
Abstract
Background
Liver ischemia–reperfusion injury (LIRI) is closely associated with immune infiltration, which commonly occurs after liver surgery, especially liver transplantation. Therefore, it is crucial to identify the genes responsible for LIRI and develop effective therapeutic strategies that target immune response. Methylation modifications in mRNA play various crucial roles in different diseases. This study aimed to identify potential methylation-related markers in patients with LIRI and evaluate the corresponding immune infiltration.
Methods
Two Gene Expression Omnibus datasets containing human liver transplantation data (GSE12720 and GSE151648) were downloaded for integrated analysis. Gene Ontology and Kyoto Encyclopedia of Genes and Genomes pathway enrichment analyses were conducted to investigate the functional enrichment of differentially expressed genes (DEGs). Differentially expressed methylation-related genes (DEMRGs) were identified by overlap** DEG sets and 65 genes related to N6-methyladenosine (m6A), 7-methylguanine (m7G), 5-methylcytosine (m5C), and N1-methyladenosine (m1A). To evaluate the relationship between DEMRGs, a protein–protein interaction (PPI) network was utilized. The core DEMRGs were screened using three machine learning algorithms: least absolute shrinkage and selection operator, random forest, and support vector machine-recursive feature elimination. After verifying the diagnostic efficacy using the receiver operating characteristic curve, we validated the expression of the core DEMRGs in clinical samples and performed relative cell biology experiments. Additionally, the immune status of LIRI was comprehensively assessed using the single sample gene set enrichment analysis algorithm. The upstream microRNA and transcription factors of the core DEMRGs were also predicted.
Results
In total, 2165 upregulated and 3191 downregulated DEGs were identified, mainly enriched in LIRI-related pathways. The intersection of DEGs and methylation-related genes yielded 28 DEMRGs, showing high interaction in the PPI network. Additionally, the core DEMRGs YTHDC1, METTL3, WTAP, and NUDT3 demonstrated satisfactory diagnostic efficacy and significant differential expression and corresponding function based on cell biology experiments. Furthermore, immune infiltration analyses indicated that several immune cells correlated with all core DEMRGs in the LIRI process to varying extents.
Conclusions
We identified core DEMRGs (YTHDC1, METTL3, WTAP, and NUDT3) associated with immune infiltration in LIRI through bioinformatics and validated them experimentally. This study may provide potential methylation-related gene targets for LIRI immunotherapy.
Similar content being viewed by others
Background
Ischemia–reperfusion injury (IRI), a pathophysiological process, involves two stages: reduction or interruption of blood supply and subsequent reperfusion and reoxygenation of the target organs [1]. Several organs, including the liver, are known to be at high risk of develo** IRI [Full size image
Screening of core DEMRGs
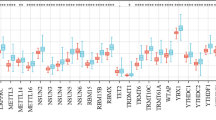
Among the 28 DEMRGs, we identified the core DEMRGs using 3 machine learning algorithms. LASSO regression algorithm was applied to identify 13 genes (Fig. 4a), RF was used to select 10 genes in descending order of importance (Fig. 4b), and SVM-RFE was employed to select 4 genes among the original 28 DEMRGs (Fig. 4c). We used a Venn diagram to determine the overlap** region of three groups of genes analyzed using the machine learning algorithms (Fig. 4d). The three algorithms identified YTH N6-methyladenosine RNA binding protein C1 (YTHDC1), METTL3, Wilms tumor 1-associated protein (WTAP), and Nudix hydrolase 3 (NUDT3) as overlap** DEMRGs at the core. Correlation analysis of the four genes is displayed in the chord diagram, which was constructed using the “circlize” package. In this diagram, the red lines represented a positive correlation, and the genes connected by green lines represented a negative correlation. Moreover, the darker the color, the stronger the correlation (Fig. 4e). To assess the predictive ability of LIRI, we constructed the gene prediction model using the “pROC” package (Fig. 4f). A higher AUC value indicates superior diagnostic performance. The ROC curves for YTHDC1, METTL3, WTAP, and NUDT3 demonstrated a reasonably satisfactory prediction accuracy, with AUCs of 0.819, 0.835, 0.933, and 0.862, respectively. As shown in Additional file 6: Fig S3, following LIRI, a notable increase was observed in the expression of YTHDC1 and WTAP, whereas the expression of METTL3 and NUDT3 decreased significantly.

