
Abstract
Molecular approaches are widely applied in species identification and taxonomic studies of minute zooplankton. One of the most focused zooplankton nowadays is from Subclass Copepoda. Accurate species identification of all life stages of the generally small sized copepods through molecular analysis is important, especially in taxonomic and systematic assessment of harpacticoid copepod populations and to understand their dynamics within the marine community. However, total genomic DNA (TGDNA) extraction from individual harpacticoid copepods can be problematic due to their small size and epibenthic behavior. In this research, six TGDNA extraction methods done on individual harpacticoid copepods were compared. The first new simple, feasible, efficient and consistent TGDNA extraction method was designed and compared with the commercial kit and modified available TGDNA extraction methods. The newly described TGDNA extraction method, “Incubation in PCR buffer” method, yielded good and consistent results based on the high success rate of PCR amplification (82%) compared to other methods. Coupled with its relatively consistent and economical method the “Incubation in PCR buffer” method is highly recommended in the TGDNA extraction of other minute zooplankton species.
Similar content being viewed by others


Background
Copepods are tiny multicellular organisms with size range between 0.1 and 1–2 mm that spread through different water bodies (Humes 1994; McKinnon et al. 2003; Pesce 2010). They even colonized harsh environment such as polar and hot spring water (Huys and Boxshall 1991). Taxonomic classification grouped copepod under Subphylum Crustacea due to the presence of two pairs of antennae, mandibles, maxillae on their heads and a pair of compound eyes (usually on stalks), a pair of appendages on each body segment for namely head, abdomen and thorax. The Subclass Copepoda is comprised of approximately 200 families, 1650 genera and 11,500 species (Humes 1994).
Currently, copepods are gaining attention in the aquaculture sector as live feed (Kahan et al. 1982; Kleppel and Hazzard 2002; Lee et al. 2005; Mckinnon et al. 2003; Peralta and Monica 2004; Watanabe and Kiron 1994; Williamson and Reid 2001). Classification of copepods in aquaculture farms is generally using conventional method based on morphological characters such as length of antenna, fifth walking leg and curvature coxa of the fifth pair of swimming legs (Huys and Boxshall 1991; Jagadeesan et al. 2009; Kabata 1979). An experienced taxonomist is needed in microscopic determination of copepods species and only copepods at late copepodite and adult stages only can be identified. The lack of precise and quantitative morphological characteristic analysis has made it difficult to classify due to their minute size (Böttger-Schnack and Machida 2011; Bucklin and Lajieunesse 1994; Dawson 2003; Huys and Boxshall 1991; Lindeque et al. 1999; McKinnon et al. 2003; Sneath and Sokal 1973; Suzuki et al. 2006; Weins 2000). Therefore, there is a need for a feasible, fast, reliable and precise technique in copepod species differentiation due to their abundances and morphological ambiguity. Molecular data such as DNA and RNA sequences provide complementary and informative data for systematic studies of copepods to determine their evolutionary relationship, taxonomy and even function of specific genes (Austin et al. 2016; Bucklin et al. 1999, 2000; Burton et al. 2007; Chow et al. 2008; Dennis et al. 2009; Lindeque et al. 1999; Machida and Tsuda 2010; Palumbi and Benzie 1991; Rhee et al. 2009; Song et al. 2008; Suzuki et al. 2006; Thum and Derry 2008; Thum and Harrison 2008).
Extraction of total genomic DNA is one of the primary steps before proceeding onto subsequent molecular studies. The total genomic DNA (TGDNA) extraction of individual larger animals and plants are easier by using conventional method and various commercial kits. But, the TGDNA extraction for the individual tiny organism (<1 mm) such as copepods proves to be difficult as they have comparatively lesser amount of target DNA to start with (Saiki et al. 1988). Schizas et al. (1997) mentioned that skills are needed in handling copepods during TGDNA extraction because copepods especially from the order Harpacticoida (epibenthic) live in close contact with sediment, fungi, bacteria and other zooplanktons. It is important to use individual copepod rather than a clump or a population for TGDNA extraction in genetics research to avoid contamination or mixed species. Various TGDNA extraction techniques (conventional methods, modified methods and commercial extraction kits) were compared in this research in order to identify a feasible, efficient and consistent TGDNA extraction method. In this study, a simple method that uses less chemicals and minimal handling of the sample while being capable of producing consistent positive results (validated by successful polymerase chain reaction (PCR) amplification) for extracting the TGDNA from individual harpacticoid copepod has been developed.
