
Abstract
Background
The recognition and delineation of morphologically indistinguishable cryptic species can have broad implications for wildlife conservation, disease ecology and accurate estimates of biodiversity. Parasites are intriguing in the study of cryptic speciation because unique evolutionary pressures and diversifying factors are generated by ecological characteristics of host-parasite relationships, including host specificity. Bat flies (Diptera: Nycteribiidae and Streblidae) are obligate, hematophagous ectoparasites of bats that generally exhibit high host specificity. One rare exception is Penicillidia fulvida (Diptera: Nycteribiidae), an African bat fly found in association with many phylogenetically distant hosts. One explanation for P. fulvida’s extreme polyxeny is that it may represent a complex of host-specific yet cryptic species, an increasingly common finding in molecular genetic studies of supposed generalist parasites.
Methods
A total of 65 P. fulvida specimens were collected at 14 localities across Kenya, from bat species representing six bat families. Mitochondrial cytochrome c oxidase subunit 1 (COI) and nuclear 28S ribosomal RNA (rRNA) sequences were obtained from 59 specimens and used to construct Bayesian and maximum likelihood phylogenies. Analysis of molecular variance was used to determine how genetic variation in P. fulvida was allocated among host taxa.
Results
The 28S rRNA sequences studied were invariant within P. fulvida. Some genetic structure was present in the COI sequence data, but this could be more parsimoniously explained by geography than host family.
Conclusions
Our results support the status of P. fulvida as a rare example of a single bat fly species with primary host associations spanning multiple bat families. Gene flow among P. fulvida utilizing different host species may be promoted by polyspecific roosting behavior in bats, and host preference may also be malleable based on bat assemblages occupying shared roosts. The proclivity of generalist parasites to switch hosts makes them more likely to vector or opportunistically transmit pathogens across host species boundaries. Consequently, the presence of polyxenous bat flies is an important consideration to disease ecology as bat flies become increasingly known to be associated with bat pathogens.
Graphical Abstract

Similar content being viewed by others

Introduction
Although the species is a basic unit of organization in biology and is of fundamental importance in the study of ecology and evolution, the definition of a species is famously contentious. At least 32 species concepts have been described [1], but these are often variations on a few themes [2]: (i) the ability to successfully reproduce (e.g. the Biological Species Concept [3]); (ii) distinctive morphology (e.g. the Morphological Species Concept [4]); and (iii) shared evolutionary descent (e.g. the Phylogenetic Species Concept [5]). An integrative approach to species delimitation addressing several species concepts is often desirable, but some criteria are difficult to satisfy in practice or categorically inapplicable to certain study systems [2].
The limited reach of species concepts is demonstrated by cryptic species that are morphologically indistinguishable but genetically and often ecologically distinct [6, 7]. The evolutionary processes associated with cryptic speciation are variable, with cryptic speciation known to occur in allopatry [8], sympatry [9] and parapatry [10]. Recent advances in molecular systematics have significantly increased the rate of discovery of cryptic species [6, 7]. The recognition and delimitation of cryptic species is not only critical to accurately quantifying biodiversity, but misidentification of cryptic species complexes as single species can have profound consequences for wildlife conservation and disease ecology [6, 7].
Parasites are compelling candidates for the study of cryptic speciation because they experience evolutionary pressure from their hosts, which may be strong enough to result in speciation [11,12,13]. The integration of molecular techniques in delimiting parasite species has uncovered substantial cryptic diversity, including the revision of 175 morphospecies of avian malaria parasites to an estimated 10,000 using mitochondrial (mt) DNA markers [14]. However, the division of nominal parasite species into cryptic species complexes has significance beyond the contribution to our knowledge of parasite biodiversity. Cryptic parasite species may inform our understanding of host-parasite coevolution and cospeciation [15], have different host-invasion pathways [16] and exhibit different degrees of host specificity [9, 17]. Where parasites are vectors of viruses or bacteria, resolving the host range of cryptic species can inform epidemiology [9].
Host specificity is a measure of the frequency with which a parasite species associates with a single host species [18]. Parasites limited to only one host species are host specialists, and less discriminate parasites found in association with multiple host species are host generalists. However, morphological conservatism looms as an alternative explanation for some host-generalist parasites identified solely by morphological attributes [9]. The value of using molecular genetic techniques to delimit levels of host specificity is increasingly recognized [19]. Genetic markers have shown several supposedly generalist parasite “species” to be complexes of cryptic, host-specific species [9, 20]. Accurately determining host specificity of parasite species is of wildlife conservation and human health concerns because generalist parasites may be more capable of vectoring pathogens to novel hosts [21]. Generalist parasites are also more competent invaders of new environments than specialists [21], a trait that warrants attention as anthropogenic change increasingly brings invasive parasites into contact with novel habitats and hosts [22].
Bat flies (Diptera: Streblidae and Nycteribiidae) are obligate, blood-feeding ectoparasites of bats. Species of Nycteribiidae are uniformly spider-like in appearance and wingless, restricting their ability to disperse independently of their hosts [23]. Bat flies reproduce via viviparous pupiparity, and gravid females generally leave the host only to adhere a single pupa to the roost substrate [24]. Bats are known reservoirs for an exceptionally diverse array of pathogens, including the ancestor of all mammalian malarias [25], as well as many zoonotic viruses relevant to human health [26,27,28]. As obligate parasites feeding exclusively on bat blood, bat flies warrant increased attention as potential vectors of these pathogens [29]. Bat flies have been identified as vectors of protozoan parasites [30] and bacterial pathogens in genus Bartonella [31, 32], and have recently been found to harbor bat-associated viruses related to medically impactful zoonoses [33,34,35].
