
Abstract
Background
The genus Rickettsia contains the lineages spotted fever group (SFG), typhus group (TG), and transitional group (TRG). The spotted fever group Rickettsia (SFGR) is transmitted by ticks. The tick species Dermacentor nuttalli is considered the main vector carrying SFGR in Inner Mongolia. Studying the genetic diversity and population structure of Rickettsia is essential for develo** effective control strategies and predicting evolutionary trends of Rickettsia.
Methods
In 2019 we collected 408 D. nuttalli in the Inner Mongolia Autonomous Region, detected the percentage of Rickettsia-positive specimens, and characterized the haplotypes. From the Rickettsia-positive ticks, the gltA and ompA genes were extracted, amplified, and sequenced.
Results
Ten haplotypes of the gltA gene and 22 haplotypes of the ompA gene were obtained. The phylogenetic analysis showed that the haplotypes G1–G7 and G9 of the gltA gene cluster with Rickettsia raoultii, while G8 and G10 cluster with Rickettsia sibirica. Haplotypes O1–O15, O18 and O20–O22 of the ompA gene cluster with R. raoultii, while O16 and O19 cluster with R. sibirica. The average haplotype diversity was 0.3 for gltA and 0.7 for ompA. The average nucleotide diversity was greater than 0.05. Neutrality tests were nonsignificant for Tajima’s D results and Fu’s Fs results. The fixation index values (FST) showed that the degree of genetic differentiation between most sampled populations was small (FST < 0.05), whereas some populations showed a medium (FST > 0.05) or large (FST > 0.15) degree of differentiation. Analysis of molecular variance (AMOVA) revealed that the variation within populations was greater than that between populations. The mismatch analysis of Rickettsia showed double peaks.
Conclusions
We found two Rickettsia spp. (R. raoultii and R. sibirica). The high genetic disparity of Rickettsia allows for easy adaption to different environments. Genetic differentiation between populations is small, and Rickettsia populations do not show a geographically differentiated structure. The high rates of retention and infection of Rickettsia in D. nuttalli together with the animal husbandry exchange in Inner Mongolia gradually led to the harmonization of genetic characteristics of Rickettsia across various regions. Overall, the significant genetic diversity and geographical structure of Rickettsia in D. nuttalli are critical for SFGR control.
Graphical Abstract

Similar content being viewed by others

Background
Rickettsiosis is an important zoonosis, which brings serious harm to the health of humans and animals [1, 2]. The species of Rickettsia belong to three monophyletic groups: spotted fever group (SFG), typhus group (TG), and transitional group (TRG). SFG is transmitted by ticks [3, 4], while TG is transmitted by lice and fleas [5]. The vast territory and diverse habitats of Inner Mongolia greatly benefit the survival of ticks. Dermacentor nuttalli is the dominant tick species and is probably the main vector carrying spotted fever group Rickettsia (SFGR) in Inner Mongolia [6,7,8]. SFGR infections occur around the world and may cause serious diseases in humans. In China, Rickettsia heilongjiangensis, Rickettsia raoultii, Rickettsia slovaca, Rickettsia sibirica, Rickettsia mongolotimonae, Rickettsia monacensis, and Candidatus Rickettsia hebeiii and Candidatus Rickettsia **gxinensis have been detected in ticks. Furthermore, Rickettsia raoultii, Rickettsia heilongjiangensis, and R. sibirica have been reported in emerging tick-borne diseases of humans. In recent years, the threat that SFGR poses to public health in China has been magnified by the increasing number of potentially novel SFGR detected in ticks [9,10,11].
Molecular biological techniques allow for the identification of Rickettsia species. 16S rRNA can accurately identify a specimen as belonging to Rickettsia. However, it is difficult to distinguish the species, because the 16S rRNA sequence is highly conserved in almost all prokaryotes. The gltA and ompA genes are used for species identification in Rickettsia [12,13,14]. The gltA gene encodes a citrate synthase, the sequence of which allows for highly reliable identification of the evolutionary distances among Rickettsia. The ompA gene is an outer membrane protein gene. It is considered the “gold standard” for species identification in Rickettsia, owing to the highly specific 5′ end [15].