Methods
Experimental organism
Live samples of harpaticoid copepod, Leptocaris canariensis were collected from pure culture maintained in laboratory of Universiti Malaysia Terengganu, Terengganu Malaysia. The use of copepods and their extraction methods in this research were approved by the Institute of Tropical Aquaculture, Universiti Malaysia Terengganu.
Comparison of TGDNA extraction methods
TGDNA of L. canariensis was extracted using six different methods, namely CTAB DNA extraction method that was modified from the method established by Winnepenninckx et al. (1993), modified phenol chloroform DNA extraction method from Pearson and Stirling (2003), KAPA Express Extract kit (KAPA Biosystems Inc, USA), Direct Boiling method designed by Vestheim et al. (2005), TGDNA extraction using “Incubation in lysis buffer and proteinase K” by Burton et al. (2007) and TGDNA extraction using “Incubation in PCR buffer”. The detection methods of the availability of TGDNA were done by using Agarose Gel Electrophoresis, spectrophotometer and PCR. 50 L. canariensis individuals were used for each TGDNA extraction methods. The efficiency and consistency of the methods used were based on the success in DNA extraction detected by agarose gel electrophoresis (AGE), spectrophotometer or PCR.
Modified CTAB
Individual L. canariensis was minced under dissecting microscope in 50 μL 2× CTAB buffer (2% w/v CTAB, 1.4 M NaCl, 0.2% v/v 2-mercaptoethanol, 20 mM EDTA, 100 mM Tris–HCl (pH 8.0), 0.1 mg/mL proteinase K) using a fine needle. The minced sample was transferred into a 1.5 mL microcentrifuge tube containing premixed 100 μL of 2× CTAB buffer with 5 μL Proteinase K (20 mg/mL) and incubated at 60 °C for 1–3 h. The incubated sample was then mixed with 60 μL Chloroform: Isoamyl alcohol (24:1) by shaking the mixture for 2 min and then centrifuged at 13,000 rpm for 10 min. Supernatant was carefully transferred into a new 1.5 mL microcentrifuge tube before repeating previous steps of adding C:IA and then centrifuged. Supernatant was transferred into a new 1.5 mL microcentrifuge tube, mixed with 60 μL of absolute ethanol and centrifuged (13,000 rpm/10 min). The supernatant was discarded. The pellet formed was washed twice by adding 50 μL of 70% ethanol and centrifuged (13,000 rpm/10 min). Pellet was air dried for about 1 h and dissolved in 50 μL sterile double distilled water (ddH2O).
Modified phenol chloroform
An individual L. canariensis was minced under dissecting microscope in 50 μL lysis buffer [10 mM NaCl, 20 mM Tris–HCl, pH 8.0, 1 mM EDTA, 1% sodium dodecyl sulfate (SDS)] using a fine needle. The minced sample was transferred into a 1.5 mL microcentrifuge tube containing premixed 60 μL of lysis buffer with 5 μL Proteinase K (20 mg/mL) and incubated at 60 °C for 1–3 h. The incubated sample was then mixed with 60 μL Phenol: Chloroform (1:1) by shaking the mixture for 2 min and centrifuged at 13,000 rpm for 10 min. The remaining supernatant was transferred into a new 1.5 mL microcentrifuge tube. 60 μL of Phenol: Chloroform: Isoamyl alcohol (25:24:1) was added and the mixture was centrifuged (13,000 rpm/10 min). Supernatant was transferred into a new 1.5 mL microcentrifuge tube, mixed with 60 μL of absolute ethanol and centrifuged (13,000 rpm/10 min). The supernatant was discarded and the pellet formed was washed twice by adding 50 μL of 70% ethanol and centrifuged (13,000 rpm/10 min). Pellet was air dried for about 1 h and dissolved in 50 μL sterile ddH2O.
KAPA Express Extract kit
KAPA Express Extract kit was used based on the procedure provided by the manufacturer on fish tissue extraction. An individual L. canariensis was minced under dissecting microscope in 10 μL sterile ddH2O using a fine needle. The minced mixture was transferred into a 0.2 mL PCR tube and 100 μL lysis solutions were added. PCR tube was sealed and transferred into BIORAD MyCycler™ thermal cycler machine and incubated (60 °C/10 min and 95 °C/5 min). The sample was centrifuged (14,000 rpm/60 s) and the supernatant was transferred into a new PCR tube.