Bat flies were historically understood to possess relatively low host specificity due to frequent host-switching opportunities, facilitated by polyspecific roosting behavior in bats [36]. They also spend a significant portion of their life-cycle off-host due to pupation on roost substrate [37]. However, most bat fly species are strictly monoxenous, or found in reliable association with only one bat species [29, 38]. In many parasites, patterns of host specificity are governed by host and parasite ecological characteristics facilitating reliable host-parasite encounters and eventual evolutionary associations. However, host specificity in bat flies seems to be unaligned with their host-independent dispersal ability (e.g. vagility) or other ecological associations that could provide host-switching opportunities [37, 39]. High host specificity in bat flies may therefore be maintained by host immunocompatibility or the decreased probability of flies encountering suitable mates on non-primary hosts (“reproductive filter” [37]). Bat flies associated with multiple host species are often stenoxenous or oligoxenous, infesting only closely related or congeneric bats [37]. Previous studies examining genetic variation in oligoxenous bat flies using mitochondrial genetic markers have uncovered little geographic or host-structured population genetic structure [40, 41].
Penicillidia fulvida (Diptera: Nycteribiidae) is an African bat fly species that exhibits unusually low host specificity [42] and is thus apparently polyxenous. Collections summarized by Theodor in 1967 associated P. fulvida with 14 host species, and noted a wide range of host associations relative to other bat fly species [42]. Penicillidia fulvida specimens collected in Kenya and currently stored in the Field Museum of Natural History Collection of Hippoboscoid Diptera (Field Museum of Natural History, Chicago, IL, USA) have been recovered from 14 putative host species spanning seven genera and six families of bat. In contrast to the general pattern of monoxenous or oligoxenous associations of bat flies with host bats [37], P. fulvida is known to parasitize bat families that diverged over 60 million years ago [43]. The rarity of polyxenous host associations among bat flies, in concert with the increasingly recognized prevalence and ecological importance of cryptic parasite species, suggests that P. fulvida may represent a complex of cryptic, and possibly more host-specific, bat fly species. Alternatively, P. fulvida may truly represent a single generalist species capable of colonizing distantly related hosts. The questions addressed in this study are: (i) do patterns of genetic differentiation exist within P. fulvida, potentially indicative of cryptic speciation? and (ii) is there genetic differentiation of P. fulvida parasitizing sympatric hosts, which might indicate cryptic host specificity?
Methods
Sampling
All P. fulvida specimens used in this study were collected during field expeditions of the “Bats of Kenya” project between 2006 and 2015. This survey was designed to inventory Kenya’s diverse habitats and rich faunas, and much remains to be learned about the limits and interrelationships of bat species. The project assayed genetic variation in 21 bat genera belonging to 10 families, identifying putative species using both mitochondrial and nuclear markers (e.g. [44,45,46,47]). To avoid adding to current taxonomic confusion, a conservative approach was purposefully taken regarding the assignment of names to clades in the analyses. Where a clade’s taxonomic identity was ambiguous or unknown, it was simply labeled as a numbered clade, and this convention is reflected in identifying P. fulvida hosts. Integrative taxonomic diagnoses of the various clades are needed to determine which (if any) existing names may apply to them.
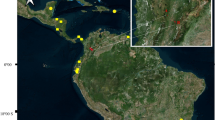
Penicillidia fulvida were recovered from 14 localities, comprising mostly tropical broadleaf forests and savannas in the southern and western regions of Kenya (Figs. 1, 2). A total of 65 P. fulvida specimens were collected (Table 1); all are referenced in this study to describe host associations (Table 2), and 59 were sequenced. During collection, bats were captured in mist nets or harp traps during foraging/commuting or in hand nets at roosting sites, and all were subsequently stored individually in clean cloth bags to minimize the risk of parasite disturbance transfers. Bats were either safely released or retained as species vouchers after processing. Vouchered bats were euthanized with halothane following guidelines by the American Society of Mammalogists [48], under the approval of the Field Museum’s Institutional Animal Care and Use Committee (2012-003). Voucher specimens were fumigated with ethyl ether to facilitate collection of their ectoparasites, but P. fulvida were also reliably detected and extracted from live bats because of their large size. Once extracted from the host, bat flies were immediately stored in 95% ethanol and later identified under a light microscope using species keys and descriptions from Theodor [42] as well as reference specimens from the Field Museum collection. All fly specimens are currently housed in the Field Museum of Natural History Collection of Hippoboscoid Diptera (currently on long-term loan to C. W. Dick at Western Kentucky University).

Map of Kenya featuring the 14 sampling localities yielding Penicillidia fulvida. A gazetteer of sampling localities is provided in Table 1

Map of Kenya featuring 14 sampling localities yielding P. fulvida and biomes as delineated by Olson et al. [82]
Two congeneric African bat flies, Penicillidia pachymela and P. leptothrinax, were also sequenced to better gauge “species-level” divergence in target genes. Penicillidia pachymela is posited to be closely related to P. fulvida based on morphological characteristics, and is also assigned to the P. fulvida group of species [42]. Penicillidia pachymela was collected in sympatry with P. fulvida but is exceedingly rare across their shared range in Kenya, with only a single specimen collected during this cumulative 9-year sampling effort. Penicillidia leptothrinax is a more distantly related and smaller-bodied species endemic to Madagascar.
DNA extraction, amplification and sequencing
One or two legs were removed from each P. fulvida specimen for genetic analysis to allow retention of morphological vouchers. Prior to DNA extraction, each leg was lacerated with sterile forceps to expose the muscle tissue beneath the exoskeleton and transferred to a 1.5-ml microcentrifuge tube. Whole genomic DNA extractions were performed according to the manufacturer’s protocol using the DNeasy Blood and Tissue Kit (Qiagen, Hilden, Germany), with the final elution divided into two steps at volumes of 35 and 65 μl, respectively, to optimize DNA concentration. All extractions were assessed for quality using a NanoDrop 2000 spectrophotometer (Thermo Fisher Scientific, Waltham, MA, USA).