Previous studies found high genetic disparity in D. nuttalli, allowing for its existence in different geographical environments [16]. However, data on the genetic diversity of Rickettsia in D. nuttalli are scarce. Therefore, in our study, we included a high number of tick specimens, and explored the percentage of Rickettsia-positive samples and the genotype distribution to identify the genetic diversity of Rickettsia. Studying the genetic diversity of Rickettsia is essential for develo** effective control strategies and predicting pathogen evolutionary trends.
Methods
Sample collection
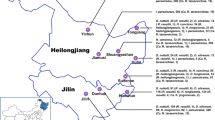
In this study, a total of 408 ticks were collected from 1078 sheep at four sampling spots in Inner Mongolia, China: Chengchuan Town, early Banner of Etoke Banner Ordos (EEDS); Siziwang Banner, Hohhot (SZWQ); the Bayan WenduSumu area Arukorqin Banner Chifeng (CF); and **nbarhu right Banner, Hulun Buir (HLBE). The locations of the sampling areas are shown in Additional file 1: Table S1 and Fig. 1.

Collection site map. Samples of D. nuttalli were collected in four regions of Inner Mongolia. Colors indicate different collection regions in Inner Mongolia, and each graphic represents the approximate geographical coordinates of each collection site
DNA extraction, amplification, and sequencing
Ticks were identified as D. nuttalli through morphological characteristics [17]. All D. nuttalli samples were individually extracted using a TIANamp Tissue and Blood Kit (TIANGEN, Bei**g, China) [18]. For amplification of Rickettsia DNA, the gltA and ompA genes were amplified by polymerase chain reaction (PCR), following the protocols described by Bermúdez et al. [19]. Specific primers targeting the gltA and ompA genes of Rickettsia from D. nuttalli were synthesized by Shanghai Sangon. DNA was amplified using a system of 40 μl, each including Taq PCR Master Mix (Sangon, Shanghai, China), 2 μl of DNA from each sample, and 1 μl of each reverse and forward primer, and filled to volume with double-distilled water. The PCR primers, amplification sizes (base pairs), and annealing temperatures are listed in Additional file 1: Table S2. Double-distilled water was used as the negative control in each PCR reaction. Prior to sequencing, the quality of the PCR products was checked with 1.5% agarose gel electrophoresis stained with GoldView (Sangon, Shanghai, China). If the quality of the PCR product was suboptimal, it was purified using the Gel DNA Recovery Kit (TIANGEN, Bei**g, China) and cloned using the pGEM-T Easy Vector System (Promega, Madison, WI, USA).
Data analysis
Sequences were edited in SeqMan 7.1 and identified by comparative analysis with sequences deposited in GenBank, using the National Center for Biotechnology Information (NCBI) BLAST search engine. Sequencing data are available at the NCBI Sequence Read Archive (https://submit.ncbi.nlm.nih.gov/about/bankit/), with accession numbers OK638141-OK638150, OL304270-OL304271, OL348251-OL348270. Multiple sequence alignment and sequence similarity calculations were done using DNAMAN 7.0. Phylogenetic trees were constructed with the neighbor-joining algorithm using MEGA 7 with 1000 bootstrap replicates to assess tree stability [20,21,22]. Sequences were analyzed in DNAsp 5.10 and Arlequin 3.5 for calculating polymorphic sites, nucleotide differences, the number of haplotypes, both haplotype and nucleotide diversity, the distribution pattern of DNA haplotype variation, and genetic variation parameters [23, 24]. The extent of genetic differentiation among and between Rickettsia populations was estimated by analysis of molecular variance (AMOVA) and FST values [25]. To determine whether genetic differentiation and population structures of Rickettsia varied among the four sampling localities in Inner Mongolia. Neutral tests were analyzed using Tajima’s D and Fu’s Fs tests using Arlequin 3.5 and DNAsp 5.10. PopART (Population Analysis with Reticulate Trees) version 1.7 software was used to evaluate the relationships between haplotypes by constructing TCS haplotype network maps.
Results
Detection of Rickettsia DNA
We collected and tested a total of 408 D. nuttalli in 2019 from four sites in Inner Mongolia: CF (n = 219), EEDS (n = 85), SZWQ (n = 30), and HLBE (n = 74) (Table 1). The quantity of amplification products of the gltA and ompA genes was regarded as the percentage of samples positive for Rickettsia in D. nuttalli [26]. Across the four regions, the average percentage of positive Rickettsia samples was 50.7%, with the highest value found in the HLBE region (85.1%).
