enriched (Fig. 1E). Due to the presence of Streptomyces sp. D1, 7 out of the 12 isolates that were enriched by ERH in SynCom19 were subsequently disfavored by ERH treatment, as indicated by a lack of bacterial growth (Fig. 1E, labelled with star). The SynCom20 was inoculated in the HC of the pot microcosm. In the presence of ERH, the relative abundance of Streptomyces sp. D1 was above 50%, while in the absence of ERH it was less than 25% (Fig. 1F). The Streptomyces sp. D1 was the most abundant in the ERH of the soil environment (Fig. 1F).
Collectively, the above results clearly showed that the genus Streptomyces was a keystone taxon in the early stage of interaction in the hyphosphere bacterial community of AM fungi under in vitro culture conditions.
Streptomyces sp. D1 showed preference for trehalose as C source
The carbon (C) assimilation profile of the 20 bacterial isolates was determined using six kinds of C sources (i.e., fructose, glucose, trehalose, inositol, citric acid, and succinic acid), which are among the major compounds found in the exudates of AM fungal hyphae [29,30,31]. Most bacteria were able to grow in the presence of at least one kind of C source and show preference for glucose, succinic acid, or citric acid (Figure S1). Streptomyces sp. D1 could use all six kinds of C sources, with a preference for trehalose (Fig. 2A). The whole-genome sequencing of Streptomyces sp. D1 showed the presence of genes encoding the proteins involved in trehalose transport (ThuE, ThuF, ThuG, and MalK) and metabolism (OtsB, TREH, TreZ, TreS, and glvA) (Fig. 2B and Table S2). The transcriptional sequence results showed that in the presence of ERH of R. irregularis MUCL 43194, the expression of genes from trehalose to glucose-6-P of Streptomyces sp. D1, including α-trehalase gene TREH (converting trehalose to glucose) and polyphosphate glucokinase gene ppgK (converting glucose to glucose-6P), was significantly increased (Fig. 2B).

A Growth curves of Streptomyces sp. D1 in M9 minimal salts medium with trehalose, fructose, glucose, succinic acid, inositol, citric acid as carbohydrate source. B Genome map of Streptomyces sp. D1 for three major C sources (fructose, glucose, and trehalose) exuded by AM fungal hyphae and metabolic pathway colored with transcriptomic genes expression data. The genes with a significant (P < 0.05) differential expression of |log2FC|> 1 are indicated with an arrow (pathway) or gene name in green (down-regulated in contact with the extraradical hyphae (ERH)), red (up-regulated in contact with the ERH). Gray arrow represents the absence of genes identified in the pathway. C Alkaline phosphatase production efficiency of 20 isolates of bacteria isolated from the ERH of Rhizophagus irregularis MUCL 43194 inoculated with a bacterial suspension of the soil BJ. Error bars represent the standard error of five independent replicates. D Numbers of genes involved in P cycling and carbohydrate-active enzymes (CAZy) genes identified in genome of Streptomyces sp. D1 and Pseudomonas sp. H2
Streptomyces sp. D1 had a strong ability to mineralize organic P in the hyphosphere
The alkaline phosphatase (AP) activity of the 20 bacterial isolates was measured under C (10-mM glucose) and P (KH2PO4, 50 mM) limited condition via the release of para-nitrophenol (pNP) from para-nitrophenol phosphate (pNPP). The bacteria were arbitrarily separated in three groups (i.e., high > 0.8, 0.4 < medium < 0.8, and low < 0.4-mM pNP hr−1 OD600−1) according to their efficacy to produce AP (Fig. 2C), with bacteria having a high AP being significantly different from those with low AP (Tukey’s HSD, P < 0.05). Streptomyces sp. D1 ranked in the highest AP production efficiency, while Pseudomonas sp. H2 had nearly the lowest AP production efficiency (Fig. 2C).
From the genome analysis, Streptomyces sp. D1 had abundant P metabolism genes, including 10 genes related to organic P mineralization, 9 genes related to inorganic P solubilization, 14 genes related to P transporters, and 2 genes related to P metabolism regulation (Fig. 2D). Streptomyces sp. D1 also had genes (e.g., GH6, PL1, PL3, and PL9) involved in the degradation of plant cell wall polysaccharides (Fig. 2D), which are absent in the genome of R. irregularis (Additional file 2). Overall, and with the exception of the regulatory genes, the number of genes involved in P metabolism (inorganic P solubilization, organic P mineralization, and P transporters), cellulose or hemicellulose degradation (glycoside hydrolases, GHs; glycosyl transferases, GTs; polysaccharide lyases, PLs; carbohydrate esterases, CEs; auxiliary activities, AAs; carbohydrate-binding modules, CBMs) was higher in the genome of Streptomyces sp. D1 compared to the genome of Pseudomonas sp. H2 (Fig. 2D, Table S3, Table S4, and Additional file 2).