A 658-bp fragment of the cytochrome c oxidase subunit I (COI) gene was amplified using the invertebrate-specific primer pair LCO1490 (5’-GGTCAACAA ATCATAA-AGATATTGG-3’) and HCO2198 (5’-TAACTTCAGGGT GACCAAAAAATCA-3’) [49]. Each PCR assay was conducted in a total reaction volume of 25 μl containing 12.5 μl GoTaq MasterMix (Promega, Madison, WI, USA), 9 μl of nuclease-free water, 1 μl each of 10 μM forward and reverse primers and 1.5 μl of DNA template. A negative control, with nuclease-free water replacing the template DNA, was included with every set of reactions. Thermal cycling conditions for COI consisted of an initial denaturing at 95 °C for 1 min, followed by 35 cycles of 95 °C for 2 min, 50 °C and 72 °C for 1 min, with a final extension at 72 °C for 5 min.
MtDNA has rapid substitution rates, making mitochondrial sequence data favorable for parsing relatively shallow, species-level phylogenetic relationships [50]. Although segments of COI have been proposed as “universal barcodes” suitable for species delimitation almost ubiquitously across taxa [50], sole reliance on mitochondrial markers disregards other modes of inheritance [51] and can obscure potential effects of introgression or infection with Wolbachia, an arthropod-associated bacterial endosymbiont capable of disrupting patterns of mitochondrial inheritance [52]. For a multilocus approach, we also amplified 28S ribosomal RNA (rRNA), a nuclear gene commonly used alongside COI for species-level analyses in arthropods [53].
The D2 region of the nuclear 28S gene was amplified using the primers F2 (5-AGAGAGAGTTCAAGAGTACGTG-3’) and 3DR (5’-TAGTTCACCATCTTTCGGGTC-3’) [54]. Reaction components and volumes were identical to those used to amplify COI. Thermal cycling conditions for 28S were consisted of an initial denaturing at 98 °C for 2 min, followed by 35 cycles of denaturing at 94 °C for 3 min, annealing at 51 °C for 30 s, with a first extension at 72 °C for 2 min and then a final extension at 72 °C for 8 min.
PCR products were verified for size and quality via electrophoresis on a 1.5% agarose gel stained with SybrSafe (Invitrogen, Thermo Fisher Scientific, Waltham, MA, USA) and visualized under a blue LED light. Sanger sequencing was performed at the North Carolina State Genomic Sciences Laboratory (Raleigh, NC, USA) using forward and reverse primers for both genes.
Phylogenetic analyses
Sequences were trimmed and assessed for ambiguous bases by eye, then aligned using the MUSCLE algorithm [55] as implemented in Geneious 6.0 [56]. A hippoboscid fly, Pseudolynchia canarensis, was included as an outgroup (sequence data retrieved from GenBank). Haplotype groups were identified using DnaSP 6 [57], and a minimum-spanning haplotype network was constructed with PopART [58].
Substitution models for Bayesian and maximum likelihood analyses were estimated with jModeltest [59]. Optimized substitution models based on Akaike’s information criterion were GTR + G for COI and HKY + I for 28S. Both concatenated and single-gene trees were created, with concatenated trees partitioned by locus. Bayesian analysis was performed in MrBayes 3.2 [60] using the default burn-in of 25% and 10 million Markov chain Monte Carlo generations. Average standard deviation in split frequencies fell below 0.01 at 380,000 then at 650,000 generations and remained under this threshold until the final generation, indicating stationarity was reached. Posterior probabilities for clade support were calculated using the trees remaining after burn-in. A maximum likelihood phylogeny was created using RAxML 8.0 [61]. The starting tree was obtained by searching for the best-scoring maximum likelihood tree in a single program run. Branch support was calculated using a random seed and 1000 rapid bootstrap replicates.
Analysis of molecular variance
A hierarchical analysis of molecular variance (AMOVA) was used to determine the extent to which genetic variation in P. fulvida is allocated among host taxa. Because AMOVA requires groups to be defined a priori, haplotypes were conservatively pooled by host family. Host families with insufficient sample sizes for statistical analysis (Emballonuridae, Nycteridae and Hipposideridae, from which only 1–3 P. fulvida specimens were recovered or sequenced) were excluded; therefore, only specimens from Miniopteridae (n = 33), Rhinolophidae (n = 11) and Vespertilionidae (n = 9) were compared. AMOVA was performed in Arlequin 3.5.2.2 [62] using a locus-by-locus method to accommodate missing data. The significance of pairwise fixation indices (Fst, an F-statistic measuring variance in allele frequency among populations) was calculated using 1023 permutations, each assigning parasite haplotypes to host families at random to generate a null distribution.
Results
Phylogenetic analyses
A total of 12 COI haplotypes were present within P. fulvida, with 11 variable sites (Fig. 3). One specimen (FMNH216012-013) could not be assigned a COI haplotype due to an ambiguous base at a variable site, and was excluded from the haplotype network. Only a single 28S haplotype was recovered from P. fulvida, but 28S haplotypes were distinct among the three described species within Penicillidia (1.8% pairwise uncorrected distance [p-distance] between P. leptothrinax and the most common fulvida haplotype, 1.6% p-distance between P. pachymela and P. fulvida and 3.2% p-distance between P. leptothrinax and pachymela). The concatenation of 28S with COI in phylogenetic analyses yielded poorly resolved relationships among the three putative species and was too conservative to inform population genetic structure within P. fulvida. Because P. pachymela and P. leptothrinax had only single specimens included for comparative purposes and, in addition, resolving relationships among putative species within Penicillidia is outside the scope of this study, only COI-based trees were included.

Minimum-spanning haplotype network of 12 unique P. fulvida cytochrome c oxidase subunit 1 (COI) haplotypes, allocated by host family
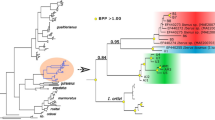
Bayesian and maximum likelihood analyses yielded incongruent topologies within P. fulvida (Figs. 4, 5). The clades that are moderately well-supported but recovered using only a single method lack apparent host-based or geographic structure, and therefore do not appear to warrant discussion. One clade was supported by both Bayesian and maximum likelihood analyses, comprising six specimens from the host family Rhinolophidae and from a single, distant locality (Marsabit; Table 1; Figs. 4, 5). Neither analysis recovered outgroups within the in-group of P. fulvida.