Detection of the core DEMRGs using machine learning methods. a The LASSO logistic regression algorithm was used to screen 13 candidate genes. b RF algorithm was used to pick the top 10 genes in importance order. c SVM-RFE was used to identify four candidate genes. d A Venn diagram was constructed by intersecting the candidate genes obtained by the three algorithms above. e The chord diagram manifested the correlation among the core DEMRGs. f The ROC curve for the verification of diagnostic efficacy verification. DEMRGs differentially expressed methylation-related genes, LASSO least absolute shrinkage and selection operator, RF random forest, SVM-RFE support vector machine-recursive feature elimination, ROC receiver operating characteristic, AUC area under the ROC curve
Validation of core DEMRG expression and cytological experiments
We examined the changes in the expression of core DEMRGs in 10 pairs of liver transplantation samples. The results were consistent with those of transcriptome analysis. Compared with liver tissue before transplantation, a significant increase was observed in the mRNA expression of YTHDC1 and WTAP post-transplantation, whereas the mRNA expression of METTL3 and NUDT3 decreased significantly (Fig. 5a). Subsequently, we isolated and cultured primary mouse hepatocytes and established a H/R model, with cells undergoing 6 h of hypoxia followed by 6 h of reoxygenation. After extracting RNA from cells, we compared the expression of the core DEMRGs before and after H/R stimulation, and the results indicated that the changes in the expression of the four DEMRGs were consistent with those observed in the clinical samples (Fig. 5b). Among the four types of methylation modifications identified in our previous analysis, only genes related to m6A or m7G showed significant changes. Therefore, we determined whether there were alterations at the level of m6A and m7G methylation in hepatocytes after LIRI. We performed dot blot assays to detect changes in m6A and m7G methylation in hepatocytes. The results revealed a significant increase in the overall levels of m6A and m7G methylation following H/R stimulation (Fig. 5c). Next, we performed functional validation of each core DEMRG. Three siRNAs were designed for each gene, and RT-qPCR was used to determine the siRNA that inhibited gene expression most effectively (Additional file 7: Fig S4). Next, we evaluated the effect of each core DEMRG on cell viability using the CCK-8 assay. We demonstrated that altering the expression of core DEMRGs within cells under normoxic conditions did not significantly affect cell viability. Subsequently, we subjected the cells to H/R stimulation and then performed CCK-8 assay. The results indicated that after reducing the expression of YTHDC1 or NUDT3, cell viability increased significantly in the siRNA group compared with that in the NC group. Conversely, decreasing the expression of METTL3 or WTAP resulted in significantly lower cell viability in the siRNA group than in the NC group (Fig. 5d).