Streptomyces sp. D1 and Pseudomonas sp. H2 were the dominant organisms in the bacterial community in the presence and absence of AM fungal hyphae, respectively, in soil BJ. Therefore, to determine the influence of AM fungi on the organic P mobilization ability of Streptomyces sp. D1 and Pseudomonas sp. H2, a comparative mRNA transcriptomic analysis was performed on both bacteria in the presence or absence of ERH of R. irregularis MUCL 43194. Overall, 768 and 1159 genes were significantly differentially expressed (P < 0.05) between these two conditions in Streptomyces sp. D1 and Pseudomonas sp. H2, respectively. Numerous genes associated with energy production were up-regulated in the glycolysis and citrate cycles in both microorganisms, indicating that a huge flow of C was transferred from the AM fungus to the bacteria (Figure S2A, B). The up-regulation of fructose phosphotransferase gene fruA and alpha-trehalase gene TREH (Fig. 3A) suggested that the hyphae released fructose and trehalose were taken up by Streptomyces sp. D1 resulting in genes up-regulated in the glycolysis and citrate cycle pathways. Furthermore, the inorganic P transporters, including pstS, pstC, and pstB, were down-regulated in Streptomyces sp. D1 (Fig. 3A). Most of the genes involved in glycolysis, citrate cycle, and pentose phosphate pathway were significantly increased in Pseudomonas sp. H2 (Fig. 3B and Figure S2B), as well as its inorganic P transporter genes (pstS, pstC, pstA, and pstB) (Fig. 3B). The inorganic P solubilization gene gcd/gdh and organic P mineralization gene phoD were not significantly changed in both strains grown in contact with the ERH (Fig. 3A, B; Figure S2A, B). Much more genes of C (glycolysis, citrate cycle, and pentose phosphate) and P (purine metabolism and pyrimidine metabolism) consumption pathways were up-regulated in Pseudomonas sp. H2 than in Streptomyces sp. D1 (Fig. 3C). The Sec secretion pathway (including genes: secA, the ATPase motor; ftsY, membrane receptor; secYEG, transmembrane SecYEG channel; secDF, yajC, and yidC, auxiliary component enhance translocation efficiency) is ubiquitous in all domains of bacteria that can secrete enzymes (e.g., AP). The genes of secY and yidC were significantly up-regulated in both bacteria (Fig. 3A, B). This was consistent with the stronger activity of AP observed in the two bacteria in contact with ERH compared to the control. Streptomyces sp. D1 showed much stronger AP (more than 37 times) than Pseudomonas sp. H2 in contact with ERH (Fig. 3D). Furthermore, 17 out of 89 cell motility genes were up-regulated in Pseudomonas sp. H2 indicating that motility was activated in the presence of ERH exudates (Table S5 and Additional file 3).

Major carbohydrate and phosphate metabolic pathways mapped with transcriptomic data in A Streptomyces sp. D1 and B Pseudomonas sp. H2. The reconstructed metabolic pathways were based on KEGG gene annotations and their relative differential gene expression profiles of triplicates (each replicate was a mix of 5 plates, 15 plates for every treatment). The genes with a significant (P < 0.05) differential expression of |log2FC|> 1 are indicated with an arrow (pathway) and gene name in green (down-regulated in contact with the extraradical hyphae (ERH)), red (up-regulated in contact with the ERH), purple (some genes down-regulated, and some genes up-regulated in contact with the ERH). C Numbers of up-regulated genes involved in carbon and phosphorus consumption in Streptomyces sp. D1 and Pseudomonas sp. H2 associated with ERH compared to controls. D Alkaline phosphatase activity of Streptomyces sp. D1 and Pseudomonas sp. H2 in contact (+ AMF) or not (− AMF) with the ERH of Rhizophagus irregularis MUCL43194 (n = 3, each replicate was a mix of five plates). Ps, Pseudomonas sp. H2; St, Streptomyces sp. D1
Streptomyces sp. D1 stimulated organic P utilization by AM fungi and gene expression of C-P exchange in plants
The phytate-P consumption of Streptomyces sp. D1 and Pseudomonas sp. H2 was significantly enhanced when the bacteria were grown in contact with the ERH of R. irregularis MUCL 43194 versus their growth in the absence of ERH (Fig. 4A). Moreover, in the presence of the ERH, the phytate consumption of Streptomyces sp. D1 was nearly doubled as compared to Pseudomonas sp. H2, and in the absence of ERH, it was six-folds greater (Fig. 4A). Gene expression of Pi-transporter (Pho84), vacuolar phosphate transporter (Pho91), and vacuolar transporter chaperone (VTC2 and VTC4) was significantly greater in the ERH inoculated with Streptomyces sp. D1 as compared to the control (i.e., the ERH without the bacteria — Fig. 4B). Reversely, in the presence of Pseudomonas sp. H2, a significant down-regulation was noticed for VTC2 and VTC4 compared to the control. No differences in gene expression in the presence/absence of Pseudomonas sp. H2 were noticed for Pho84 and Pho91 compared to the control (Fig. 4B).

