
Abstract
Background
Thermotolerance is widely acknowledged as a pivotal factor for fungal survival across diverse habitats. Heat stress induces a cascade of disruptions in various life processes, especially in the acquisition of carbon sources, while the mechanisms by which filamentous fungi adapt to heat stress and maintain carbon sources are still not fully understood.
Results
Using Trichoderma guizhouense, a representative beneficial microorganism for plants, we discover that heat stress severely inhibits the lignocellulases secretion, affecting carbon source utilization efficiency. Proteomic results at different temperatures suggest that proteins involved in the poly ADP-ribosylation pathway (TgPARP and TgADPRase) may play pivotal roles in thermal adaptation and lignocellulose utilization. TgPARP is induced by heat stress, while the deletion of Tgparp significantly improves the lignocellulose utilization capacity and lignocellulases secretion in T. guizhouense. Simultaneously, the absence of Tgparp prevents the excessive depletion of ATP and NAD+, enhances the protective role of mitochondrial membrane potential (MMP), and elevates the expression levels of the unfolded protein response (UPR)-related regulatory factor Tgire. Further investigations reveal that a stable MMP can establish energy homeostasis, allocating more ATP within the endoplasmic reticulum (ER) to reduce protein accumulation in the ER, thereby enhancing the lignocellulases secretion in T. guizhouense under heat stress.
Conclusions
Overall, these findings underscored the significance of Tgparp as pivotal regulators in lignocellulose utilization under heat stress and provided further insights into the molecular mechanism of filamentous fungi in utilizing lignocellulose.
Similar content being viewed by others

Background
The Trichoderma genus represents one of the most widely distributed beneficial microbes in the global agricultural system, engaging in robust interactions with plant roots and soil microbes [1]. Nowadays, Trichoderma has been extensively utilized as a common bio-agent, playing a crucial role in promoting plant growth and controlling soil-borne diseases [2], thus greatly contributing to advancements in agricultural production. During the colonization process in diverse habitats, including soil and root systems, Trichoderma typically acquires sufficient energy resources for growth by secreting lignocellulases to decompose dead fungi or plant debris [3]. Furthermore, Trichoderma can adapt to local environmental changes and compete with other organisms by secreting lignocellulases, thereby occupying new ecological niches [4]. In recent years, the characteristic of Trichoderma secreting a large number of lignocellulases has been extended to the field of chemical engineering, facilitating the processing of agricultural waste and the development of green fuels [5, 6]. Therefore, in-depth research into the mechanism of Trichoderma secreting cellulases, aiming to maximize lignocellulases production, holds significant importance for promoting Trichoderma colonization and the industrial application of enzymes.
The regulation of lignocellulases secretion primarily involves three mechanisms, including direct transcriptional regulation, upstream regulation through nutritional sensing pathways, and feedback regulation from the secretion pathway [7]. Despite the involvement of different regulatory modes, the fine-tuning cooperation of transcription factors (TFs) appears to be relatively conserved among various filamentous fungi [8]. Currently, five main classes of TFs have been identified in filamentous fungi, primarily including the positive regulators XYR1, ACE2, and the HAP2/3/5 complex, as well as the repressor protein ACE1 and the carbon catabolite repression inhibitor CRE1 [9]. The post-translational regulation of cellulase is another extensively studied mechanism. Following transcription and translation, newly synthesized lignocellulases are transported to the endoplasmic reticulum (ER) for sorting and processing before secretion. This targeting may lead to an increased protein flux in the ER, resulting in heightened demands for protein folding, disulfide bond formation, glycosylation, and sorting, consequently causing significant ER stress and unfolded protein response (UPR) [10, 11]. In S. cerevisiae, the UPR activated HAC1 by initiating IRE1, and the efficiently translated HAC1 enhanced the transcription of a set of genes, thereby strengthening ER folding capacity and protein transport [12]. During the degradation of lignocellulose, the scarcity of favored carbon sources (such as sugars) can also induce ER stress and UPR in filamentous fungi [7]. However, there still needs more in-depth research on how to alleviate ER stress and UPR to sustain the secretion of cellulolytic enzymes.
