
Abstract
Background
Colombia is ranked very high among countries with the highest numbers of endemic Leishmania species (n = 9) causing human disease. Although much effort has been devoted to generating simple and specific tools for Leishmania species identification, challenges remain in the discrimination of species belonging to the Leishmania (Viannia) guyanensis complex: L. (V.) guyanensis and L. (V.) panamensis.
Methods
A set of seven reference strains of species belonging to the L. (Leishmania) and L. (Viannia) subgenera, clinical strains from human cases of cutaneous leishmaniasis (CL; n = 26) and samples collected from sylvatic mammals and sand flies (n = 7) from endemic areas in Colombia were analyzed in this study. The heat-shock protein 70 gene (hsp70) was amplified by PCR from DNA extracted from logarithmic-phase promastigotes or tissue samples, and the PCR products were sequenced. Sequence alignment was performed against a set of previously published and curated sequences, and phylogenetic analysis based on the maximum-likelihood and Bayesian inference approaches was conducted. Haplotype diversity among strains and species of the L. (V.) guyanensis complex was explored using a median-joining network.
Results
Sequencing of the hsp70 gene for L. (Viannia) spp. ty** was comparable to species identification using isoenzyme electrophoresis or monoclonal antibodies. Complete species matching was found, except for one sylvatic sample with an identity yet unsolved. Among the L. (V.) panamensis clinical strains, two distinctive phylogenetic clusters were found to correlate with two different zymodemes: L. (V.) panamensis Z2.2 and Z2.3. Analysis of samples from sylvatic environments identified novel records of naturally infected wild mammal and sand fly species.
Conclusions
Our results support the adequacy of hsp70 gene sequencing as a single-locus approach for discrimination of L. (Viannia) spp., as well as for exploring the genetic diversity within the L. (V.) guyanensis complex.
Graphical Abstract

Similar content being viewed by others
Background
The leishmaniases are a group of diseases caused by parasites of the genus Leishmania, which are transmitted through the bite of infected sand flies to humans and domestic and wild animals [1]. This spectrum of diseases is a global public health problem, with > 1.3 million new cases reported each year across 98 countries and an estimated 350 million people at risk of infection [2]. The most usual transmission cycles involve a wide, yet unspecified, range of vectors, vertebrate hosts and parasite strains, which at the same time are modulated by an uncertain number of human and environmental-related factors [3]. The leishmaniases are endemic in many areas of Central and South America [4], where usually two or more Leishmania species occur in sympatry [5]. Spatial co-occurrences and phylogenetic proximity of clinically relevant species pose an important challenge for species identification in cases of human infection, especially as advances in molecular biology continue to reveal a taxonomic mismatch between genetic sequence similarity and species delimitation [6].
Species belonging to the Viannia subgenus are the primary etiological agents of American cutaneous leishmaniasis (CL) [7]. Colombia ranks among those countries with the highest numbers of endemic Leishmania species (n = 9) causing disease in humans. These species are widely distributed across the country [8], with a predominance of L. (Viannia) panamensis in the north and southwest (Andean, Caribbean and Pacific regions) [8,9,10], causing close to 65% of all CL cases in the country, followed by L. (Viannia) braziliensis in the southeast (Orinoquia and Amazon regions), responsible for 30% of annual cases. Infections with L. (Viannia) guyanensis are less frequent (approx. 1% annually), with the exception of critical outbreaks, such as the 2003–2004 outbreak in Chaparral, Tolima, which affected > 2000 people [11]. The eco-epidemiological dynamics of some of these species has changed over time from sylvatic to domestic cycles [8]; (ii) the lower susceptibility of L. (V.) braziliensis to miltefosine compared to L. (V.) panamensis or L. (V.) guyanensis [14]; and (iii) L. (V.) guyanensis infections being more likely to be completely resolved after antimonial treatment compared to infections caused by L. (V.) braziliensis and L. (V.) panamensis [15]. However, it is also true that phenotypic diversity within a species occurs, as demonstrated by the range of clinical and therapeutic outcomes associated with L. (V.) panamensis or L. (V.) braziliensis infections [16,17,18].
During the last three decades, multilocus enzyme electrophoresis (MLEE) has been used as the gold standard for the identification of Leishmania species [19]. This technique has proven reliable for defining interspecific boundaries in Leishmania, as well as intraspecific variations, such as those found in strains of the L.(V.) guyanensis and L. (V.) braziliensis complexes, by constraining specific populations based on their isoenzyme profile (zymodeme) [20, 21]. The L.(V.) guyanensis complex constitutes a monophyletic complex in which L. (V.) panamensis and L. (V.) guyanensis species assemble in two sub-clusters. However, phenetic and phylogenetic analyses performed on MLEE and random amplified polymorphic DNA (RAPD) data [20, 22] have not shown strict boundaries between these species, introducing a challenge for molecular-based ty**.
Assessment of Leishmania species diversity in Colombia has focused on human host isolates, as studies in naturally infected vectors and vertebrates from endemic or sylvatic areas are scarce [23]. This is primarily due to the difficulty in capturing and isolating parasites from infected vectors or mammalian reservoirs due to low infection rates [24]. Therefore, tools that allow Leishmania ty** directly from either clinical or biological (vector and reservoir) samples are urgently needed. The aim of the study reported here was to evaluate the performance of heat-shock protein 70 gene (hsp70) sequencing as an alternative for Leishmania species genoty** of strains isolated from cases of human CL in Colombia, and to provide proof-of-concept of its utility in species ty** from primary tissue samples from sylvatic mammals and sand flies collected in areas of Colombia with high endemicity of CL.