Rickettsia identification
We detected 10 haplotypes of gltA sequences and 22 haplotypes of ompA sequences. The sequences had the highest similarity with R. raoultii and R. sibirica, as registered in GenBank, with 98% and 99% identity, respectively. In the gltA phylogenetic trees, haplotypes G1–G7 and G9 were clustered with R. raoultii, while G8 and G10 were clustered with R. sibirica. Furthermore, we found high similarity with Candidatus Rickettsia uralica, with 98% identity, and distinctly lower similarity in Rickettsia asembonensis. In the ompA phylogenetic trees, haplotypes O1–O15, O18, and O20–O22 clustered with R. raoultii, while O16 and O19 clustered with R. sibirica. The highest similarity was found with Candidatus R. uralica, with 98% identity, and substantially lower similarity with Rickettsia montana. The phylogenetic trees for both genes showed that the haplotypes of Rickettsia clustered into one branch with the ingroup, which contained the genotypes of R. raoultii and R. sibirica (Fig. 2a, b).

a Phylogenetic tree based on tbe gltA gene of R. raoultii. b Phylogenetic tree based on the ompA gene of R. raoultii. c TCS haplotype network of R. raoultii based on the gltA gene from four different populations in Inner Mongolia. d TCS haplotype network of R. raoultii based on the ompA gene from four different populations in Inner Mongolia
Rickettsia genetic diversity by gltA gene
In the gltA sequences, the final alignment consisted of 1318 base pairs, with 842 variable sites. Of 10 haplotypes recovered, four were shared haplotypes (G1, G2, G3, G10). Numerically, the most common haplotype was G1, with 167 sequences (80.7% of all sequences) (Additional file 1: Table S3). G1 turned out to be the dominant haplotype. It was placed in the center of the haplotype network and was found in four geographically separate populations (Fig. 2c). The average haplotype diversity was 0.335. The average nucleotide diversity was 0.04922. EEDS was the region with the highest haplotype diversity (h = 0.909). Neutrality analysis revealed nonsignificant values of Tajima's D and Fu's Fs results, confirming that the population had not experienced recent expansion (Table 2). Wright’s F index was calculated to measure the levels of genetic differentiation among the four geographical populations, indicating the allelic variation between populations, which correlated negatively with gene flow. Comparing pairwise FST indices showed that the FST value between HLBE and SZWQ was greater than 0.25, indicating high genetic differentiation among populations, probably on account of low gene flow. The FST values among the other regions were less than 0.05, indicating that the genetic differentiation among these populations was very small, with a high degree of gene flow (Additional file 1: Table S4). The AMOVA showed that the variability in Rickettsia mainly arose from within each population, and the genetic differentiation between populations was very small (Additional file 1: Table S5). The mismatch analysis presented double peaks, indicating that the four geographical populations did not experience rapid population expansion (Fig. 3a).

a Mismatch distribution analysis for the R. raoultii groups based on gltA. b Mismatch distribution analysis for the R. raoultii groups based on ompA
Rickettsia genetic diversity by ompA gene
In the ompA sequences, the final alignment consisted of 738 base pairs, with 466 variable sites. Of the 22 haplotypes that were found, four were shared haplotypes (O1, O2, O3, O16). Numerically, the most common haplotypes were O1, O2, and O3, with 170 sequences (82.1% of all sequences) (Additional file 1: Table S6). O2 was the dominant haplotype. It was placed in the center of the haplotype network and was found in four different populations (Fig. 2d). The total average haplotype diversity was 0.735 and the total nucleotide diversity was 0.07308. SZWQ was the locality with the highest haplotype diversity (h = 1). Neutrality results were the same as those for gltA, indicating that the population had not experienced expansion recently (Table 3). The FST value between CF and HLBE was greater than 0.25, indicating that there was high genetic differentiation between populations. The FST values between HLBE, SZWQ, and EEDS were all greater than 0.05, confirming a moderate genetic differentiation between populations, with a small extent of gene flow. The FST values between the other regions were less than 0.05, confirming a very small genetic differentiation between these populations, likely on account of high gene flow (Additional file 1: Table S7). The results of the AMOVA and mismatch analyses were consistent with the gltA results (Additional file 1: Table S8, Fig. 3b).