Temperature, one of the most ubiquitous environmental factors, regulates many physiological and morphological processes in fungi [13]. Optimal temperature is crucial for fungal growth and development, and an increase in temperature generally leads to attenuation and ultimately results in organism death [14]. Due to global warming, the sustained increase in surface temperature has various adverse effects on soil microbes, including the colonization of Trichoderma in the soil [15]. On the other hand, as an efficient cellulolytic fungus and beneficial plant symbiont [16], Trichoderma is widely used in China for solid-state fermentation of crop straw to produce biofertilizers [17]. However, the stacking and sealing of substrates, coupled with the enhanced metabolic activity of fungi, typically result in a rapid increase in system temperature. Excessive temperatures significantly limit the biomass of Trichoderma. Moreover, precise temperature control and human intervention significantly increase costs. Therefore, enhancing the ability of Trichoderma to degrade lignocellulose under heat stress becomes particularly important.
Conventional wisdom suggests that one of the most effective adaptation mechanisms for filamentous fungi under heat stress is the heat stress response (HSR). This gene expression regulatory circuit, which is highly conserved, induces the expression of a series of cytoprotective genes that encode heat shock proteins (HSPs) [18]. In addition, maintaining energy homeostasis by preserving ATP levels is imperative because many pivotal proteins are ATP-dependent under heat stress [19]. For example, protein folding, refolding, and the UPR, which limit the accumulation of unfolded proteins in the ER, are all energy-requiring processes. Hence, the question arises as to whether there is a potential mechanism in filamentous fungi that links heat stress to the lignocellulases secretion, possibly by maintaining ATP homeostasis or alleviating ER stress. In this study, employing enzymatic assays, omics analyses, and genetic manipulations, we provided further evidence to demonstrate that Trichoderma could alleviate ER stress and enhance lignocellulases secretion by elevating cellular ATP levels. This study is expected to provide a better understanding of the lignocellulose catabolism mechanism of filamentous fungi and provide more references for exploring lignocellulase regulatory networks.
Results
Evaluation of lignocellulases secretion at different temperatures in Trichoderma guizhouense
Temperature is a critical parameter influencing the growth and metabolism of microbes. We studied the carbon utilization efficiency of Trichoderma guizhouense (T. guizhouense) at various temperatures, revealing an optimal growth temperature around 28 °C, with significant inhibition observed above 35 °C using rice straw as the sole carbon source (Additional file 1: Fig. S1). Based on this phenotype, 28 °C and 37 °C were selected as representative temperatures to investigate the relationship between lignocellulases secretion and heat stress (Fig. 1a). The systematic assessment of various lignocellulases activities is the preferred method for determining fungal lignocellulases secretion level [20]. Therefore, different lignocellulases, including endoglucanase, cellobiohydrolase, xylanase, and filter paper activity (FPA), were determined at 28 °C (T28) and 37 °C (T37) during solid-state fermentation (SSF). As expected, different lignocellulases activities exhibited consistent trends during continuous SSF, showing significant reductions in T37 compared to T28, indicating lower lignocellulose utilization efficiency in T. guizhouense at heat stress (Fig. 1b–e). The CO2 evolution rate is a critical parameter to evaluate the growth and metabolism condition of soil fungi [21]. Not surprisingly, the maximum respiration rate occurred on the fourth day, with a close to 30% decrease in T37 compared to T28 (Fig. 1f). Besides, SEM was also employed to assess the extent of lignocellulose biodegradation, and the result indicated that the degradation of rice straw was significantly lower in T37, with almost no exposure of the lower thick-walled tissue compared to T28 (Additional file 1: Fig. S1). These findings suggested that the ability to secrete lignocellulases was severely inhibited in T. guizhouense under heat stress, thereby limiting the efficiency of carbon sources utilization and metabolic rates.