Methods
Leishmania reference strains
Seven Leishmania reference strains obtained from the CIDEIM BioBank were used to confirm the accuracy of the ty** protocol: Leishmania (V.) panamensis (MHOM/PA/71/LS94), Leishmania (V.) guyanensis (MHOM/BR/75/M4147), Leishmania (V.) braziliensis (MHOM/BR/75/M2903), Leishmania (L.) infantum (MHOM/BR/74/PP75), Leishmania (L.) amazonensis (MHOM/BR/73/M2269), Leishmania (L.) mexicana (MHOM/BZ/82/BEL21), Leishmania (L.) donovani (MHOM/IN/80/DD8) and Leishmania (V.) naiifi (MDAS/BR/79/M5533). Promastigotes were maintained at 25 °C in complete RPMI medium (supplemented with 10% heat-inactivated fetal bovine serum [Gibco™, Thermo Fisher Scientific, Waltham, MA, USA], 1% glutamine, 100 U/ml penicillin and 100 μg/mL streptomycin). Logarithmic-phase promastigotes were harvested by centrifugation, washed in phosphate-buffered saline and solubilized in lysis buffer for DNA extraction.
Selection of clinical strains
Clinical strains isolated from 26 patients with CL were obtained from the CIDEIM BioBank. All strains had been previously typed as L. (V.) panamensis or L. (V.) guyanensis with either monoclonal antibodies or by isoenzyme electrophoresis. Additional details for these strains, including collection sites and MLEE or antibody species ty**, are described in Table 1.
Sand flies and wild mammals
Leishmania DNA was obtained from samples previously collected and stored at the Research Center for Microbiology and Tropical Parasitology (CIMPAT) at the Universidad de los Andes (Uniandes). Five pools (n = 20 specimens per pool) of Lutzomyia gomezi from Córdoba department collected in 2016 [25], one pool of Psychodopygus panamensis from Buenaventura collected in 2019 and a tissue DNA sample of one sylvatic rodent, Oecomys sp., from San Joaquin, Cundinamarca collected in 2019 were included (see Table 2 for sampling sites). Leishmania detection in these samples was previously achieved by PCR analysis of kinetoplast DNA (kDNA) for the P. panamensis [26] pool and by analysis of the internal transcribed spacer 1 (ITS1) for the L. gomezi and Oecomys sp. samples [27].
DNA extraction, PCR amplification of hsp70 gene and amplicon purification
Genomic DNA was isolated with the DNeasy extraction kit (Qiagen, Hilden, Germany) according to the manufacturer's protocol. Next, 2.5 μl of extracted DNA was used in PCR reactions performed in a final reaction volume of 25 μl, following the ty** protocol and sequencing algorithm designed by [28], with the PCR parameters adapted for the used of Platinum® Taq DNA Polymerase High Fidelity (Life Technologies™, Thermo Fisher Scientific). We used the F25 and R1310 primers to amplify a 1286-bp fragment of the hsp70 gene (PCR-F). Samples for which no amplification product was detected were re-tested with two additional PCRs: (i) PCR-N (500-bp fragment), using primers F25 and R617; and (ii) PCR-T (700-bp fragment), using primers 6F and R1310. The amplified products were analyzed by electrophoresis in Sybr Safe-stained 2% agarose gels, the band size was confirmed and PCR products were purified using the QIAquick Gel Extraction Kit (Qiagen) following the manufacturer's protocol.
Sequence editing and alignment
Bidirectional sequencing of the amplification products was conducted by Macrogen Genomics Laboratories (Macrogen Inc., Seoul, South Korea) using 4F and 6R primers [28]. The resulting sequences were edited and aligned in the DNA STAR program (DNASTAR, Inc., Madison, WI, USA). We performed a search of Leishmania sequences in GenBank using the Basic Local Alignment Search Tool (BLAST) to locate additional related strains. A first alignment was made with the sequences generated in this study alongside 59 reference sequences (Additional file 1: Table S1) retrieved from NCBI (https://www.ncbi.nlm.nih.gov) [29,30,31,32,33,34,35]. All sequences were aligned and edited using Muscle implemented in MegAlign Pro within (DNASTAR, Inc.). For haplotype diversity analysis, a second alignment was constructed that included only sequences of L. (V.) panamensis.
Phylogenetic analysis
Phylogenies were inferred using maximum likelihood (ML) and Bayesian inference (BI). The ML tree was built using IQ-TREE [36]. The best-fitting model of molecular evolution for the ML tree was selected based on the Bayesian information criterion (BIC) using the ModelFinder command in IQ-TREE. To assess branch support, the IQ-TREE analyses used the ultrafast bootstrap approximation (UFboot) with 1000 replicates and the SH-like approximate likelihood ratio test (SH-aLRT) also with 1000 bootstrap replicates [37]. For the BI-inferred tree, we used the program PartitionFinder v2.1.1 [38] and Jmodeltest to select the most appropriate substitution model. Bayesian analyses were carried out using the program MrBayes v3.2 [39]. Two parallel sets of four simultaneous Monte Carlo Markov chains (3 hot and 1 cold) were run for 10,000,000 generations, and the trees were sampled for every 1000 generations. Temperature burn-in was set to 25% (burn-in frac = 0.25). To speed up convergence, we employed the ML tree as the starting value (‘starting tree’) for the tree parameter (tau) and the branch length parameter (V) with the MrBayes v3.2 commands: ‘startvals tau = mystarttree V = mystarttree.’ The maximum clade credibility tree (MCC) was displayed and edited in the online tool ITOL.