A Phytate-P consumption of Streptomyces sp. D1 and Pseudomonas sp. H2 in the absence (− AMF) or presence (+ AMF) of the ERH of Rhizophagus irregularis MUCL 43194. The least significant difference (LSD) test was used to identify the differences in P consumption at 0.05 significance level. Error bars represent the standard error of three independent replicates. B Gene expression of Pho84, Pho91, VTC2, and VTC4 in the ERH of R. irregularis MUCL 43194 in the absence of bacteria (Ctrl) or inoculated with Streptomyces sp. D1 or Pseudomonas sp. H2 (n = 3 biologically independent samples, ***P < 0.001, **P < 0.01, *P < 0.05, LSD). C Summary of the significantly up-regulated genes in R. irregularis MUCL 43194 involved in phosphate, polyphosphate, and energy turnover inoculated with Streptomyces sp. D1 as compared to the fungus grown in the absence of the bacteria. The genes with a significant (P < 0.05) differential expression of log2FC > 1 are indicated with gene name in red (up-regulated in contact with Streptomyces sp. D1). D Differentially expressed gene number (P < 0.05, |log2FC|> 1) in cellular components, biological processes, and molecular functions of the ERH of R. irregularis MUCL 43194 inoculated with Streptomyces sp. D1 as compared to the fungus grown in the absence of the bacterium. E Gene expression of WRI5a, MtFatM, STR2, and MtHA1 in Medicago truncatula associated with the ERH of R. irregularis MUCL 43194 in the absence of bacteria (Ctrl) or inoculated with Streptomyces sp. D1 or Pseudomonas sp. H2 (n = 4 biologically independent samples, ***P < 0.001, **P < 0.01, *P < 0.05, LSD)
A comparative mRNA transcriptome was conducted on triplicate ERH samples (each replicate was a mix of 5 plates, 15 plates for each treatment) of R. irregularis MUCL 43194 associated or not to Streptomyces sp. D1. More than 1.6% of the AM fungal gene repertoire (434 out of 26143 genes) was affected by the bacterium. Twenty genes, known to be necessary for phosphate and polyphosphate transport and metabolism in R. irregularis, were scrutinized. A general increase of expression of genes involved in phosphate limitation regulated gene (pho80) phosphate transport (pho84) and polyphosphate synthesis/decomposition (VTC2, VTC4, and PPN1) was noticed in the presence of Streptomyces sp. D1 (Fig. 4C). In addition, the expression of polyphosphate-related ion transporter genes including ZRT1 (zinc-regulated transporter 1), MatA (magnesium transporting ATPase), and VIT1 (vacuolar iron transporter 1) was up-regulated in the presence of the bacterium (Fig. 4C). A general increase in NAD(P)H synthesis and oxidoreduction was also observed (Fig. 4C).
The differentially expressed genes were divided into Gene Ontology (GO) functional categories. This included 112 genes associated to cellular components, 49 genes to biological processes, and 136 genes to molecular functions (Fig. 4D). The genes most up- or down-regulated belong to the integral component of membrane, the ATP binding, and protein kinase activity. This included 46 genes up-regulated and 48 genes down-regulated (integral component of membrane), 24 genes up-regulated and 21 genes down-regulated (ATP binding), and 17 genes up-regulated and 20 genes down-regulated (protein kinase activity) in the ERH of the AM fungus inoculated with Streptomyces sp. D1 as compared to the fungus grown in the absence of the bacterium, respectively (Fig. 4D).