Characterization of the lignocellulose utilization efficiency of T. guizhouense at different temperatures. a Growth of T. guizhouense using rice straw as the sole carbon source at 28 °C and 37 °C. b-e Lignocellulase activities, including endoglucanase, cellobiohydrolase, xylanase, and filter paper activities, between T28 and T37 under SSF. f Released amount of carbon dioxide (CO2 evolution rate) of T. guizhouense in T28 and T37 under SSF. The results are presented as the mean of at least three replicates, and the bars indicate the standard error of replicates
Quantitative analysis of the proteomes at different temperatures
To investigate the potential mechanisms underlying the impact of heat stress on the lignocellulases secretion, the proteomes of T. guizhouense in T28 and T37 were identified and quantified using the SWATH method. Data quality analysis indicated that the unique protein counts and biological replicates of the samples met the required standards (Additional file 1: Fig. S2). Based on GO (Gene Ontology) analysis, a total number of 1464 differentially expressed proteins were quantified between T28 and T37, mainly involved in binding (36.54%), catalytic activity (52.82%), and structural molecule activity (5.12%) (Additional file 1: Fig. S2). Concurrently, all the identified proteins were classified based on their locations, in which 1226 proteins were annotated as intracellular proteins and 333 proteins were in different detailed intracellular regions, including ribosome (39), ER (21), cytoplasm/cytosol (143), Golgi apparatus (6), mitochondria (33), and nucleus (91) (Fig. 2a). Notably, most intracellular organelles had spindle morphology in the abundance of proteins, and the differences in functional enrichment revealed the different corresponding strategies of T. guizhouense under SSF at different temperatures. Besides, with criteria of |log2Fold Change|≥ 0.58 and P-value < 0.05, 125 and 138 proteins were significantly upregulated and downregulated in T37 compared to T28, respectively (Fig. 2b).

Quantification of the intracellular proteins and carbohydrate-active enzyme (CAZy) family proteins of T. guizhouense in different treatments based on SWATH analysis. a Location and total abundances for each grouped protein identified by SWATH analysis. RI: ribosome, ER: endoplasmic reticulum, CY: cytoplasm/cytosol, NU: nucleus, GA: Golgi apparatus, Mit: mitochondria, CW: cell wall, CS: cytoskeleton, PM: plasma membrane, VA: vacuole, PE: peroxisome, VE: vesicle. b Volcano plot of all the identified proteins with different abundances between T37 and T28. The P-value was plotted against the expression fold change of all detected proteins within the different groups. Data points in the lower center area of the plots (cyan circles) display unchanged or proteins with no significant fold change. Data points in the upper quadrants indicate proteins (filled circles) with significant negative (blue circles) and positive (red circles) changes in protein abundance. c Protein–protein interaction (PPI) analysis in T37 compared with T28. The nodes are proteins (circles) or KEGG/CAZy categories (yellow diamonds); the edges are protein interactions defined by KEGG or CAZy databases. Protein node sizes indicate the log10 of protein expression values. Node colors represent the fold change (red, upregulated; blue, downregulated) of protein expression values between different treatments. Numbers 1 to 15 indicate different pathways. 1: Glycoside hydrolase, 2: starch and sucrose metabolism, 3: oxidative phosphorylation, 4: fatty acid biosynthesis, 5: citrate cycle (TCA cycle), 6: auxiliary activities, 7: proteasome, 8: purine metabolism, 9. Ribosome, 10: Glycolysis/Gluconeogenesis, 11: Carbohydrate esterases, 12: Spliceosome, 13: Transport, 14: Protein processing in the ER, 15: Other glycan degradation. d Abundance ratio of the proteins involved in the critical pathway based on SWATH (T37/T28). Nodes represent the fold change (red, upregulated significantly; blue, downregulated significantly; yellow and green, no significant difference) of protein content values between different treatments. RI: ribosome, GG: glycolysis/gluconeogenesis, MM: microbial metabolism in diverse environments, PM: pyruvate metabolism, PP: protein processing in the ER, FA: fatty acid metabolism, TCA: TCA cycle. e, Hierarchical cluster analysis of the CAZy family proteins between T28 and T37. Red and blue indicate relatively high and low protein abundance, respectively, and white indicates equal median abundance