Test of monophyly
Bayes factor (BF) comparisons of constrained and unconstrained tree topologies were used to test for monophyly of the L. (V.) panamensis strains belonging to the zymodemes 2.2 and 2.3 based on the hsp70 sequencing. To do this, we compared an unconstrained hypothesis (no changes at all) with a constrained hypothesis in which all L. (V.) panamensis zymodemes were constrained to monophyly. We used MrBayes to calculate the harmonic mean estimator of marginal likelihood. BFs were calculated as the difference of harmonic mean estimators of the two models (constrained vs unconstrained) in log units; a log difference of 3–5 is considered to be strong evidence, and a difference > 5 is considered to be very strong evidence in favor of the better model [40]. BI of constrained topologies was run with the same settings as described above for unconstrained phylogenetic analysis.
Genetic diversity
To quantify diversity among co-occurring L. (V.) panamensis strains in each department, 31 sequences (25 from clinical isolates, 5 from sand flies and 1 from a rodent) plus L. (V.) panamensis strains from Miranda et al. [41] were analyzed to search for polymorphisms using the DnaSP 6.10.03 software package. Gaps/missing bases were not considered, and invariable sites were removed. Genetic diversity was described by estimating the number of segregating sites (S), nucleotide diversity (µ), haplotype number (Nh) and haplotype diversity (Hd). The neutrality test-based Tajima’s D was calculated based on segregation sites, using the same software. The haplotype network was constructed based on a median-joining model with 1000 iterations and default parameters using the PopArt software.
Results
Sequencing and sequence analysis
Amplification of the 1.2-kb PCR-F fragment was achieved for all seven reference strains and for 16 of the 26 clinical strains. For the remaining samples, two additional PCR reactions for amplification of the PCR-N and PCR-T fragments were performed, followed by sequence assembly. All amplification products derived from the wild samples were obtained by re-amplification, over the first product of PCR-N and PCR-T using the same primer set for each sample.
Species ty** and phylogenetic relationships
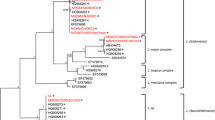
Using previously published and curated sequences (n = 59; Additional file 1: Table S1) in addition to the 41 sequences from this work, we assembled a final alignment of 100 Leishmania strains. The whole alignment is shown in Additional file 2: Figure S1, including the consensus sequence (at 50% conservation) and residue conservation that were calculated. Phylogenetic trees were generated using ML and BI (model of substitutions for the ML tree was HKY + F + I and for the BI tree, HKY + I). As expected, the phylogenetic analysis separated the Viannia from Leishmania clades (Fig. 1). Both trees corroborated the distinctive division between the Leishmania and Viannia subgenera, as well as the different complexes and species (Additional file 3: Figure S2). Based on hsp70 sequences, the identity of all of the clinical isolates analyzed (n = 26) was 100% congruent to the species previously defined by monoclonal antibodies or MLEE ty** (Table 1). This result was corroborated by the assignment of all seven sequences to the reference strains analyzed (Fig. 1).

Phylogeny of Leishmania spp. from partial nucleotide sequences of the hsp70 gene. The tree was constructed with IQ-TREE using maximum-likelihood bootstrap support (1000 replicates). The new sequences contributed by this study are indicated in bold. Branch support values are represented by a color gradient. Leishmania (Viannia) panamensis strains are marked with blue dots, L. (Viannia) guyanensis with green dots and L. (Viannia) braziliensis with red dots. Reference sequences are labeled with strain codes and abbreviated species names. Sylvatic strains are marked with an asterisk. Abbreviations: hsp70, Heat-shock protein 70,
Two different clusters of L. (V.) panamensis strains were observed in the phylogenetic trees. In one cluster, nine strains (codes 12581, 12615, 12578, 2183, 12563, 2198, 2168, 2173 and 2169) isolated from patients with CL from the South Pacific coast of Colombia typed as L.(V.) panamensis clustered with a group of reference sequences of Leishmania sp. from Panama [41]. Five of those nine strains were also typed as L.(V.) panamensis zymodeme 2.3 using MLEE (Fig. 1). In the other cluster, 11 L.(V.) panamensis clinical strains (codes 2363, 5967, 2277, 7123, 2420, 2348, 2330, 12567, 2350, 12985 and 12637) grouped with the reference sequences of L. (V.) panamensis, including the strain LS94, in the phylogenetic tree. Of these 11 strains, eight had been previously typed by MLEE as L.(V.) panamensis zymodeme 2.2. Leishmania (V.) guyanensis clinical and reference strains were grouped with lower support values than L. (V.) panamensis and did not display intraspecific separations.