Direct boiling
An individual L. canariensis was minced under dissecting microscope in 10 μL sterile ddH2O using fine needle. The minced sample was transferred into a 0.2 mL PCR tube containing 30 μL ddH2O and boiled at 100 °C for 5 min.
Incubation in lysis buffer and proteinase K
An individual L. canariensis was minced under dissecting microscope in 10 μL sterile ddH2O using a fine needle. The minced sample was transferred into a 1.5 mL microcentrifuge tube containing 40 μL of lysis buffer (10 mM NaCl, 20 mM Tris–HCl, pH 8.0, 1 mM EDTA, 1% SDS), 10 μL Proteinase K (20 mg/mL) and incubated at 60 °C for 60 min. The incubated sample was then mixed with 60 μL of cold absolute ethanol for 30 min in 4 °C and centrifuged (13,000 rpm/15 min). The supernatant was discarded and the pellet formed was washed by 100 μL of 70% ethanol was added and centrifuged (13,000 rpm/10 min) twice. The pellet was then air dried for about 1 h and dissolved in 50 μL sterile ddH2O.
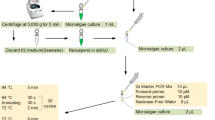
Incubation in PCR buffer
The PCR buffer used in this study was PCR buffer A (500 mM KCl, 100 mM Tris–HCl and 0.1% Triton™X-100) (VIVANTIS Technologies, MY). An individual L. canariensis was minced under dissecting microscope in 5 μL sterile ddH2O using a fine needle. The minced mixture was transferred into a 0.2 mL PCR tube containing premixed 5 μL of sterile ddH2O and 2.5 μL VIVANTIS PCR buffer A. The mixture was incubated for 15 min at room temperature. The availability of TGDNA was only assessed by PCR.
Detection of TGDNA
AGE
A 1% agarose gel [1× Tris-acetate-EDTA (TAE) buffer pre-stained with ethidium bromide] was prepared. The samples were run at 70 V for 45 min.
Spectrophotometer
Optical density (OD) readings at 260 and 280 nm of TGDNA extraction products were carried out to determine the availability, quantity and quality of obtained DNA using spectrophotometer.
PCR
Amplifications were done in a thermal cycler (BIO-RAD MyCycler™). The primers used in were ITS1a and ITS1r designed by Dennis et al. (2009) and LCO-1490 and HCO-2198 design by Folmer et al. (1994). The PCR reactions carried out with 40 cycles of a 25 μL reaction volume containing 5.6 μL mixture of sterile ddH2O, 2.5 μL of 10× PCR buffer, 1.0 μL of dNTP (10 mM each), 1.5 μL of each primer (2.5 µM), 12.5 μL of DNA template and 0.4 μL of Taq Polymerase manufactured by Vivantis Technologies Sdn. Bhd. (5 μ/μL). The thermal cycle profile was as follows: denaturation at 94 °C for 45 s, annealing at 52 °C for 45 s (for both primers), and extension at 72 °C for 60 s. The PCR products were run on a 1% AGE pre-stained with ethidium bromide for band characterization using ultraviolet trans-illumination. The negative control was prepared without the DNA template in PCR mixture and provided in each amplification reaction.
Results
The detection of TGDNA of an individual L. canariensis was not applicable by using AGE and spectrophotometer due to the minute concentration of extracted TGDNA. Out of the three detection methods, only PCR was the most practical approach to detect the extracted TGDNA from individual L. canariensis (Table 1).
Three out of six methods successfully extracted TGDNA from individual copepod, L. canariensis, as indicated by the presence of PCR products. Comparatively, “Incubation in PCR buffer” method was the most feasible, consistent and efficient method with the highest success rate (82%) (Figs. 1, 2), followed by KAPA Express Extract Kit (46%) and “Incubation in lysis buffer and proteinase K” method (18%) (Table 1). The “Incubation in PCR buffer” method successfully and consistently amplifies the partial ITS1 gene of nuclear DNA and partial COI gene of mitochondrial DNA. This indicated that this method was able to extract nuclear and mitochondrial genome itself. Direct sequencing of the partial COI gene of L. canariensis of this study showed 77% similarities with partial mitochondrial COI gene region of calanoid copepods, Boeckella brasiliensis. The mitochondrial COI sequence was submitted to GenBank, NCBI, with Accession Number JF707331 (Waiho et al. 2013).