Bayesian phylogenetic tree (P. fulvida labeled with voucher identifications (IDs), congeners Penicillidia pachymela and P. leptothrinax and outgroup Pseudolynchia canarensis) constructed using COI, a GTR + G substitution model and 10 million generations. Posterior probabilities for clade support are presented on a scale of 0–100 for ease of comparison to maximum likelihood bootstrap values

Maximum likelihood tree (P. fulvida labeled with voucher IDs, congeners P. pachymela and P. leptothrinax and outgroup Pseudolynchia canarensis) generated using COI and a GTR + G substitution model. Branch support was calculated using 1000 rapid bootstrap replicates, and only support values > 60% are displayed
Analysis of molecular variance
Hierarchical AMOVA rejected the null hypothesis of haplotype homogeneity among host families (P < 0.001; Table 3). The significance of pairwise Fst values indicated that allele frequencies were unique in P. fulvida haplotype groups from rhinolophid bats (Yinpterochiroptera: Rhinolophoidea) when compared to haplotype groups from both miniopterid and vespertilionid bats (Yangochiroptera: Vespertilionoidea) (Table 4). However, this taxonomic association appears artefactual when regarded alongside phylogenetic structure and sampling composition. Genetic structure associated with Rhinolophidae aligns more reliably with sampling locality (Table 1; Fig. 4); 7 of 11 P. fulvida recovered from rhinolophid bats were recovered from Marsabit, and 6/6 of the “rhinolophid” clade were from Marsabit bats). Viewed in this light, the results do not suggest host-based genetic differentiation is present within P. fulvida.
Discussion
Parasites are susceptible to placement in artificial species groups because of their morphological conservatism relative to their hosts [9]. The advent of molecular genetic techniques has provided a valuable strategy for scrutinizing morphologically indistinguishable but ecologically differentiated parasites, and can result in the division of host-generalist nominal species into more host-specific cryptic species complexes [9, 14, 15, 17]. Using two genetic markers (mitochondrial COI and nuclear 28S), we have uncovered no such evidence of host-mediated genetic structure in the polyxenous bat fly P. fulvida. This finding is concordant with other studies evaluating patterns of population genetic structure in non-monoxenous (oligoxenous) nycteribiid bat flies [40, 41], but to our knowledge represents the first such investigation of a truly polyxenous bat fly species. These results suggest inter-host gene flow may be occurring in P. fulvida, and support its recognition as a single species under both morphological and phylogenetic species concepts. Unlike most bat flies, it is capable of parasitizing phylogenetically disparate bat species, and in Kenya parasitizes both suborders (Yinpterochiroptera and Yangochiroptera) and six different families of bats.
Although no genetic structure could be reliably attributed to host identity, there is some evidence P. fulvida is not uniformly panmictic across Kenya. Both maximum likelihood and Bayesian phylogenetic analyses supported a clade of 6/7 P. fulvida from a single remote sampling site in Marsabit Forest (Figs. 3, 4). Isolation by distance alone is insufficient to explain this geographic differentiation, as P. fulvida is essentially panmictic (with respect to the localities sampled) across the comparable 700 km distance between Kakamega and Fikirini (Figs. 1, 2). Kenya is composed of diverse biomes, and Marsabit Forest is well-representative of this mosaic: a tropical broadleaf forest sustained by volcanic soil and orographic precipitation that is completely surrounded by swaths of rocky desert (Fig. 2). Further, all P. fulvida from Marsabit Forest were collected from horseshoe bats (5/6 from Rhinolophus fumigatus [clade 2–3; see [45]], and 1/6 from Rhinolophus cf. landeri); all have wings with relatively low aspect ratios, marking them as relatively weak fliers [63]. Marsabit Forest’s geographic isolation may stymie gene flow in P. fulvida by restricting the dispersal of its bat hosts. However, given the small sample size and the lack of intervening sampling sites that might firmly implicate the surrounding desert as a barrier to gene flow, the origin of this site-related genetic structure remains speculative.
One specimen of Rhinolophus fumigatus from Marsabit hosted two distinct haplotypes of P. fulvida: the typical Marsabit-associated clade and a haplotype also found on a Miniopterus host at Kakamega Forest over 400 km distant. This suggests lack of barriers between Marsabit and other populations, but absent additional data this finding cannot be explored in more detail.
An alternative explanation for the lack of genetic structure in P. fulvida is insufficient variety and variability in the genes used. Nuclear 28S was conservative among the three Penicillidia species sequenced and invariant in P. fulvida. Consequently, only mitochondrial COI was used to assess patterns of intraspecific, inter-host variation in P. fulvida. Drawing conclusions from one mitochondrial gene can be precarious, as potential mito-nuclear discordance and the small proportion of the overall genome represented in single-gene phylogenies can cause incongruence [64]. Further, reliance on mtDNA is unfavorable in some insect taxa, including bat flies, due to possible infection with bacterial Wolbachia [65]. Wolbachia is maternally transmitted and can cause disruptions in mitochondrial inheritance, resulting in the overrepresentation of certain haplotypes in a population or the absence of polymorphism altogether [52, 66]. The use of more variable nuclear genetic markers in future studies, alongside increased research efforts investigating the ability for Wolbachia to influence inheritance in nycteribiid bat flies, could help clarify the possibility of Wolbachia infection obscuring genetic structure in P. fulvida.