The effect of bacteria on AM fungi could be extended to the plant. After inoculation of Streptomyces sp. D1 in the hyphosphere, a significant increase in the expression of the ATPase gene MtHA1 that is essential for P transport from the symbiotic interface to the plant was observed. Similarly, the expression of genes related to fatty acid synthesis (WRI5a and MtFatM) and transport (STR2) from plants to symbiotic interface was significantly increased (Fig. 4E). The inoculation of Pseudomonas sp. H2 in the hyphosphere also significantly increased the expression of WRI5a and STR2 in the plant, while no differences were noticed for the expression of genes MtFatM and MtHA1 (Fig. 4E).
Streptomyces sp. D1 impacted the bacterial community in the hyphosphere
Exudates of Streptomyces sp. D1 were tested for growth inhibition on 19 bacteria from the surface of ERH grown with soil BJ bacterial suspension. Fourteen out of 19 isolates, including Pseudomonas sp. H2, were inhibited by the exudates (Fig. 5A). Notably, six isolates with higher AP production efficiency (> 0.4 mM pNP h−1 OD600−1) were not impacted or minimally inhibited (Fig. 5B). A negative correlation was found between Streptomyces sp. D1 and Pseudomonas sp. H2 in soil BJ. qPCR showed a significant decrease in the absolute abundance ratio of Streptomyces sp. D1 and Pseudomonas sp. H2 in the absence of ERH. Streptomyces sp. D1 had greater colony development in ERH treatment in single or dual culture than in the control treatment (Fig. 5C, D). Both the culture cells and exudates of Streptomyces sp. D1 were able to inhibit the normal growth of Pseudomonas sp. H2 (Fig. 5E).

A Growth inhibition of Streptomyces sp. D1 exudates on 19 bacteria isolated from the surface of the ERH cultured in contact with the bacterial suspension of soil BJ. B Correlation of growth inhibition and phosphatase producing efficiency of 19 bacteria isolated from the surface of the extraradical hyphae (ERH) of Rhizophagus irregularis MUCL 43194. C Absolute abundance of Streptomyces sp. D1 and Pseudomonas sp. H2 growing singly or dually, in contact (+ AMF) or not (− AMF) with the ERH of R. irregularis MUCL 43194 (n = 4 biologically independent samples, ***P < 0.001, **P < 0.01, *P < 0.05, LSD). D Ratio of the absolute abundance of Streptomyces sp. D1 and Pseudomonas sp. H2 in the absence (left bar) or presence (right bar) of ERH in dual culture conditions. E Impact of Streptomyces sp. D1 (top) or its zymotic liquid (down) on Pseudomonas sp. H2 (Ps) on solid medium (0.5% peptone, 0.3% yeast extract, 0.5% NaCl, 0.8% agar). Ctrl is the control with 0.9% NaCl (w:v) solution or TSB medium. F Albaflavenone biosynthesis gene cluster detected by antiSMASH6 in genome of Streptomyces sp. D1. G Reconstruction of the Streptomyces sp. D1 terpenoid backbone biosynthesis and sesquiterpenoid and triterpenoid biosynthesis pathways mapped with transcriptomic data. Reconstructed metabolic pathway of the Streptomyces sp. D1 based on KEGG gene annotations and its relative differential gene expression profiles of triplicates (each replicate was mixed by 5 plates, 15 plates for every treatment). The pathways with genes significant (P < 0.05) differential expression of log2FC > 1 are indicated with pathway name in red (up-regulated in contact with the ERH). H UPLC-MSMS analysis of metabolites extracted from the 1/2 TSB medium cultured Streptomyces sp. D1. UPLC-MSMS chromatograms of albaflavenone standard (top) and from the growth medium (TSB, tryptone soy broth) of Streptomyces sp. D1 (down)
The Streptomyces sp. D1 harbored the entire albaflavenone (bactericidal antibiotic) synthesis pathway genes (Fig. 5F, G and Table S6). Consistently, a general increase of genes involved in terpenoid backbone biosynthesis in Streptomyces sp. D1 was observed, which were necessary for synthesis of albaflavenone precursor substances (Fig. 5G and Figure S3). Additionally, albaflavenone (C15H22O) at the concentration of 0.93 µg L−1 (R2 = 0.998) was identified by UPLC-MSMS against the standard (CAS: 157078–47-2) on the 1/2 TSB medium cultured Streptomyces sp. D1 (Fig. 5H).