AGE photograph showing partial ITS1 gene bands of L. canariensis extracted using “incubation in PCR Buffer” method. AGE was run using 1% TAE agarose gel at 70 V for 45 min. L 100 kb Ladder; L1–L8 PCR product from samples 1–8; N negative control

AGE photograph showing partial COI gene band of L. canariensis canariensis extracted using “incubation in PCR Buffer” method. AGE was run using 1% TAE agarose gel at 70 V for 45 min. L 100 kb Ladder; L1–L5 PCR product from sample 1–5; N negative control
Discussion
The amplified TGDNA product, even in initial low concentration, gave positive result in PCR reaction as expected (Grunenwald 2003; Raven and Johnson 2002). The best concentration of TGDNA required to yield satisfying results in PCR is between 100 and 500 ng, although concentration as low as 50 ng was reported to yield positive results as well (Creighton 1999). In this study, the observable PCR product bands in gel electrophoresis showed that the TGDNA from L. canariensis was successfully extracted although the initial TGDNA concentration from a single copepod is too low to be quantified via AGE or spectrophotometer. The applied methods to detect the presence of DNA in present study, i.e. spectrophotometer, gel electrophoresis and PCR are common DNA detection methods used by most researchers (Chen et al. 2010; de Oliveira et al. 2014; Wang and Wang 2012; Yoganandhan et al. 2003). In addition, due to the high sensitivity of PCR compared to the other two DNA detection methods, researchers also have resorted to use only PCR to detect the presence of DNA and directly sequence the targeted gene (Dashti et al. 2009; Englen and Kelley 2000; Vasuki et al. 2001). Currently available DNA extraction kits such as DNAMITE DIRECT DNA extraction kit (Microzone Ltd., UK), MightyPrep reagent for DNA (Takara Bio Company, USA), Phire animal tissue direct PCR kit (Thermo Fisher Scientific Inc, USA) and Extract-N-Amp tissue PCR kit (Sigma-Aldrich Co., Germany) also focused on the sensitivity of PCR and the inconsistency of spectrophotometer and gel electrophoresis, and directly skip DNA quantification steps prior PCR in their kit manual (extracted DNA are PCR-ready), thereby saving time and cost.
The ability to extract TGDNA using Vivantis PCR buffer A is attributed to the components incorporated in it. The Vivantis PCR buffer A contains 500 mM KCl, 100 mM Tris–HCl (pH 9.1 at 20 °C) and 0.1% Triton™X-100. According to Sha et al. (2008), Triton™X-100 is normally used as a detergent in lysis buffer and has the ability to denature membrane protein of the cell, resulting in the release of cell components including TGDNA. Combination of detergent and salts was reported to increase DNA extraction ability in cells as salts such as KCl provides a hypotonic environment that promotes cell lysis (Raven and Johnson 2002). Apart from 10× buffer A used in this study, almost all commercial PCR buffers contain detergent and salt as well that are essential in the lysis of cell membrane (i.e. KCl, Tris–HCl, Triton™X-100 and ammonium sulfate).
The primary advantages of TGDNA extraction using “Incubation in PCR buffer” method over other extraction methods used in this study are simplicity and consistency. This method is also relatively cheaper in comparison to the other two methods (KAPA Express Extract Kit and “Incubation in lysis buffer and proteinase K” method) that were able to yield positive PCR results. “Incubation in PCR buffer” method involves only a few simple and short procedures, thereby minimizing potential exposure time of DNA to other contaminants and risk of being degraded. Mishandling or improper pipetting technique can be avoided as the reaction involves only one chemical (buffer A) during extraction. In addition, the DNA reduction can be avoided as well because due to the limited TGDNA obtainable in copepod, conventional TGDNA extraction methods with longer procedures and more chemicals involved will tend to reduce the quantity and quality of TGDNA along with their extraction steps. The overnight incubation at 4 °C done in this study was proven to be able to extract sufficient amount of DNA for subsequent PCR amplification. Long incubation period was needed for chemicals in PCR buffer A to react and lyse cells, releasing TGDNA as no proteinase K was applied. Previous research done on copepods used PCR buffers for brief pre-incubation, before subjecting copepod samples to commercial DNA extraction kit (GeneReleaser™) containing proteinase K (Easton et al. 2010; Schizas et al. 1997), “Incubation in PCR buffer” method avoids the need to use either proteinase K or expensive commercial DNA extraction kit, thereby saving cost yet did not compromise the outcome.