Hsp70 sequences obtained from vertebrate tissues and insect pools fitted as well as the clinical strains within the whole sequence alignment, with the same resolution at a specific taxon level (Fig. 1). The five sequences of L. (V.) panamensis obtained from Lutzomyia gomezi from Córdoba Department clustered with L.(V.) panamensis zymodeme 2.2 clinical strains. The Leishmania hsp70 sequence obtained from Oecomys sp. was also found in this cluster and is congruent with the geographical distribution of L. (V.) panamensis in Colombia. Lastly, the hsp70 sequence from Psychodopygus panamensis (L500) did not group with any specific cluster but was closely related to strains of the L. guyanensis complex. This sequence also remained undetermined typed by ITS1 and Mini-exon techniques (data not shown).
Monophily of L. (V.) panamensis strains
Regarding the phylogenetic topology among L. (V.) panamensis strains, a comparison of marginal likelihood estimates from topologically constrained and unconstrained trees rejected the monophyly of the group based on hsp70 sequencing. The likelihood value of the constrained tree topology (− 2820.74) was smaller than that of the unconstrained tree topology (− 2811.30). The value of the test statics (BF) was 18.88, indicating very strong evidence in favor of the better model. Therefore, the BF test indicates that the constrained tree topology is not supported, thus bringing into question the monophyly of L. (V.) panamensis based on this marker. Detailed information on the obtained likelihood is given in Additional file 1: Table S2.
Median-joining network
Seven segregating sites and five parsimony-informative sites were found among the sequences of L. (V.) panamensis, and five haplotypes (H) were defined (Fig. 2). To compare the diversity detected among the strain panel using this genetic region with other published data, we also tested the nucleotide diversity (µ = 0.0019) and Tajima’s D values (0.8671, p[D > 0.8671] = 0.4020). Twelve of the L. (V.) panamensis sequences from clinical samples, four of the sequences from vectors and the sequence from the reservoir were grouped in haplotype H2. All of the hsp70 sequences obtained from L. (V.) panamensis zymodeme 2.2 strains clustered in the H2 haplotype. Haplotypes H1, H3 and H4 corresponded to L. (V.) panamensis sequences generated from L. (V.) panamensis zymodeme 2.3 strains in addition to other clinical strains obtained from patients with CL on the Pacific coast of Colombia (Fig. 3).

Haplotype of L. (V.) panamensis network based on hsp70 sequences from this study and reference strains from Panama. Each observed haplotype is indicated by a circle, sized according to its frequency and by number of colored slices according to the localities associated to these. Haplotype relationships are indicated by lines; mutational steps between haplotypes are represented by the number of lines

Map of Leishmania guyanensis complex presenting the distribution and diversity of the hsp70 haplotype. The number in each circle corresponds to the total samples analyzed for each location. The haplotypes are color coded distinctively
Discussion
Co-circulation of different Leishmania species has been reported in Colombia, with L. (V.) panamensis and L. (V.) braziliensis species accounting for most of the human CL cases [42, 43]. We evaluated the performance of hsp70 gene sequencing for Leishmania Viannia genoty** in a panel of clinical strains of L. (V.) panamensis and L. (V.) guyanensis, and for species identification directly from tissue and DNA samples obtained from sand flies and a wild mammal captured in endemic areas of CL transmission in Colombia.
Previous studies in Neotropical countries, such as Ecuador, French Guiana, Brazil, Peru and Panama, have reported the usefulness of hsp70 sequence analysis for Leishmania species identification [41, 37,38,39,47]. Our results expand these findings, lending support to hsp70 sequencing for discriminating closely related species within the L. (V.) guyanensis complex. Our study provides the research community with an additional number of strains and species from Colombia, thereby enhancing the discriminatory power of sequence-based Leishmania species ty**, which supports the feasibility of implementing this tool in a wide and heterogeneous geographical range. This characteristic is of great importance in beginning to define a unified method for regional species identification, one of the priorities of local and regional public health programs [48, 49]. By using this approach, we achieved species identification from clinical isolates, mammalian hosts and vectors, highlighting the potential of hsp70 sequencing in multidisciplinary studies with an ecological and epidemiological scope [49].
In our study, L. (V.) panamensis was over-represented, including clinical strains and DNA samples from a sylvatic mammal and sand flies. Two discrete clusters of L. (V.) panamensis were identified based on the tree topology, BF and haplotype data: one cluster is represented by L. (V.) panamensis strains of zymodeme 2.2 and the other cluster is represented by zymodeme 2.3. MLEE analysis brought into question the distinction between L. (V.) panamensis and L. (V.) guyanensis since data did not indicate distinct monophyletic lines [50]. In addition, the zymodeme 2.3 strains sequenced in this work clustered with high support values together with strains sequenced in Panama by Miranda et al. [41] and identified by the authors as Leishmania sp.1. Therefore, a close, if not identical, taxonomic identity is suggested. Interestingly, the authors of previous studies of Leishmania (Viannia) species circulating in the South Pacific coast region of Colombia concluded that the genetic similarity between the L. (V.) guyanensis and L. (V.) panamensis zymodeme 2.2 was greater than that between the L. (V.) panamensis zymodemes 2.2 and 2.3 [51]. Previous findings [52] demonstrated the distinction of three well-supported clades in a tested panel of L. (V.) panamensis from Colombia and Panama, and a possible geographic distinction between these intraspecific clades. These findings provide evidence of the need for the revision of the taxonomic status of the L. (V.) guyanensis complex.