To reveal the relationships between the different proteins that participate in specific metabolic pathways, protein–protein interaction (PPI) analyses were performed between T28 and T37 with Cytoscape software based on the KEGG analysis results (Fig. 2c). Compared to T37, the proteins in T28 clustered in glycoside hydrolase and starch and sucrose metabolism, and most of the proteins that were involved in these pathways were significantly upregulated, which clarified that heat stress could significantly restrict the lignocellulases secretion. Proteins involved in energy metabolism (glycolysis/Gluconeogenesis, TCA cycle, pyruvate metabolism, and fatty acid metabolism) and protein synthesis (ribosome and protein processing in ER) were undoubtedly more abundant and vital when responding to complex conditions [22]. The expression of these proteins, closely related to energy metabolism and protein synthesis, was correspondingly suppressed under heat stress (Fig. 2d). The underrepresentation of energetic core proteins in T37 and the decrease in the specific enzymes involved in energy generation likely contributed to the slow growth and low biomass that were observed. Simultaneously, a lack of protein synthesis and quality control might explain the lower secretion of lignocellulases under heat stress. These results suggested that retaining energy stability and improving the synthesis and processing of proteins are essential steps to promote the production of lignocellulases in T. guizhouense under heat stress. Additionally, 238 proteins were annotated as extracellular proteins, mainly including glycoside hydrolases, carbohydrate esterases, auxiliary activities enzymes, proteases, and small secreted cysteine-rich proteins (SSCRPs), in which most proteins were directly involved in lignocellulose degradation and exhibited significant differences. It was worth noting that different hydrolytic enzymes also showed differential expression in T37, which might be due to differences in the transcription levels of genes encoding these enzymes. Additionally, there might be functional similarities among different hydrolytic enzymes, which could also affect protein expression levels (Fig. 2e and Additional file 1: Table S1).
To comprehensively explore the potential regulatory mechanisms, we conducted in-depth PPI analysis of differentially expressed proteins based on the interolog method and the domain interaction-based method [23, 24]. Based on scoring criteria, five proteins were identified as critical proteins responding to the lignocellulases secretion under different temperatures, including NRPS protein (OPB39084), P5CDH (OPB43669), ALDH (KKP01310), ADPRase (OPB41268), and PARP (OPB37503) (Additional file 1: Fig. S3). Surprisingly, it was found that ALDH, PARP, and ADPRase were all involved in the metabolism of NAD+ and ATP, and the latter two were present in the poly ADP-ribosylation pathway (Additional file 1: Fig. S3). This conveyed a message: whether NAD+ and ATP metabolism were key factors linking heat stress to the lignocellulases secretion in T. guizhouense. Although the functions of homologous protein of PARP and ADPRase involved in abiotic stress have been previously addressed in plant [25, 26], the internal regulatory mechanism of growth and lignocellulases secretion in filamentous fungi under heat stress has not been stated clearly. Therefore, we attempted to explore the roles of TgPARP (Tgparp) and TgADPRase (Tgadprase) of T. guizhouense in responding to heat stress and lignocellulases secretion.
Gene Tgparp and Tgadprase respond to lignocellulases secretion of T. guizhouense under heat stress
Based on the above speculation, we constructed ΔTgparp (deletion of gene Tgparp) and OE-Tgadprase (overexpression of gene Tgadprase), and the mutants were confirmed by the southern blot and quantitative RT-PCR analysis (Additional file 1: Figs. S4–S5), respectively. Both mutants exhibited no difference in vegetative growth compared to wild type (wt), while showing lusher biomass compared to wt using rice straw as the sole carbon source under heat stress. However, the growth of ΔTgparp complemented strain (ΔTgparp-C) was restored to wt (Fig. 3a). Moreover, the carbon utilization characterizations of wt, ΔTgparp, and OE-Tgadprase were investigated by the Biolog FF microplates analysis, which indicated that OE-Tgadprase increased the utilization capacity of some carbon source substrates, while ΔTgparp exhibited a similar efficiency as that of wt (Additional file 1: Fig. S6). This would exclude the effect of the metabolic discrepancy in ΔTgparp on the carbon transformation of the primary carbon source. Meanwhile, the lignocellulases activity assays revealed that, in comparison to wt, FPA (filter paper activity), EG (endoglucanase), CBH (cellobiohydrolase), and XYL (xylanase) activities were increased by 1.93-, 1.84-, 4.39-, and 3.30-fold in ΔTgparp, respectively, and they were also increased by 1.94-,1.76-,1.18-, and 1.39-fold in OE-Tgadprase, respectively (Fig. 3b). Consistent with the enzyme activities, the biomass of ΔTgparp and OE-Tgadprase achieved copy numbers of 1 × 108.83 and 1 × 107.66 on the fourth day during SSF under heat stress, respectively, which were both greater than those of wt (1 × 105.95) and ΔTgparp-C (1 × 105.97) (Fig. 3c).