The “Incubation in lysis buffer and proteinase K” method produced inconsistent results and is not recommended to be used in the TGDNA extraction of copepods. On the other hand, the KAPA Express Extract kit can be considered as an alternative method to extract the TGDNA of copepods, as it is simple in procedure and can yield at least 42% success rate in PCR amplification.
Conclusions
We report in this study a simple, feasible, efficient and consistent TGDNA extraction method, i.e. “Incubation in PCR buffer” method, to extract TGDNA from individual zooplankton. This study also shows that when only a single zooplankton was used, the TGDNA extracted was undetected using normal TGDNA detection methods (i.e. AGE and spectrophotometer) due to their very low concentration. The “Incubation in PCR buffer” method described in this study is highly applicable in future research on the molecular aspects of zooplankton such as molecular identification or population ecological studies.
Abbreviations
- AGE:
-
agarose gel electrophoresis
- ddH2O:
-
double distilled water
- min:
-
minute
- PCR:
-
polymerase chain reaction
- SDS:
-
sodium dodecyl sulfate
- TAE:
-
Tris-acetate-EDTA
- TGDNA:
-
total genomic DNA
References
Austin CM, Tan MH, Lee YP, Croft LJ, Meekan MG, Pierce SJ, Gan HM (2016) The complete mitogenome of the whale shark parasitic copepod Pandarus rhincodonicus norman, Newbound & Knott (Crustacea; Siphonostomatoida; Pandaridae)—a new gene order for the copepoda. MDN 27(1):694–695. doi:10.3109/19401736.2014.913147
Böttger-Schnack R, Machida RJ (2011) Comparison of morphological and molecular traits for species identification and taxonomic grou** of oncaeid copepods. Hydrobiologia 666:111–125. doi:10.1007/s10750-010-0094-1
Bucklin A, Lajieunesse TC (1994) Molecular genetic variation of Calanus pacificus (Copepoda: Calanoida): preliminary evaluation of genetic structure and subspecific differentiation based on mtDNA sequences. Cal Coop Ocean Fish 35:45–51
Bucklin A, Guarnieri M, Hill RS, Bentley AM, Kaartvedt S (1999) Taxonomic and systematic assessment of planktonic copepods using mitochondrial COI sequence variation and competitive species-specific PCR. Hydrobiologia 401:239–254
Bucklin A, Kaartved S, Guarnieri M, Gowsami U (2000) Population genetics of drifting (Calanus spp.) and resident (Acartia clausi) plankton in Norwegian fjords. J Plankton Res 22:1237–1251. doi:10.1093/plankt/22.7.1237
Burton RS, Byrne RJ, Rawson PD (2007) Three divergent mitochondrial genomes from California populations of the copepod Tigriopus californicus. Gene 403:53–59. doi:10.1016/j.gene.2007.07.026
Chen H, Rangasamy M, Tan SY, Wang H, Siegfried BD (2010) Evaluation of five methods for total DNA extraction from western corn rootworm beetles. PLoS ONE. doi:10.1371/journal.pone.0011963
Chow S, Ueno Y, Toyokowa M, Oohara I, Takeyama H (2008) Preliminary analysis of length and GC content variation in the ribosomal First Internal Transcribed Spacer (ITS1) of marine animals. Mar Biotechnol 2:301–306. doi:10.1007/s10126-008-9153-2
Creighton T (1999) Encyclopedia of molecular biology. Wiley-lnterscience, London
Dashti AA, Jadaon MM, Abdulsamad AM, Dashti HM (2009) Heat treatment of bacteria: a simple method of DNA extraction for molecular techniques. Kuwait Med J 41(2):117–122
Dawson MN (2003) Macro-morphological variation among cryptic species of the moon jellyfish, Aurelia (Cnidaria Scyphozoa). Mar Biol 143(2):369–379. doi:10.1007/s00227-003-1070-3
de Oliveira CF, da Silva Paim TG, Reiter KC, Rieger A, D’azevedo PA (2014) Evaluation of four different DNA extraction methods in coagulase-negative Staphylococci clinical isolates. Rev Inst Med Trop Sao Paulo 56(1):29–33