Understanding host specificity and the processes responsible for its evolution and maintenance across parasite communities is crucial to understanding the evolutionary context of host-parasite associations and the role of parasites in disease transmission. High host specificity in parasites has historically been regarded as the default trend in parasite evolution; specialization is also regarded as an evolutionary “dead end” because morphological specialization required for a highly specific parasite to efficiently exploit its host makes a specialist lineage’s “return” to a generalist strategy improbable [67]. However, the evolution of ecological resource specialization is not characterized by fixed directionality: different degrees of host specificity can arise multiple times in the evolutionary history of a parasite, with generalist species sometimes emerging from specialist lineages [67,68,69]. Extensive biodiversity surveys have found bat flies to possess considerably high host specificity as a group, and some instances of low specificity in bat flies may be attributable to poorly understood species boundaries [29]. Using molecular techniques, this study demonstrates that, although apparently rare, polyxenous bat fly species do exist. If P. fulvida evolved from specialist ancestors, the ecological precedent for a decrease in specificity across evolutionary time may be resource breadth-associated performance trade-offs [70, 71] or interspecific competition [69].
Host ecology serves as an important evolutionary driver of patterns of host specificity in parasite communities. Multiple bat species often aggregate closely in a single roost, a behavior which may confer anti-predator benefits or simply reflect limited availability of suitable roosts [72, 73]. Mixed-species groups present host-switching opportunities for parasites that depend on their hosts for dispersal; as a result, polyspecific bat roosts were a historical precedent for proposing universally low host specificity in bat flies [36, 74]. Penicillidia fulvida was collected from multiple host species in eight of 14 roosts and from multiple host families in seven of 14 roosts in Kenya, indicating polyspecific roosting behavior may be a proponent of P. fulvida’s broad host range and high genetic structure within hosts. Further, although it is now recognized that specialist bat flies can maintain their high specificity despite host-switching opportunities [37], bat assemblages in shared roosts may influence host preference in generalist bat flies [75]. Accordingly, there is some indication that parasitism by P. fulvida is based on host availability. Penicillidia fulvida occasionally parasitizes the African sheath-tailed bat Coleura afra (4/63 total host associations in the Bats of Kenya survey; Table 2), but associations with C. afra were only recorded in the absence of potential miniopterid and rhinolophid hosts (Table 1). Coleura afra was present in three shared roosts containing Miniopterus hosts of P. fulvida, but P. fulvida was never recovered from C. afra when these alternative hosts were available. This pattern suggests that although host selection in P. fulvida is unconstrained by phylogenetic distance, P. fulvida may still demonstrate tiers of host preference, which could function to increase fitness by mitigating local competition or selecting optimally nutritious or compatible host blood [29].
Vector ecology is a valuable determinant of pathogen spread and potential zoonotic spillover [76]. Low host specificity in parasites may promote exposure to a wider range of infectious agents and facilitate disease transmission to new hosts and geographic areas [22]. Bats are reservoirs for a broad spectrum of pathogens, a product of immunological or ecological predisposal [77] or a reflection of their high species diversity [28]. Nycteribiid bat flies, including P. fulvida, can serve as vectors facilitating the transmission of several bat pathogens: hemosporidian parasites in genus Polychromophilus [25, 30] and bacterial bartonellae [31, 32]. Moreover, research linking nycteribiid bat flies to impactful bat-associated viral zoonoses is expanding [33, 35, 78]. Within the scope of this study, Kenyan hosts of P. fulvida are known reservoirs of paramyxoviruses (Miniopterus spp. [79]), SARS coronaviruses (Rhinolophus fumigatus and R. landeri [80]) and lyssaviruses (Miniopterus spp. [81]).
Because this study failed to detect significant genetic differentiation among P. fulvida parasitizing bats across three superfamilies in both suborders, the results clearly demonstrate that P. fulvida is a rare polyxenous species of bat fly. However, it is important to acknowledge this study is not comprehensive with respect to P. fulvida’s geographic and host range. Penicillidia fulvida has a broad range outside Kenya, comprising much of sub-Saharan Africa [42] and possibly Madagascar (C.W. Dick, unpublished data). Additionally, P. fulvida was collected from three host families in Kenya with relatively uninformative sample sizes (Emballonuridae, Rhinonycteridae and Nycteridae) and has previously been recorded from Pteropodidae, although this association is tenuously based on a single record (Eidolon helvum in South Africa; [42]). Based on associations recorded in this study and past surveys, it seems probable that bats from Miniopteridae, Rhinolophidae and Vespertilionidae represent P. fulvida’s primary host range, whereas other, infrequently observed hosts are non-primary associations indicative of natural host-switching events. Increased, targeted sampling efforts may be useful for further parsing host associations and for testing hypotheses associated with geographic differentiation in P. fulvida.
Conclusions
Host specificity is one of the most basic ecological characteristics of parasites and likely subject to strong selection. Accurate measurement of host specificity is vital to understanding parasite biodiversity, vulnerability to population decline and the role of parasites in disease transmission. Moreover, accurately estimating host specificity relies on acknowledging the presence of cryptic diversity and using integrative approaches to delimiting parasite species. This study provides molecular genetic evidence that the nycteribiid bat fly P. fulvida does not exhibit cryptic host specificity, and instead represents a single species with a wide range of phylogenetically distant bat hosts.
Availability of data and materials
All sequence data are available in the GenBank repository (COI accession numbers ON704663-ON70421; 28S accession numbers ON693298-ON693355).
References
Zachos FE. Species concepts in biology: historical development, theoretical foundations and practical relevance. Berlin: Springer; 2016.
Perkins SL. Species concepts and malaria parasites: detecting a cryptic species of Plasmodium. Proc Biol Sci. 2000;267:2345–50.
Mayr E. Systematics and the origin of species. New York: Columbia University Press; 1942.
Cronquist A. Once again, what is a species? In: Romberger JA, editor. Biosystematics in agriculture. Monclair: Allenheld & Osmun; 1978. p. 3–20.
Cracraft J. Species concepts and speciation analysis. In: Johnson RF, editor. Current ornithology, vol. 1. New Yori: Plenum Press; 1983. p. 159–87.