Promotion of lignocellulose utilization efficiency of T. guizhouense by deleting Tgparp or overexpressing Tgadprase under heat stress. a Growth comparison of the wt and mutants inoculated on PDA medium for 72 h at 28 °C and rice straw medium for 48 h at 28 °C and 37 °C. b Hydrolase activities, including FPA, EG, CBH, and XYL, of wt and mutants using rice straw as the sole carbon source under heat stress. c, Biomass of T. guizhouense by quantifying the copies of a 300 bp fragment from T. guizhouense in different treatments under heat stress (values are shown as the log10 conversion of the fragment copies·g−1 FW rice straw-induced cultures). d, e Transcriptional levels of some representative lignocellulase genes and regulatory factors in different treatments based on SWATH analysis. The representative lignocellulase genes included r-Tgegl (endo-1,4-β-glucanase, OPB37031), r-Tgcbh (cellobiohydrolase, OPB45635), r-Tgbgl (β-glucosidase, KKP01743), and r-Tgxyl (endo-1,4-β-xylanase, OPB45659). The regulatory factors included Tgace1 (OPB46882.1), Tgcre1 (OPB38360.1), Tgxyr1 (OPB38038.1), and Tgire (OPB43384.1). Data were calculated from three biological replicates, and error bars represent ± SDs. * P < 0.05, ** P < 0.01, *** P < 0.001. A P-value < 0.05 was regarded as statistically significant
To elucidate the molecular mechanisms underlying the mediation of lignocellulases secretion by genes Tgparp and Tgadprase, a targeted subset of lignocellulase genes and regulatory factors (Tgcre1, Tgace1, Tgxyr1, and Tgire) were initially quantified after 72 h of cultivation using straw as the sole carbon source, aiming to probe potential transcriptional regulatory processes. Remarkably, the expression levels of lignocellulase genes and regulatory factors (Tgcre1, Tgace1, and Tgxyr1) exhibited no significant distinctions among the ΔTgparp, OE-Tgadprase, and wt (Fig. 3d and Additional file 1: Fig. S7). However, a pronounced upregulation of Tgire was observed, with a 4.44-fold increase in ΔTgparp and a 3.12-fold increase in OE-Tgadprase, respectively (Fig. 3e). Given the well-established link between severe ER stress induced by heat stress and the role of IRE in orchestrating the UPR [27], the regulatory mechanism of Tgire assumes paramount importance. Thus, we hypothesized that genes Tgparp and Tgadprase potentially modulated lignocellulases secretion by influencing the transcriptional regulation of Tgire, thereby bolstering the ER stress under heat stress.
The functions of Tgparp in activating mitochondrial activity under heat stress
The functional genes parp and adprase are mainly involved in protein poly(ADP-ribosyl)ation (PARylation), and the recycling of ADP-ribose may represent an important pathway to re-establish ATP that can be utilized by the cell [28, 29]. Besides, it is well documented that PARP inhibitors could protect against oxidative stress-induced cell death and prevent the oxidative stress-induced depolarization of MMP [30]. In light of the functions above of PARP and the superior lignocellulases secretion capacity observed in ΔTgparp compared to OE-Tgadprase under heat stress, strain ΔTgparp was chosen as the experimental material to scrutinize the intricate relationship between gene Tgparp, ATP content, and MMP.