Dennis F, Ravallec R, Pavillon JF, Van Wormhoudt A (2009) Genetic differentiation of Atlantic populations of the intertidal copepod Tigriopus brevicornis. Sci Mar 73(3):579–587. doi:10.3989/scimar.2009.73n3579
Easton EE, Thistle D, Spears T (2010) Species boundries in Zausodes-complex species (Copepoda: Harpacticoida: Harpacticidae) from the north-eastern Gulf of Mexico. Invertebr Syst 24:258–270. doi:10.1071/IS09038
Englen MD, Kelley LC (2000) A rapid DNA isolation procedure for the identification of Campylobacter jejuni by the polymerase chain reaction. Lett Appl Microbiol 31:421–426
Folmer O, Black M, Hoeh W, Lutz R, Vrijenhoek R (1994) DNA primers for amplification of mitochondrial cytochrome c oxidase subunit I from diverse metazoan invertebrates. Mol Mar Biol Biotechnol 3(5):294–299
Grunenwald H (2003) Optimization of polymerase chain reactions. In: Bartlett JMS, Stirling D (eds) Methods in molecular biology: PCR protocols, 2nd edn. Humana Press, Totowa, pp 89–99
Humes AG (1994) How many copepods? Hydrobiologia 292–293:1–7
Huys R, Boxshall AG (1991) Copepod evolution. The Ray Society, London
Jagadeesan L, Perumal P, Thangaraj M (2009) Molecular identification of marine calanoid copepod Paracalanus parvus (Claus 1863) using RFLP. World J Fish Mar Sci 1(3):239–242
Kabata Z (1979) Parasitic copepoda of British fishes. The Ray Society, London
Kahan D, Uhlig G, Schwenzer D, Horowitz L (1982) A simple method for cultivating harpacticoid copepods and offering them to fish larvae. Aquaculture 26:303–310
Kleppel GS, Hazzard SE (2002) The significance of zooplankton nutrition in the aquatic sciences. Outcomes of an international workshop on zooplankton nutrition. University of South Carolina, Columbia
Lee CS, O’Bryen PJ, Marcus NH (2005) Copepods in aquaculture. Blackwell, Oxford
Lindeque PK, Hariss RP, Jones M, Smerdon GR (1999) Simple molecular method to distinguish the identity of Calanus species (Copepoda: calanoida) at any developmental stages. Mar Biol 133:91–96. doi:10.1007/s002270050446
Machida RJ, Tsuda A (2010) Dissimilarity of species and forms of planktonic Neocalanus copepods using mitochondrial COI, 12S, nuclear ITS, and 28S gene sequences. PLoS ONE 5(4):e10278. doi:10.1371/journal.pone.0010278
McKinnon AD, Duggan S, Nichols PD, Rimmer MA, Semmens G, Robino B (2003) The potential of tropical Paracalanid copepods as live feeds in aquaculture. Aquaculture 223(1–4):89–106. doi:10.1016/S0044-8486(03)00161-3
Palumbi SR, Benzie J (1991) Large mitochondrial DNA differences between morphologically similar penaeid shrimp. Mol Mar Biol Biotechnol 1:27–34
Pearson H, Stirling D (2003) DNA extraction from tissue. In: Bartlett JMS, Stirling D (eds) Methods in molecular biology: PCR protocols, 2nd edn. Humana Press, Totowa, p 33
Peralta M, Monica H (2004) Biology and culture of a tropical harpacticoid copepod, Nitocra affinis. Dissertation, Universiti Putra Malaysia, Malaysia
Pesce GL (2010) Copepod web portal. Available from: http://www.luciopesce.net/copepods/intro.htm. Accessed 5 Nov 2011
Raven PH, Johnson GB (2002) Biology, 6th edn. McGraw Hill Higher Education, Washington
Rhee JS, Raisuddin S, Lee KW, Seo JS, Ki JS, Kim IC, Park HG, Lee JS (2009) Heat shock protein (Hsp) gene responses of the intertidal copepod Tigriopus japonicus to environmental toxicants. Comp Biochem Physiol C Toxicol Pharmacol 149(1):104–112. doi:10.1016/j.cbpc.2008.07.009
Saiki RK, Gelfand DH, Stoffel S, Scharf SJ, Higuchi R, Horn GT, Mullis KB, Erlich HA (1988) Primer-directed enzymatic amplification of DNA, with a thermo-stable DNA polymerase. Science 239:487–491
Schizas NV, Street GT, Coull BC, Chandler GT, Quattro JM (1997) An efficient DNA extraction method for small metazoans. Mol Mar Biol Biotechnol 6(4):381–383