Bickford D, Lohman DJ, Sodhi NS, Ng PK, Meier R, Winker K, et al. Cryptic species as a window on diversity and conservation. Trends Ecol Evol. 2007;22:148–55.
de Leon GP, Nadler SA. What we don’t recognize can hurt us: a plea for awareness about cryptic species. J Parasitol. 2010;96:453–64.
Norman MD, Cameron HE, Strugnell JM. Allopatric speciation within a cryptic species complex of Australasian octopuses. PLoS ONE. 2014;9:1–13.
Whiteman NK, Sánchez P, Merkel J, Klompen H, Parker PG. Cryptic host specificity of an avian skin mite (Epidermoptidae) vectored by louseflies (Hippoboscidae) associated with two endemic Galápagos bird species. J Parasitol. 2006;92:1218–28.
Dennis AB, Hellberg ME. Ecological partitioning among parapatric cryptic species. Mol Ecol. 2010;19:3206–25.
Hafner MS, Nadler SA. Phylogenetic trees support the coevolution of parasites and their hosts. Nature. 1988;332:258–9.
Light JE, Hafner MS. Cophylogeny and disparate rates of evolution in sympatric lineages of chewing lice on pocket gophers. Mol Phylogenetics Evol. 2007;45:997–1013.
Tortosa P, Dsouli N, Gomard Y, Ramasindrazana B, Dick CW, Goodman S. Evolutionary history of Indian Ocean nycteribiid bat flies mirroring the ecology of their hosts. PLoS ONE. 2013;8:1–8.
Bensch S, Perez-Tris J, Waldenstrom J, Hellgren O. Linkage between nuclear and mitochondrial DNA sequences in avian malaria parasites: multiple cases of cryptic speciation? Evolution. 2004;58:1617–21.
Engelbrecht A, Matthee CA, Ueckermann EA, Matthee S. Evidence of cryptic speciation in mesostigmatid mites from South Africa. J Parasitol. 2014;141:1322–32.
Miura O, Torchin ME, Kuris AM, Hechinger RF, Chiba S. Introduced cryptic species exhibit different invasion pathways. Proc Natl Acad Sci USA. 2006;103:19818–23.
Smith MA, Woodley NE, Janzen DH, Hallwachs W, Hebert PDN. DNA barcodes reveal cryptic host-specificity within the presumed polyphagous members of a genus of parasitoid flies (Diptera: Tachinidae). Proc Natl Acad Sci USA. 2005;103:3657–62.
Poulin R. Evolutionary ecology of parasites. 2nd ed. Princeton: Princeton University Press; 2007.
Poulin R, Keeney DB. Host specificity under molecular and experimental scrutiny. Trends Parasitol. 2008;24:24–8.
McCoy KD, Boulinier T, Tirard C, Michalakis Y. Host specificity of a generalist parasite: genetic evidence of sympatric host races in seabird tick Ixodes uriae. J Evol Biol. 2001;14:395–405.
Tompkins DM, Poulin R. Parasites and biological invasions. In: Lee W, Burns B, editors. Biological invasions in New Zealand. Ecological studies, vol. 186. Berlin: Springer; 2006. p. 67–84.
Daszak P, Cunningham AA, Hyatt AD. Emerging infection diseases—threats to biodiversity and human health. Science. 2000;287:443–9.
Dick CW, Patterson BD. Bat flies: Obligate ectoparasites of bats. In: Morand S, Krasnov BR, Poulin R, editors. Micromammals and macroparasites: from evolutionary ecology to management. Berlin Heidelberg: Springer; 2006. p. 179–94.
Ching L, Marshall A. Breeding biology of the bat-fly Eucampsipoda sundaicum Theodor, 1955 (Diptera, Nycteribiidae). Malay Nat J. 1968;21:171–80.
Lutz HL, Patterson BD, Peterhans JCK, Stanley WT, Webala PW, Gnoske TP, et al. Diverse sampling of East African haemosporidians reveals chiropteran origin of malaria parasites in primates and rodents. Mol Phylogenet Evol. 2016;99:7–15.
Calisher CH, Childs JE, Field HE, Homes KV, Shountz T. Bats: important reservoir hosts of emerging viruses. Clin Microbiol Rev. 2006;19:521–45.
Olival KJ, Hosseini PR, Zambrana-Torrelio C, Ross N, Bogich TL, Daszak P. Host and viral traits predict zoonotic spillover from mammals. Nature. 2017;546:646–50.
Mollentze N, Streicker DG. Viral zoonotic risk is homogeneous among taxonomic orders of mammalian and avian reservoir hosts. Proc Natl Acad Sci USA. 2020;117:9423–30.
Dick CW, Dittmar K. Parasitic bat flies (Diptera: Streblidae and Nycteribiidae): host specificity and potential as vectors. In: Klimpel S, Mehlhorn S, editors. Bats (Chiroptera) as vectors of disease and parasites. Berlin: Springer; 2014. p. 131–55.
Megali A, Yannic G, Christe P. Disease in the dark: molecular characterization of Polychromophilus murinus in temperate zone bats revealed a worldwide distribution of this malaria-like disease. Mol Ecol. 2010;20:1039–48.
Morse SF, Olival KJ, Kosoy M, Billeter S, Patterson BD, Dick CW, et al. Global distribution and genetic diversity of Bartonella in bat flies (Hippoboscoidea, Streblidae, Nycteribiidae). Infect Genet Evol. 2012;12:1717–23.
Wilkinson DA, Duron O, Cordonin C, Gomard Y, Ramasindrazana B, Mavingui P, et al. The bacteriome of bat flies (Nycteribiidae) from the Malagasy region: a community shaped by host ecology, bacterial transmission mode, and host-vector specificity. Appl Environ Microbiol. 2016;82:1778–88.
Bennett AJ, Paskey AC, Kuhn JH, Bishop-Lilly KA, Goldberg TL. Diversity, transmission, and cophylogeny of Ledanteviruses (Rhabdoviridae: Ledantevirus) and nycteribiid bat flies parasitizing Angolan soft-furred bats in Bundibugyo District, Uganda. Microorganisms. 2020;8:750.