Expression and function validation of core DEMRGs. a The mRNA expression level of core DEMRGs in clinical liver transplantation samples. b The mRNA expression level of core DEMRGs in hepatocytes after treated by H/R. c m6A and m7G level detection by using Dot Blot in vitro. d The viability of cells in each group was evaluated by the CCK-8 assay. The P-values were shown as: *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. ns non-significant, DEMRGs differentially expressed methylation-related genes, H/R hypoxia/reoxygenation
Assessment of infiltration of immune cells
The ssGSEA method from the “GSVA” R package was used to assess the correlation among immune cells, with a darker red color representing a greater association between any two types of cells (Fig. 6a). We compared immune cell infiltration between pre- and post-transplant samples according to ssGSEA scores. As shown in the box plot, activated CD4 T cells, activated dendritic cells, mast cells, CD65dim natural killer cells, natural killer T cells, regulatory T cells, T helper 1 cells, T helper 2 cells, T helper 17 cells, eosinophils, and neutrophils were more abundant in the post-transplant group than in the pre-transplant group (Fig. 6b). We then investigated the potential association between the expression levels of each of the core DEMRGs and 23 types of immune cells (Fig. 6c). The lollipop diagrams indicated that the expression of WTAP and YTHDC1 was significantly positively correlated with the infiltration levels of activated CD4 T cells, mast cells, eosinophils, and CD65dim natural killer cells. In contrast, the expression of NUDT3 was negatively correlated with the infiltration levels of CD65dim natural killer cells, mast cells, eosinophils, and neutrophils. Additionally, the expression of METTL3 was negatively correlated with the infiltration levels of activated CD4 T cells, eosinophils, T helper 1 cells, mast cells, and activated dendritic cells.

The normalized data set for the evaluation of the degree of immune cell infiltration. a Correlation coefficient diagram showing the relationship among immune cells. The b box plot showed the differences in immune cell infiltration between the pre-transplant and post-transplant groups. c Lollipop diagrams showed the correlation between the core DEMRGs and immune cells. *P < 0.05, only immune cells with P < 0.05 are shown. DEMRGs differentially expressed methylation-related genes
Further research on core DEMRGs
To explore the possible function of core DEMRGs in LIRI, including the interrelated pathways, we performed a correlation analysis between each of the core DEMRGs and the rest of the genes. Then, we revealed the top 50 genes most positively associated with the core DEMRGs by constructing heatmaps (Fig. 7). GSEA based on REACTOME was performed to determine the significant pathways mainly associated with the mechanisms of LIRI. Ridgeline plots of the top 20 pathways obtained using the “clusterProfiler” R package are displayed in Fig. 8. With an adjusted P-value of 0.01823362, the enrichment of Interleukin-10 signaling, Interleukin-4, and Interleukin-13 signaling, Signaling by interleukins, and Cytokine signaling in the immune system was negatively associated with the expression of METTL3. Similarly, the enrichment of Toll-like receptor 2 cascade, Toll-like receptor 4 cascade, interleukin-10 signaling, interleukin-4, and interleukin-13 signaling was significantly negatively associated with the expression of NUDT3. In contrast, the enrichment of interleukin-10 signaling, interleukin-4, interleukin-13 signaling, cytokine signaling in the immune system, Toll-like receptor 4 cascade, and Toll-like receptor 3 cascade was significantly associated with the expression of WTAP. Furthermore, the enrichment of mitochondrial translation initiation, mitochondrial translation, mitochondrial translation termination, and respiratory electron elongation was significantly negatively associated with the expression of YTHDC1. These results suggest that the core DEMRGs are associated with many pathways in the progression of LIRI, especially immune-related and oxidative metabolism pathways. We then predicted the upstream miRNAs and transcription factors of the four core DEMRGs using the RegNetwork data repository and constructed the network using Cytoscape (Additional file 8: Fig S5). As shown in the network, activating the transcription factor family might be the crucial upstream transcription factor of YTHDC1 and NUDT3. Additionally, the E2F family showed potential as the upstream transcription factors of YTHDC1 and METTL3. Seven miRNAs, including hsa-miR-181a, hsa-miR-181b, hsa-miR-181c, hsa-miR-543, hsa-miR-141, hsa-miR-200a, and hsa-miR-451, showed the possibility of being present upstream of WTAP and YTHDC1. Among all miRNAs and transcription factors, MYC showed the best match as the predictive factor, which could be an upstream transcription factor of YTHDC1, NUDT3, and METTL3.

Heat map of the top 50 positively correlated genes in the core DEMRGs. DEMRGs differentially expressed methylation-related genes

Gene set enrichment analysis of single-core DEMRGs based on the REACTOME database. DEMRGs differentially expressed methylation-related genes