Firstly, ATP levels were quantified in both wt and ΔTgparp as the temperature progressively increased from 28 °C to 37 °C. The outcomes revealed a gradual elevation in ATP content within the ΔTgparp compared to wt when the temperature reached 34 °C (Additional file 1: Fig. S8). This result might offer insights into the substantial growth inhibition experienced by T.guizhouense beyond 34 °C (Additional file 1: Fig. S1), suggesting a potential correlation between gene Tgparp activation and ATP consumption in response to heightened heat stress. To further elucidate the compensatory role of ATP under heat stress, exogenous ATP-Na2 was introduced into the culture medium at varying concentrations, suggesting the addition of 500–1000 μM ATP-Na2 significantly ameliorated the growth of T.guizhouense (Additional file 1: Fig. S9).
MMP provides the driving force for ATP synthesis in mitochondria, and MMP stability is considered a requisite for energy homeostasis and normal cell functioning [31]. Here, monitoring of MMP was conducted using a membrane potential-dependent fluorescence dye, which can form red fluorescent aggregates in healthy mitochondria (indicative of higher MMP) and predominantly generates green fluorescence in unhealthy mitochondria (indicative of lower MMP) [43]. Besides, it has been reported that the folding of many secretory proteins could be inhibited by depleting cellular ATP levels and then strengthened by increasing ATP levels [44,45,46]. This work revealed that lignocellulases secretion might be broadly modulated by regulating cellular ATP content under heat stress. This ATP regulation could reduce the negative effect of heat stress on MMP through building energy homeostasis, which could relieve the ER stress when sufficient ATP is gathered in the ER. Besides, we proposed that ER stress might be mitigated through an energy homeostasis state by deleting Tgparp, which could contribute to the proper folding of various lignocellulases and avoid the aggregation in the ER to promote their secretion into the extracellular space using rice straw as the sole carbon source under heat stress.
Inevitably, DNA repair through the base excision repair pathway is always mentioned when studying the functions of PARP family members in different species [47, 48]. In P. Anserina, the PaParp gene seemed necessary for the proliferation and metabolism, and extensively attempted to delete the gene via homologous recombination had never been successful [49]. This result was in concordance with that in mice, in which knockout of parp-1 and parp-2 separately was found to be lethal [50], and the same results were also obtained in A. nidulans [51]. The deletion of the Npo gene encoding the ortholog with parp in Neurospora crassa had succeeded, and the researchers found that the Npo gene was optional and not required for heterochromatin formation [52]. These results clearly indicated that the PARP family proteins had diverse functions and exhibited a certain degree of specificity across different species. To avoid the loss of some unknown functions owing to the complete absence of Tgparp, whose suppressor mutants (Tgparp-RNAi) were generated, and various relevant enzyme activities indicated that more efficient lignocellulases secretion was detected than that in wt (Additional file 1: Fig. S12). Besides, the quantitative results of transcription levels for three DNA repair genes (Tgalkb, Tgrad50, and Tgrhp54) revealed significant upregulation of the Tgalkb gene in the ΔTgparp compared to wt, whereas this upregulation was not observed in Tgparp-RNAi (Additional file 1: Fig. S12). These results further suggested that, similar to other species, the DNA repair function of the Tgparp gene existed in T. guizhouense but was not essential. Therefore, appropriately inhibiting the Tgparp transcription level might be crucial for establishing a balance between DNA repair and ATP consumption, thereby promoting cellular resistance to heat stress and maintaining the lignocellulases secretion.