Ramírez-Martínez MM, Bennett AJ, Dunn CD, Yuill TM, Goldberg TL. Bat flies of the family Streblidae host relatives of medically and agriculturally important “bat-associated” viruses. Viruses. 2021;13:860.
Kemenesi G, Tóth GE, Mayora-Neto M, Scott S, Temperton N, Wright E, et al. Isolation of infectious Lloviu virus from Schreiber’s bats in Hungary. Nat Commun. 2022;13:1–11.
Theodor O. Parasitic adaptation and host-parasite specificity in the pupiparous Diptera. In: Mayr E, editor. First symposium on host specificity among parasites of vertebrates. Neuchâtel: Institut de Zoologie, Université de Neuchâtel; 1957. p. 50–63.
Dick CW, Patterson BD. Against all odds: explaining high host specificity in dispersal-prone parasites. Int J Parasitol. 2007;37:871–6.
Hiller T, Brändel S, Honner B, Page R, Tschapka M. Parasitization of bats by bat flies (Streblidae) in fragmented habitats. Biotropica. 2020;52:488–501.
ter Hofstede HM, Fenton BM, Whitaker JO. Host and host-site specificity of bat flies (Diptera: Streblidae and Nycteribiidae) on Neotropical bats (Chiroptera). Can J Zool. 2004;82:616–26.
Olival KJ, Dick CW, Simmons NB, Morales JC, Melnick DJ, Dittmar K, et al. Lack of population genetic structure and host specificity in the bat fly, Cyclopodia horsfieldi, across species of Pteropus bats in southeast Asia. Parasit Vectors. 2013;6:231.
Wilson GM, Byrd KS, Caire W, Bussche RAVD. Lack of population genetic structure in the bat fly (Trichobius major) in Kansas, Oklahoma, and Texas based on DNA sequence data for the cytochrome oxidase I (COI) and NADH dehydrogenase 4 (ND4) genes. Proc Okla Acad Sci. 2007;87:31–6.
Theodor O. An illustrated catalogue of the Rothschild collection of Nycteribiidae in the British Museum (Natural History), with keys and short descriptions for the identification of subfamilies, genera, species and subspecies. London: British Museum of Natural History; 1967.
Teeling EC, Jones G, Rossiter SJ. Phylogeny, genes and hearing: implications for the evolution of echolocation in bats. In: Fenton MB, Grinnell AD, Popper AN, Fay RR, editors. Bat bioacoustics. New York: Springer; 2016. p. 25–54.
Demos TC, Webala PW, Lutz HL, Kerbis Peterhans JC, Goodman SM, Cortés-Delgado N, et al. Multilocus phylogeny of a cryptic radiation of African long-fingered bats (Chiroptera, Miniopteridae). Zool Scripta. 2019;49:1–13.
Demos TC, Webala PW, Goodman SM, Kerbis Peterhans JC, Bartonjo M, Patterson BD. Molecular phylogenetics of the African horseshoe bats (Chiroptera: Rhinolophidae): expanded geographic and taxonomic sampling of the Afrotropics. BMC Evol Biol. 2019;19:166.
Monadjem A, Demos TC, Dalton DL, Webala PW, Musila S, Kerbis Peterhans JC, et al. A revision of pipistrelle-like bats (Mammalia: Chiroptera: Vespertilionidae) in East Africa with the description of new genera and species. Zool J Linn Soc. 2020;191:1–33.
Patterson BD, Webala PW, Havery TH, Agwanda BR, Goodman SM, Kerbis Peterhans JC, et al. Evolutionary relationships and population genetics of the Afrotropical leaf-nosed bats (Chiroptera, Hipposideridae). Zookeys. 2020;929:117–71.
Sikes RS, The Animal Care and Use Committee of the American Society of Mammalogists. 2016 guidelines of the American Society of Mammalogists for the use of wild mammals in research and education. J Mammal. 2016;97:663–688.
Folmer O, Black M, Hoeh W, Lutz R, Vrijenhoek R. DNA primers for amplification of mitochondrial cytochrome c oxidase subunit I from diverse metazoan invertebrates. Mol Mar Biol Biotechnol. 1994;3:294–9.
Hebert PDN, Cywinska A, Ball SL, deWaard JR. Biological identifications through DNA barcodes. Proc R Soc Lond. 2003;270:313–21.
Rubinoff D. Utility of mitochondrial DNA barcodes in species conservation. Conserv Biol. 2006;20:1026–33.
Jiggins FM. Male-killing Wolbachia and mitochondrial DNA: selective hybrid introgression and parasite population dynamics. Genetics. 2003;164:5–12.
Kuhlmann M, Almeida E, Laurenne N, Quicke D. Molecular phylogeny and historical biogeography of the bee genus Colletes Latreille, 1802 (Hymenoptera: Apiformes: Colletidae), based on mitochondrial COI and nuclear 28S sequence data. Insect Syst Evol. 2009;40:290-318
Belshaw R, Lopez-Vaamonde C, Degerli N, Quicke DLJ. Paraphyletic taxa and taxonomic chaining: evaluating the classification of braconine wasps (Hymenoptera: Braconidae) using 28S, D2–3 rDNA sequences and morphological characters. Biol J Linn Soc. 2011;73:411–24.
Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792–7.
Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, Sturrock S, et al. Geneious basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics. 2012;2012:1647–9.
Rozas J, Ferrer-Mata A, Sánchez-DelBarrio JC, Guirao-Rico S, Librado P, Ramos-Onsins SE, et al. DnaSP 6: DNA sequence polymorphism analysis of large data sets. Mol Biol Evol. 2017;34:3299–302.
Leigh JW, Bryant D. PopART: full-feature software for haplotype network construction. Methods Ecol Evol. 2015;6:1110–6.
Posada D. jModelTest: phylogenetic model averaging. Mol Biol Evol. 2008;25:1253–6.