Discussion
For planning effective control measures against Rickettsia infection, it is of utmost importance to continuously monitor the emergence of new species, study the population structure, and investigate the genetic diversity of the pathogen. In this study, two Rickettsia species were identified in Inner Mongolia, R. raoultii and R. sibirica, both belonging to the SFGR. In recent years, human infection with R. raoultii has led to tick-borne lymphadenitis in many countries [27,28,29,30]. To strengthen appropriate detection and treatment measures in endemic regions, public health workers and physicians should pay close attention to the high risk of human infection by R. raoultii.
This study analyzed the genetic diversity of a region within gltA and ompA genes of Rickettsia from four localities of Inner Mongolia. The aim of the investigation was to find new possibilities for controlling the transmission and reproduction of Rickettsia. Our results showed that the gltA and ompA genes have shared haplotypes in four regions. These are dominant haplotypes characterized as primitive and stable. Shared haplotypes indicate that the degree of genetic communication is high in Rickettsia populations. Furthermore, the lowest genetic diversity was found in HLBE in the gltA gene. The diversity of the ompA was higher in HLBE. The different results for the two genes indicate that the genetic diversity of species is affected by many factors, such as geographical distribution and population size [31]. Different gene markers are under different selective pressure during the evolution of species, leading to inconsistent genetic diversity [32]. Overall, the high genetic diversity of Rickettsia is in accord with different environments. The high rate of genetic communication in Rickettsia populations not only leads to a higher positive rate of Rickettsia in Inner Mongolia, but also increases the probability of Rickettsia transmission among and between humans and livestock.
Regarding the four geographical populations investigated, there is genetic differentiation between populations from AMOVA results. Further studies on FST values revealed that the degree of genetic differentiation was highest between HLBE and the other three regions. Obviously, HLBE is not only a beneficial habitat for ticks but also an important pastoral region in Inner Mongolia [33]. It also has the highest Rickettsia-positive rate and the highest genetic differentiation in Rickettsia populations in Inner Mongolia.
The neutrality test values of Tajima’s D and Fu’s Fs are used to test the historical dynamics of populations. If both are significantly negative, it indicates that the D. nuttalli population has historically experienced rapid population expansion. In this study, we did not find evidence of a recent rapid expansion of the population. Furthermore, the mismatch analysis for the two genes gltA and ompA showed genetic differentiation with no population expansion. The haplotypes of Rickettsia did not branch in relation to the clustering of geographical regions, indicating that the Rickettsia populations did not form a geographical differentiation structure. Therefore, in recent years, Rickettsia populations have not experienced excessive outbreaks, and the phenomenon of high genetic diversity may be a historical effect.
Since Rickettsia can be transmitted either vertically or horizontally, the high carry and infection rates of Rickettsia in D. nuttalli, together with the animal husbandry exchange in Inner Mongolia, gradually led to the harmonization of genetic characteristics of Rickettsia across various regions. The National Center for Disease Control and Prevention should strengthen the monitoring of tick-borne Rickettsia in Inner Mongolia and develop effective control measures.
Conclusion
The gltA and ompA genes were used to study the genetic diversity of Rickettsia from four geographical localities in Inner Mongolia. This study provides a reference for detecting new genotypes and complex genetic structures of Rickettsia populations. It also indicates that although Rickettsia species in Inner Mongolia have adapted to different environments, effective control measures of tick-borne Rickettsia transmission is crucial, especially in the HLBE region.
Availability of data and materials
All datasets have been included with this article and our sequences have been deposited within GenBank (accession number OK638141-OK638150 for gltA, accession number OL304270-OL304271, OL348251-OL348270 for ompA).
Abbreviations
- PCR:
-
Polymerase chain reaction
- D. nuttalli :
-
Dermacentor nuttalli
- R. raoultii :
-
Rickettsia raoultii
- R. sibirica :
-
Rickettsia sibirica
- AMOVA:
-
Analysis of molecular variance
- BLAST:
-
Basic Local Alignment Search Tool
References
Parola P, Raoult D. Ticks and tickborne bacterial diseases in humans: an emerging infectious threat. Clin Infect Dis. 2001;32:897–928.
Černý J, Buyannemekh B, Needham T, Gankhuyag G, Oyuntsetseg D. Hard ticks and tick-borne pathogens in Mongolia-a review. Ticks Tick Borne Dis. 2019;10:101268.