Sha Y, Pandit L, Zeng S, Eissa NT (2008) A critical role for CHIP in the aggresome pathway. Mol Cell Biol 29(1):116–128
Sneath PHA, Sokal RR (1973) Numerical taxonomy: the principals and practice of numerical classification. Freeman, San Francisco
Song Y, Wang GT, Yao WJ, Gao Q, Nie P (2008) Phylogeny of freshwater parasitic copepods in the Ergasilidae (Copepoda: Poecilostomatoida) based on 18S and 28S rDNA sequences. Parasitol Res 102:299–306. doi:10.1007/s00436-007-0764-8
Suzuki H, Matsutani T, Tamate H (2006) Early detection of the parasitic Copepod Pectenophilus ornatus in the gills of Japanese scallop (Patinopecten yessoensis) by using Polymerase Chain Reaction with species-specific primers. Aquacult sci 54(3):353–357. doi:10.11233/aquaculturesci1953.54.353
Thum RA, Derry AM (2008) Taxonomic implications for diaptomid copepods based on contrasting patterns of mitochondrial DNA sequence divergences in four morphospecies. Hydrobiologia 614:197–207. doi:10.1007/s10750-008-9506-x
Thum RA, Harrison RG (2008) Deep genetic divergences among morphologically similar and parapatric Skistodiaptomus (Copepoda: Calanoida: Diaptomidae) challenge the hypothesis of Pleistocene speciation. Biol J Linnean Soc 96:150–165. doi:10.1111/j.1095-8312.2008.01105.x
Vasuki V, Patra KP, Hoti SL (2001) A rapid and simplified method of DNA extraction for the detection of Brugia malayi infection in mosquitoes by PCR assay. Acta Trop 79:245–248
Vestheim H, Edvardsen B, Kaartvedt S (2005) Assessing feeding of a carnivorous copepod using species-specific PCR. Mar Biol 147:381–385. doi:10.1007/s00227-005-1590-0
Waiho K, Fazhan H, Shahreza MS, Zaleha K (2013) Isolation and characterization of partial mitochondrial CO1 gene from harpacticoid copepod, Leptocaris canariensis (Lang, 1965). Afr J Biotechnol 12(50):6901–6906. doi:10.5897/AJB11.2064
Wang Q, Wang X (2012) Comparison of methods for DNA extraction from a single chironomid for PCR analysis. Pak J Zool 44(2):421–426
Watanabe T, Kiron V (1994) Prospects in larval fish dietetics. Aquaculture 124:223–251
Weins JJ (2000) Coding morphological variation within species and higher taxa for phylogenetic analysis. In: Weins JJ (ed) Phylogenetic analysis of morphological data. Smithsonian Institution Press, Washington, pp 115–145
Williamson CE, Reid JW (2001) Copepoda. In: Thorp JH, Covich AP (eds) Ecology and classification of North American freshwater invertebrates, 2nd edn. Academic Press, New York, pp 915–954
Winnepenninckx B, Backeljau T, De Wachter R (1993) Extraction of high molecular weight DNA from mollusks. Trends Genet 9:407
Yoganandhan K, Sathish S, Narayanan RB, Sahul Hameed AS (2003) A rapid non-enzymatic method of DNA extraction for PCR detection of white spot syndrome virus in shrimp. Aquacult Res 34:1093–1097
Authors’ contributions
HF and KW conceived the study, worked on the study design/methodology and drafted the manuscript. MSS worked on the study design/methodology, provided institutional support and edited/oversaw the manuscript. All authors read and approved the final manuscript.
Acknowledgements
The authors would like to thank Institute of Tropical Aquaculture, Universiti Malaysia Terengganu for providing the research facilities, samples and funding for this project.
Competing interests
The authors declare that they have no competing interests.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Fazhan, H., Waiho, K. & Shahreza, M.S. A simple and efficient total genomic DNA extraction method for individual zooplankton. SpringerPlus 5, 2049 (2016). https://doi.org/10.1186/s40064-016-3724-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40064-016-3724-x