Among the diverse L. (V.) panamensis strains and parasite populations circulating in Colombia, L. (V.) panamensis zymodeme 2.3 has been associated with higher in vitro profiles of drug tolerance/resistance to pentavalent antimony [53]. HSPs have been previously implicated in antimony resistance in clinical L. donovani strains [54]. Moreover, parasite exposure to antimony results in differential expression of hsp70 [55], suggesting that environmental (or clinical) exposure to metals may exert a selection pressure over HSPs, which could result in population genetic diversity and phenotypic associations. Whether any association exists between the drug-susceptible phenotype and the hsp70 haplotype remains to be determined and corroborated in a larger number of samples.
The implementation of species ty** tools based on DNA or complementary DNA (cDNA) targets (such as 18S ribosomal RNA [rRNA] or 7SL RNA) is further supported by its potential usefulness in cases where parasites are difficult to isolate, such as those coming from infected sand flies or asymptomatic mammalian reservoirs. Here, we provide proof-of-concept of the applicability of hsp70 sequencing for Leishmania ty** directly from DNA samples collected from naturally infected sand flies and mammals. Of the seven sylvatic samples analyzed, six were classified as L. (V.) panamensis, which is in line with the predominant species circulating in the endemic areas where these samples were collected (the Caribbean and Andean regions). Here w report for the first time a natural infection of Oecomys sp. with L. (V.) panamensis in Colombia, expanding the number of known natural interactions [3]. Ocampo et al. [56] registered the natural infection of Oecomys trinitatus with L. (Viannia) spp. in a transmission area of L. (V.) guyanensis in Colombia; however, these authors did not confirm the parasite identity at the species level. The role of sylvatic mammals in the transmission cycles is key since it is known that transmission rates in certain areas are related to the densities of mammals acting as reservoirs [3], which for L. (V.) panamensis has been traditionally linked to the presence of sloths and spiny rats in the transmission areas [57, 58].
One sequence obtained from a sand fly pool of Psy. panamensis was ‘undetermined’ since it did not group closely with any other strains but appeared as an outgroup of the panamensis/guyanensis complex. This is quite interesting, especially as clinical samples collected in nearby areas were typed as L. (V.) panamensis strains. It is important to note here the possible existence of a sampling bias which leads to the parasites that successfully spread in vertebrate hosts with clinical signs, such as dogs, being more frequently captured, which would have the potential consequence of missing novel sylvatic strains [59].
The haplotype and nucleotide diversity found in this study are similar to those previously reported by Patiño et al. [50] and Van der Auwera et al. [60], indicating the existence of intraspecific variability using the hsp70 locus. Implementing similar species ty** methodologies in future work and on broader geographic scales can increase our ability to offer reliable and comparable information to define the current status of Leishmania diversity in Colombia and neighboring countries.
CL outbreaks in Colombia have a dissimilar spatial structure [12], and the strains included in this work represent some of these spatial clusters located in endemic transmission areas. We report here a panel of hsp70 sequences from Colombian strains belonging to five different geographical areas with distinct environmental conditions; five haplotypes represented among them provide confirmatory evidence of genetically different strains co-occurring in close geographical clusters.
Availability of data and materials
The nuclear DNA sequences of Leishmania spp. obtained in this study were deposited in GenBank database (Table 1).
Abbreviations
- BF:
-
Bayes factor
- BI:
-
Bayesian inference
- CL:
-
Cutaneous leishmaniasis
- Hsp70:
-
Heat-shock protein 70
- ITS1:
-
Internal transcribed spacer 1
- kDNA:
-
Kinetoplast DNA
- ML:
-
Maximum likelihood
- MLEE:
-
Multilocus enzyme electrophoresis
- RAPD:
-
Random amplified polymorphic DNA
References
Bates PA. Revising Leishmania’s life cycle. Nat Microbiol. 2018;3:529–30.
Burza S, Croft SL, Boelaert M. Leishmaniasis. Lancet. 2018;392:951–70.
Roque AL, Jansen AM. Wild and synanthropic reservoirs of Leishmania species in the Americas. Int J Parasitol Parasites Wildl. 2014;3:251–62.
Kevric I, Cappel MA, Keeling JH. New World and Old World Leishmania infections: a practical review. Dermatol Clin. 2015;33:579–93.
Hashiguchi Y, Velez LN, Villegas NV, Mimori T, Gomez EAL, Kato H. Leishmaniases in Ecuador: a comprehensive review and current status. Acta Trop. 2016;166:299–315.
Kaufer A, Ellis J, Stark D, Barratt J. The evolution of trypanosomatid taxonomy. Parasit Vectors. 2017;10:287.
Dutari LC, Loaiza JR. American cutaneous leishmaniasis in panama: a historical review of entomological studies on anthropophilic Lutzomyia sand fly species. Parasit Vectors. 2014;7:218.
Ramírez JD, Hernández C, León CM, Ayala MS, Flórez C, González C. Taxonomy, diversity, temporal and geographical distribution of cutaneous leishmaniasis in Colombia: a retrospective study. Sci Rep. 2016;6:28266.
Ovalle-Bracho C, Londoño-Barbosa D, Salgado-Almario J, González C. Evaluating the spatial distribution of Leishmania parasites in Colombia from clinical samples and human isolates (1999 to 2016). PLoS ONE. 2019;3:e0214124.