Guillotte ML, Chandler CE, Verhoeve VI, Gillespie JJ, Driscoll TP, Rahman MS, et al. Lipid A structural divergence in Rickettsia pathogens. mSphere. 2021. https://doi.org/10.1128/mSphere.00184-21.
Satjanadumrong J, Robinson MT, Hughes T, Blacksell SD. Distribution and ecological drivers of spotted fever group Rickettsia in Asia. EcoHealth. 2019;16:611–26.
Raoult D, Roux V. Rickettsioses as paradigms of new or emerging infectious diseases. Clin Microbiol Rev. 1997;10:694–719.
Fischer T, Myalkhaa M, Krücken J, Battsetseg G, Batsukh Z, Baumann MPO, et al. Molecular detection of tick-borne pathogens in bovine blood and ticks from Khentii. Mongolia Transbound Emerg Dis. 2020;67:111–8.
Song S, Chen C, Yang M, Zhao S, Wang B, Hornok S, et al. Diversity of Rickettsia species in border regions of northwestern China. Parasit Vectors. 2018;11:634.
Yin X, Guo S, Ding C, Cao M, Kawabata H, Sato K, et al. Spotted fever group Rickettsiae in inner Mongolia, China, 2015–2016. Emerg Infect Dis. 2018;24:2105–7.
Wang Q, Guo WB, Pan YS, Jiang BG, Du CH, Que TC, et al. Detection of novel spotted fever group Rickettsiae (Rickettsiales: Rickettsiaceae) in ticks (Acari: Ixodidae) in Southwestern China. J Med Entomol. 2021;58:1363–9.
Fang LQ, Liu K, Li XL, Liang S, Yang Y, Yao HW, et al. Emerging tick-borne infections in mainland China: an increasing public health threat. Lancet Infect Dis. 2015;15:1467–79.
Liu H, Liang X, Wang H, Sun X, Bai X, Hu B, et al. Molecular evidence of the spotted fever group Rickettsiae in ticks from Yunnan Province. Southwest China Exp Appl Acarol. 2020;80:339–48.
Miranda J, Mattar S. Molecular detection of Rickettsia bellii and Rickettsia sp. strain colombianensi in ticks from Cordoba Colombia. Ticks Tick Borne Dis. 2014;5:208–12.
Pillay AD, Mukaratirwa S. Genetic diversity of Rickettsia africae isolates from Amblyomma hebraeum and blood from cattle in the eastern cape province of South Africa. Exp Appl Acarol. 2020;82:529–41.
Qin XR, Han HJ, Han FJ, Zhao FM, Zhang ZT, Xue ZF, et al. Rickettsia japonica and novel Rickettsia species in ticks. China Emerg Infect Dis. 2019;25:992–5.
Roux V, Rydkina E, Eremeeva M, Raoult D. Citrate synthase gene comparison, a new tool for phylogenetic analysis, and its application for the Rickettsiae. Int J Syst Bacteriol. 1997;47:252–61.
Gui Z, Wu L, Cai H, Mu L, Yu JF, Fu SY, et al. Genetic diversity analysis of Dermacentor nuttalli within inner Mongolia, China. Parasit Vectors. 2021;14:131.
Filippova NA, Plaksina MA. Some aspects of intraspecific variability of the closely related species of the Dermacentor marginatus complex (Acari: Ixodidae) as demonstration of microevolutionary process. Parazitologiia. 2005;39:337–64.
Halos L, Jamal T, Vial L, Maillard R, Suau A, Le Menach A, et al. Determination of an efficient and reliable method for DNA extraction from ticks. Vet Res. 2004;35:709–13.
Bermúdez S, Martínez-Mandiche J, Domínguez L, Gonzalez C, Chavarria O, Moreno A, et al. Diversity of Rickettsia in ticks collected from wild animals in panama. Ticks Tick Borne Dis. 2021;12:101723.
Kumar S, Stecher G, Tamura K. MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol Biol Evol. 2016;33:1870–4.
Li K, Stanojević M, Stamenković G, Ilić B, Paunović M, Lu M, et al. Insight into diversity of bacteria belonging to the order Rickettsiales in 9 arthropods species collected in Serbia. Sci Rep. 2019;9:18680.
Mendell NL, Reynolds ES, Blanton LS, Hermance ME, Londoño AF, Hart CE, et al. Detection of rickettsiae, borreliae, and ehrlichiae in ticks collected from Walker County, Texas, 2017–2018. Insects. 2019;10:10.