Ronquist F, Huelsenbeck JP. MRBAYES 3: bayesian phylogenetic inference under mixed models. Bioinformatics. 2003;19:1572–4.
Stamatakis A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 2014;30:1312–3.
Excoffier L, Lischer HEL. Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Mol Ecol Resour. 2010;10:564–7.
Norberg U, Rayner JMV. Ecological morphology and flight in bats (Mammalia: Chiroptera): wing adaptations, flight performance, foraging strategy and echolocation. Phil Trans R Soc B. 1987;316:335–427.
Rubinoff D, Holland BS. Between two extremes: mitochondrial DNA is neither the panacea nor the nemesis of phylogenetic and taxonomic inference. Syst Biol. 2005;54:952–61.
Szentiványi T, Christe P, Glaizot O. Bat flies and their microparasites: current knowledge and distribution. Front Vet Sci. 2019;6:115.
James AC, Ballard JW. Expression of cytoplasmic incompatibility in Drosophila simulans and its impact on infection frequencies and distribution of Wolbachia pipientis. Evolution. 2000;54:1661–72.
Poulin R, Krasnov BR, Shenbrot GI, Mouillot D, Khokhlova IS. Evolution of host specificity in fleas: Is it directional and irreversible? Int J Parasitol. 2006;36:185–91.
Thompson JD. The coevolutionary process. Chicago: University of Chicago Press; 1994.
Johnson KP, Malenke JR, Clayton DH. Competition promotes the evolution of host generalists in obligate parasites. Proc Biol Sci. 2009;276:3921–6.
Jaenike J. Host specialization in phytophagous insects. Annu Rev Ecol Evol Syst. 1990;21:243–73.
Caley MJ, Munday PL. Growth trades off with habitat specialization. Proc Biol Sci. 2003;270:S175–7.
Kunz TH. Roosting ecology. In: Kunz TH, editor. Ecology of bats. New York: Plenum Press; 1982. p. 1–55.
Stensland E, Angerbjörn A, Berggren P. Mixed species groups in mammals. Mammal Rev. 2003;33:205–23.
Jobling B. Host-parasite relationship between the American Streblidae and the bats, with a new key to the American genera and a record of the Streblidae from Trinidad, British West Indies (Diptera). J Parasitol. 1949;39:315.
Seneviratne SS, Fernando HC, Udagama-Randeniya PV. Host specificity in bat ectoparasites: a natural experiment. Int J Parasitol. 2009;2009:995–1002.
Plowright RK, Parrish CR, McCallum H, Hudson PJ, Ko AI, Graham AL, et al. Pathways to zoonotic spillover. Nat Rev Microbiol. 2017;15:502–10.
Brook CE, Dobson AP. Bats as “special” reservoirs for emerging zoonotic pathogens. Trends Microbiol. 2015;23:172–80.
Jansen van Vuren P, Wiley M, Palacios G, Storm N, McCulloch S, Markotter W, et al. Isolation of a novel fusogenic orthoreovirus from Eucampsipoda africana bat flies in South Africa. Viruses 2016;8:65.
Conrardy C, Tao Y, Kuzmin IV, Niezgoda M, Agwanda B, Breiman RF, et al. Molecular detection of adenoviruses, rhabdoviruses, and paramyxoviruses in bats from Kenya. Am J Trop Med Hyg. 2014;91:258–66.
Warahiu C, Ommeh S, Obanda V, Agwanda B, Gakuya F, Ge X, et al. Molecular detection of viruses in Kenyan bats and discovery of novel astroviruses, caliciviruses and rota viruses. Virol Sin. 2017;32:101–14.
Kuzmin IV, Niezgoda M, Franka R, Agwanda B, Markotter W, Beagley JC, et al. Possible emergence of West Caucasian Bat Virus in Africa. Emerg Infect Dis. 2008;14:1887–9.
Olson DM, Dinerstein E, Wikramanayake ED, Burgess ND, Powell GVN, Underwood EC, et al. Terrestrial ecoregions of the world: a new map of life on Earth. Bioscience. 2001;51:933–8.
Acknowledgements
We thank Bernard Agwanda, Michael Bartonjo, Beryl Makori, Ruth Makena, David Wechuli, the late Richard Yego, Aziza Zuhura and all of the National Museums of Kenya Mammalogy Section for help in obtaining specimens in the field. We are also grateful to Samuel Kasiki and Robert Mwasya, formerly of the Kenya Wildlife Service, for access to field sites and to specimens. We thank Steven M. Goodman and associates for access to specimens from Madagascar.
Funding
For financial support, we thank Western Kentucky University’s Biodiversity Center, Biology Department and Graduate School, and NSF-DEB grant 2127292 to CWD. We thank the Grainger Bioinformatics Center for partial funding of this study. Field collections were funded by several agencies in cooperation with the Field Museum. The JRS Biodiversity Foundation, Field Museum’s Council on Africa, Marshall Field III Fund and Barbara E. Brown Fund for Mammal Research were critical to fieldwork and analyses, as was the support of Bud and Onnolee Trapp and Walt and Ellen Newsom.
Author information
Authors and Affiliations
Contributions
TBV contributed to the study design, conducted laboratory work and systematic analyses and drafted the initial manuscript. CWD contributed to the study design, specimen collection, bat fly identification and manuscript writing. BDP and PWW contributed to the original Bats of Kenya Survey, specimen collection, bat identification and manuscript writing. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
All protocols for bat work were conducted under the approval of the Field Museum’s Institutional Animal Care and Use Committee (2012-003).
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Verrett, T.B., Webala, P.W., Patterson, B.D. et al. Remarkably low host specificity in the bat fly Penicillidia fulvida (Diptera: Nycteribiidae) as assessed by mitochondrial COI and nuclear 28S sequence data. Parasites Vectors 15, 392 (2022). https://doi.org/10.1186/s13071-022-05516-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13071-022-05516-z