Herrera G, Teheran A, Pradilla I, Vera M, Ramirez JD. Geospatial-temporal distribution of Tegumentary Leishmaniasis in Colombia (2007–2016). PLOS Negl Trop Dis. 2018;12:e0006419.
Rodríguez-Barraquer I, Góngora R, Prager M, Pacheco R, Montero LM, Navas A, et al. Etiologic agent of an epidemic of Cutaneous Leishmaniasis in tolima, Colombia, the American journal of tropical medicine and hygiene. Am J Trop Med Hyg. 2008;78:276–82.
Hernández AM, Gutierrez JD, **ao Y, Branscum AJ, Cuadros DF. Spatial epidemiology of Cutaneous Leishmaniasis in Colombia: socioeconomic and demographic factors associated with a growing epidemic. Trans R Soc Trop Med Hyg. 2019;113:560–8.
Ferro C, López M, Fuya P, Lugo L, Cordovez JM, González C. Spatial distribution of sand fly vectors and eco-epidemiology of Cutaneous Leishmaniasis transmission in Colombia. PLoS ONE. 2015;10:e0139391.
Rugani JN, Quaresma PF, Gontijo CF, Soares RP, Monte-Neto RL. Intraspecies susceptibility of Leishmania (Viannia) braziliensis to antileishmanial drugs: antimony resistance in human isolates from atypical lesions. Biomed Pharmacother. 2018;108:1170–80.
Arevalo J, Ramirez L, Adaui V, Zimic M, Tulliano G, Miranda-Verástegui C, et al. Influence of Leishmania (Viannia) species on the response to antimonial treatment in patients with American tegumentary leishmaniasis. J Infect Dis. 2007;195:1846–51.
Weigle KA, Santrich C, Martinez F, Valderrama L, Saravia NG. Epidemiology of cutaneous leishmaniasis in Colombia: a longitudinal study of the natural history, prevalence, and incidence of infection and clinical manifestations. J Infect Dis. 1993;168:699–708.
Soto J, Toledo J, Gutierrez P, Nicholls RS, Padilla J, Engel J, et al. Treatment of American cutaneous leishmaniasis with miltefosine, an oral agent clinic. Infect Dis. 2001;33:5761.
Rojas R, Valderrama L, Valderrama M, Varona MX, Ouellette M, Saravia N. Resistance to antimony and treatment failure in human Leishmania (Viannia) infection. J Infect Dis. 2006;193:1375–83.
Schönian G, Kuhls K, Mauricio IL. Molecular approaches for a better understanding of the epidemiology and population genetics of Leishmania. Parasitol. 2011;138:405–25.
Bañuls AL, Jonquieres R, Guerrini F, Le Pont F, Barrera C, Espinel I, et al. Genetic analysis of Leishmania parasites in Ecuador: are Leishmania (Viannia) panamensis and Leishmania ((V)) Guyanensis distinct taxa. Am J Trop Med Hyg. 1999;61:838–45.
Bañuls AL, Dujardin JC, Guerrini F, De Doncker S, Jacquet D, Arevalo J, et al. Is Leishmania (Viannia) peruviana a distinct species? A MLEE/RAPD evolutionary genetics answer. J Eukaryot Microbiol. 2000;47:197–207.
Banũls AL, Hide M, Tibayrenc M. Molecular epidemiology and evolutionary genetics of Leishmania parasites. Int J Parasitol. 1999;29:1137–47.
Castillo RL, Ovalle BC, Díaz JD, Sánchez VG, Muvdi AS, Castañeda OC. Cost-effectiveness analysis of Mucosal Leishmaniasis diagnosis with PCR-based vs parasitological tests in Colombia. PLoS ONE. 2019;14:e0224351.
Akhoundi M, Downing T, Votýpka J, Kuhls K, Lukeš J, Cannet A, et al. Leishmania infections: molecular targets and diagnosis. Mol Aspects Med. 2017;57:1–29.
González C, León C, Paz A, López M, Molina G, Toro D, et al. Diversity patterns, Leishmania DNA detection, and bloodmeal identification of Phlebotominae sand flies in villages in northern Colombia. PLoS ONE. 2018;13:e0190686.
Rosales-Chilama M, Gongora RE, Valderrama L, Jojoa J, Alexander N, Rubiano LC, et al. Parasitological confirmation and analysis of Leishmania diversity in asymptomatic and subclinical infection following resolution of Cutaneous Leishmaniasis. PLoS Negl Trop Dis. 2015;12:e0004273.
Cupolillo E, Grimaldi JG, Momen H, Beverley SM. Intergenic region ty** (IRT): a rapid molecular approach to the characterization and evolution of Leishmania. Mol Biochem Parasitol. 1995;73:145–55.
Van der Auwera G, Maes I, De Doncker S, Ravel C, Cnops L, Van Esbroeck M, et al. Heat-shock protein 70 gene sequencing for Leishmania species ty** in European tropical infectious disease clinics Eur. Surveill. 2014;18:30.
Fraga J, Montalvo AM, De Doncker S, Dujardin JC, Van der Auwera G. Phylogeny of leishmania species based on the heat-shock protein 70 gene. Infect Genet Evol. 2010;10:238–45.