Librado P, Rozas J. DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics. 2009;25:1451–2.
Rozas J. DNA sequence polymorphism analysis using DnaSP. Methods Mol Biol. 2009;537:337–50.
Meirmans PG, Hedrick PW. Assessing population structure: F(ST) and related measures. Mol Ecol Resour. 2011;11:5–18.
Santibáñez S, Portillo A, Santibáñez P, Palomar AM, Oteo JA. Usefulness of rickettsial PCR assays for the molecular diagnosis of human rickettsioses. Enferm Infec Micr Cl. 2013;31:283–8.
Dong Z, Yang Y, Wang Q, **e S, Zhao S, Tan W, et al. A case with neurological abnormalities caused by Rickettsia raoultii in northwestern China. BMC Infect Dis. 2019;19:796.
Li H, Zhang PH, Huang Y, Du J, Cui N, Yang ZD, et al. Isolation and identification of Rickettsia raoultii in human cases: a surveillance study in 3 medical centers in China. Clin Infect Dis. 2018;66:1109–15.
Seo MG, Kwon OD, Kwak D. High prevalence of Rickettsia raoultii and associated pathogens in Canine ticks. South Korea Emerg Infect Dis. 2020;26:2530–2.
Wen J, Jiao D, Wang JH, Yao DH, Liu ZX, Zhao G, et al. Rickettsia raoultii, the predominant Rickettsia found in Dermacentor silvarum ticks in China-Russia border areas. Exp Appl Acarol. 2014;63:579–85.
Feng X, Liu J, Chiang YC, Gong X. Investigating the genetic diversity, population differentiation and population dynamics of Cycas segmentifida (Cycadaceae) endemic to Southwest China by multiple molecular markers. Front Plant Sci. 2017;8:839.
Tang QY, Shi LX, Liu F, Yu D, Liu HZ. Evolution and phylogenetic application of the MC1R gene in the Cobitoidea (Teleostei: Cypriniformes). Zool Res. 2016;37:281–9.
Jiao J, Lu Z, Yu Y, Ou Y, Fu M, Zhao Y, et al. Identification of tick-borne pathogens by metagenomic next-generation sequencing in Dermacentor nuttalli and Ixodes persulcatus in Inner Mongolia, China. Parasit Vectors. 2021;14:287.
Acknowledgements
We are very grateful to the Inner Mongolia Center for Disease Control and Prevention for providing tick samples. We are very grateful to the Molecular Biology Research Center of Inner Mongolia Medical University for providing experimental facilities and conditions. We are very grateful to the Research Foundation of Ningbo Institute of Life and Health Industry for financial support.
Funding
This work received financial support from the Medical Scientific Research Foundation of Zhejiang Province, China (Grant no. 2021423808), Research Foundation of Ningbo Institute of Life and Health Industry, University of Chinese Academy of Sciences (Grant no. 2020YJY0214 & 2021YJY1008), Achievement Transformation Project of Inner Mongolia Medical University (Grant no. YKD2020CGZH001), and Zhiyuan Talent Project of Inner Mongolia Medical University (Grant no. ZY0201027).
Author information
Authors and Affiliations
Contributions
ZG, HC, and DDQ performed laboratory analysis, analyzed data, and wrote the first draft. RM, TC, and JFY revised the manuscript. XYS and DDQ participated in sample collection. SYF directed the experiment and helped to revise the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
No ethical permissions were necessary for this study as the parasites were collected from the environment of public places.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
13071_2022_5387_MOESM1_ESM.docx
Additional file 1: Table S1. Sample information of D. nuttalli populations. Table S2. Primers of PCR and amplification conditions. Table S3 Haplotype distribution of Rickettsia based on the gltA gene. Table S4. FST values among different groups of Rickettsia based on the gltA gene. Table S5. AMOVA of gltA gene of Rickettsia population. Table S6 Haplotype distribution of Rickettsia based on the ompA gene. Table S7 FST values among different groups of Rickettsia based on the ompA gene. Table S8 AMOVA of the ompA gene of the Rickettsia population.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Gui, Z., Cai, H., Qi, DD. et al. Identification and genetic diversity analysis of Rickettsia in Dermacentor nuttalli within inner Mongolia, China. Parasites Vectors 15, 286 (2022). https://doi.org/10.1186/s13071-022-05387-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13071-022-05387-4