Bock JH, Langer PJ. Sequence and genomic organization of the HSP70 genes of Leishmania amazonensis. Mol Biochem Parasitol. 1993;62:187–97.
Espada CR, Ortiz PA, Shaw JJ, Barral AMP, Costa JML, Uliana SRB, et al. Identification of Leishmania (Viannia) species and clinical isolates of Leishmania (Leishmania) amazonensis from Brazil using PCR-RFLP of the heat-shock protein 70 gene reveals some unexpected observations. Diagn Microbiol Infect Dis. 2018;91:312–8.
Ramírez CA, Requena JM, Puerta CJ. Identification of the HSP70-II gene in Leishmania braziliensis hsp70 locus: genomic organization and UTRs characterization. Parasit Vectors. 2011;4:1.
Fernández-Arévalo A, El Baidouri F, Ravel C, Ballart C, Abras A, Lachaud L, et al. The Leishmania donovani species complex: a new insight into taxonomy. Int J Parasitol. 2020;50:1079–88.
Van der Auwera G, Davidsson L, Buffet P, Ruf MT, Gramiccia M, Varani S, et al. Surveillance of leishmaniasis cases from 15 European centres, 2014 to 2019: a retrospective analysis. Euro Surveill. 2022;27:2002028.
Montalvo Alvarez AM, Nodarse JF, Goodridge IM, Fidalgo LM, Marin M, Van Der Auwera G, et al. Differentiation of Leishmania (Viannia) panamensis and Leishmania (V.) guyanensis using BccI for hsp70 PCR-RFLP. Trans R Soc Trop Med Hyg. 2010;104:364–7.
Nguyen LT, Schmidt HA, von Haeseler A, Minh BQ. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol Biol Evol. 2015;32:268–74.
Hoang DT, Chernomor O, von Haeseler A, Minh BQ, Vinh LS. UFBoot2: improving the ultrafast bootstrap approximation. Mol Biol Evol. 2017;35:518–22.
Lanfear R, Frandsen PB, Wright AM, Senfeld T, Calcott B. PartitionFinder 2: new methods for selecting partitioned models of evolution for molecular and morphological phylogenetic analyses. Mol Biol Evol. 2017;34:772–3.
Ronquist F, Teslenko M, van der Mark P, Ayres DL, Darling A, Höhna S, et al. MrBayes 32: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst Biol. 2012;61:539–42.
Kass RE, Raftery AE. Bayes factors. J Am Stat Assoc. 1995;90:773–95.
Miranda ADC, González KA, Samudio F, Pineda VJ, Calzada JE, Capitan-Barrios Z, et al. Molecular identification of parasites causing Cutaneous Leishmaniasis in Panama. Am J Trop Med Hyg. 2021;104:1326–34.
Correa CA, Perez J, Patiño LH, Ramírez JD, Duque MC. Distribution, treatment outcome and genetic diversity of Leishmania species in military personnel from Colombia with cutaneous leishmaniasis. BMC Infect Dis. 2020;20:938.
Montalvo AM, Fraga J, Montano I, Monzote L, Van der Auwera G, Marin M, et al. Identificación molecular de aislamientos clínicos de Leishmania spp. procedentes de Colombia con base en el gen hsp70. Biomédica. 2016;36:3744.
Kato H, Bone AE, Mimori T, Hashiguchi K, Shiguango GF, Gonzales SV, et al. First human cases of Leishmania (Viannia) lainsoni infection and a search for the vector sand flies in Ecuador. PLoS Negl Trop Dis. 2016;10:5.
Ducharme O, Simon S, Ginouves M, Prévot G, Couppie P, Demar M, et al. Leishmania naiffi and lainsoni in French Guiana: clinical features and phylogenetic variability. PLoS Negl Trop Dis. 2020;14:8.
da Silva LA, de Sousa C, da Graça GC, Porrozzi R, Cupolillo E. Sequence analysis and PCR-RFLP profiling of the hsp70 gene as a valuable tool for identifying Leishmania species associated with human leishmaniasis in Brazil. Infect Genet Evol. 2010;10:77–83.
Odiwuor S, Veland N, Maes I, Arévalo J, Dujardin JC, Van der Auwera G. Evolution of the Leishmania braziliensis species complex from amplified fragment length polymorphisms, and clinical implications. Infect Genet Evol. 2012;12:1994–2002.
Maurício IL. Leishmania taxonomy. In: Bruschi F, Gradoni L, editors. The leishmaniases: old neglected tropical diseases. Cham: Springer; 2018.
Courtenay O, Peters NC, Rogers ME, Bern C. Combining epidemiology with basic biology of sand flies, parasites, and hosts to inform leishmaniasis transmission dynamics and control. PLoS Pathog. 2017;13:10.
Patino LH, Mendez C, Rodriguez O, Romero Y, Velandia D, Alvarado M, et al. Spatial distribution, Leishmania species and clinical traits of Cutaneous Leishmaniasis cases in the Colombian army. PLOS Negl Trop Dis. 2017;11:8.
Saravia NG, Segura I, Holguin AF, Santrich C, Valderrama L, Ocampo C. Epidemiologic, genetic, and clinical associations among phenotypically distinct populations of Leishmania (Viannia) in Colombia. Am J Trop Med Hyg. 1998;59:86–94.
Patino LH, Muñoz M, Muskus C, Méndez C, Ramírez JD. Intraspecific genomic divergence and minor structural variations in Leishmania (Viannia) panamensis. Genes. 2020;11:252.
Fernández OL, Diaz Y, Ovalle C, Valderrama L, Muvdi S, Rodríguez I, et al. Miltefosine and antimonial drug susceptibility of Leishmania Viannia species and populations in regions of high transmission in Colombia. PLoS Negl Trop Dis. 2014;8:e2871.
Maharjan M, Madhubala R. Heat shock protein 70 (hsp70) expression in antimony susceptible/resistant clinical isolates of Leishmania donovani. Nepal J Biotech. 2015;3:22–8.
Vacchina P, Norris-Mullins B, Carlson ES, Morales MA. A mitochondrial hsp70 (HSPA9B) is linked to miltefosine resistance and stress response in Leishmania donovani. Parasit Vectors. 2016;9:621.
Ocampo CB, Ferro MC, Cadena H, Gongora R, Pérez M, Valderrama-Ardila CH, et al. Environmental factors associated with American cutaneous leishmaniasis in a new Andean focus in Colombia. Trop Med Int Health. 2012;10:1309–17.
González K, Calzada JE, Saldaña A, Rigg CA, Alvarado G, Rodríguez B, et al. Survey of wild mammal hosts of cutaneous leishmaniasis parasites in Panama and Costa Rica. Trop Med Health. 2015;43:75–8.
Travi BL, Arteaga LT, León AP, Adler GH. Susceptibility of spiny rats (Proechimys semispinosus) to Leishmania (Viannia) panamensis and Leishmania (Leishmania) chagasi. Mem Inst Oswaldo Cruz. 2002;97:887–92.
d’Avila CM, Boucinha C, Kostygov A, Santos HL, Morelli KA, Grybchuk A, et al. Exploring the environmental diversity of kinetoplastid flagellates in the high-throughput DNA sequencing era. Mem Inst Oswaldo Cruz. 2015;110:956–65.
Van der Auwera G, Bart A, Chicharro C, Cortes S, Davidsson L, Di Muccio T, et al. Comparison of Leishmania ty** results obtained from 16 European clinical laboratories in 2014. Euro Surveill. 2016;21:304–18.
Acknowledgements
We wish to acknowledge Monica Oviedo and Isabel Guasaquillo in CIDEIM Biobank for their work in propagating and phenoty** of all reference and clinical strains processed in this study. We also wish to thank Universidad del Sinú for the sample collection in Córdoba.
Funding
This research was funded in part by the NIAID/NIH U19AI129910 and Wellcome Trust 107595/Z/15/Z grants, and Universidad de Los Andes, Faculty of Sciences INV-2019-87-1793. Sylvatic sample collection was funded by Colciencias project number 63302 (807-2018), the General System of Royalties Contract Number 754-2013 and the Government of Córdoba. JH was awarded a full scholarship as a Master’s student from the Department of Biological Sciences, Faculty of Sciences, Universidad de Los Andes. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health or other agencies.
Author information
Authors and Affiliations
Contributions
MAG, JH, MRC and CG conceptualized the study. JH, MRC and CL performed the laboratory work. JH, CL and CG collected the sylvatic samples. JH analyzed the data and built the visualizations. JH, MAG and CG drafted the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
The use of the clinical strains analyzed in this study was approved and monitored by the Institutional Review Board for Ethical Conduct of Research Involving Human Subjects of the Centro Internacional de Entrenamiento e Investigaciones Médicas (CIDEIM) under project code CIEIH 1291, following national and international guidelines for the conduct of clinical studies. Vector and reservoir sampling was approved by CICUAL and the ethics committee at Uniandes. All specimens were collected and processed as a part of the collection permit provided by the Colombian Ministry of Environment to Uniaversidad de los Andes.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Table S1.
Reference sequencesof Leishmania spp. obtained from GenBank database for phylogeneticanalysis. Table S2. Summary of Bayesfactor comparisons for phylogenetic hypotheses tested: monophyly (constrained)and non-monophyly (unconstrained) of the strains typed as L. (V.)panamensis. All the strains previously typed as L. (V.)panamensis by monoclonal antibodies, MLEE and ITS1 sequencing were included.
Additional file 2: Figure S1.
Phylogenetic tree andmultiple-sequence alignment analysis for the species ty** of Leishmania spp. Thealignment was generated using Geneious Prime. The numbers at the branches are confidencevalues (percentage) calculated based on the bootstrap method. Consensussequence (at 50% conservation) and residue conservation were calculated oniTOL.
Additional file 3: Figure S2
. HSP70gene majority-rule consensus tree inferred from Bayesian inference by usingMrBayes v.3.2. Posterior probability values from the Bayesian analysis fornodes are indicated below or above branches. Sequences belonging to subgenus Leishmaniaand Viannia are shown in green and blue color, respectively.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

{kind=link}
{kind=link}
Cite this article
Hoyos, J., Rosales-Chilama, M., León, C. et al. Sequencing of hsp70 for discernment of species from the Leishmania (Viannia) guyanensis complex from endemic areas in Colombia. Parasites Vectors 15, 406 (2022). https://doi.org/10.1186/s13071-022-05438-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13071-022-05438-w