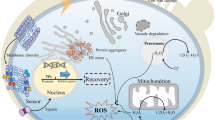
Usually, PARP activity depends on NAD+ levels, and a large amount of energy consumption accompanies the generation of NAD+ [53]. In the course of this investigation, two additional proteins identified in the protein PPI analysis, namely, P5CDH, and ALDH, were found to exhibit a preference for participating in NAD+ metabolism. P5CDH and ALDH are two nicotinamide adenine dinucleotide (phosphate) (NAD(P))-dependent enzymes involved in detoxification, biosynthesis, antioxidant functions, and regulatory mechanisms. These biological processes are typically accompanied by the conversion from NAD+ to NADH [54]. Importantly, these proteins exhibited significant downregulation in T37 compared to T28, potentially preventing excessive NAD+ depletion and redirecting energy consumption toward the resynthesis of NAD+. This hypothesis was further substantiated by in situ detection of NAD+, employing constructed sensors for in situ tracking of NAD+ (mCherry-cpYFP and mCherry-FiNad). As the control, the mCherry-cpYFP fluorescence ratio did not significantly change in wt (1.22) and ΔTgparp (1.32). Notably, compared to the mCherry-FiNad fluorescence ratio of wt (2.41), a lower ratio (1.75) was detected in ΔTgparp. Normalized to the control, the NAD+ ratio decreased by approximately 1.5-fold in ΔTgparp compared to wt (Additional file 1: Fig. S13 and S14). Therefore, we boldly speculated that more NAD+ was synthesized to be the substrate for TgPARP activation and was accompanied by tremendous ATP consumption. In fact, this process was a “waste” of ATP in T. guizhouense under heat stress. Furthermore, Persson et al. suggested that slowing some cellular processes could conserve energy under stress conditions [37]. Thus, a large amount of energy expenditure caused by the overactivation of Tgparp was more pernicious compared with the replaceable function of DNA repair under heat stress. On the other hand, communication between mitochondria and the ER has been reported to be primarily associated with the transfer of ions and lipids, signal transduction, and membrane dynamics [55]. Here, we propose an alternative connection: avoiding excessive consumption of NAD+ and ATP by inhibiting Tgparp activation can lead to healthier mitochondria. Stable mitochondria can establish energy homeostasis, allocating more ATP in the ER to reduce protein overaccumulation, thereby enhancing the lignocellulases secretion of T. guizhouense under heat stress (Fig. 7).

Proposed model for enhancing the lignocellulases secretion in T. guizhouense under heat stress by deleting the gene Tgparp. ER stress and gene Tgparp expression could be activated by heat stress and nutrition stress. Deletion of Tgparp maintains energy homeostasis, allocating more ATP to the ER for protein folding or refolding. This prevents excessive protein aggregation in the ER, thereby promoting the lignocellulases secretion of T. guizhouense under heat stress
Conclusion
This study provided insights into the significant impact of heat stress on the lignocellulases secretion in T. guizhouense and elucidated the potential mechanisms underlying this process. TgPARP was activated under heat stress, leading to accelerated depletion of intracellular ATP. Deletion of gene Tgparp led to a significant increase in intracellular ATP levels and maintained a higher MMP. Simultaneously, knocking out Tgparp promoted the transcription level of an ER stress response factor (Tgire), and its transcription level significantly correlated with intracellular ATP levels. Further investigations indicated that the absence of Tgparp regulated the intracellular distribution of ATP, leading to increased ATP accumulation in the ER, and enhancing protein folding and repair. This alleviated the accumulation of lignocellulases in the ER and enhanced their extracellular secretion. In conclusion, this study elucidated a novel pathway and mechanism for enhancing lignocellulose utilization of filamentous fungi under heat stress.
Materials and methods
Strains and culture conditions
Trichoderma guizhouense NJAU4742 was used as the wild-type strain for all experiments, and its genome sequence was already published in the NCBI database (accession No. LVVK00000000.1) and T. guizhouense genome website (https://bioinfo.njau.edu.cn/tgn4742/). To assess the lignocellulose utilization capacity and lignocellulases secretion, 2% (w/v) rice straw powder was added to the Mandels' salt solution without organic components and thoroughly mixed to generate straw agar plates and solid fermentation medium. Fresh spores of T. guizhouense were collected from PDA plates and filtered through four layers of cheesecloth. The spore concentration was quantified using a hemocytometer and adjusted to 1.0 × 107 spores·mL−1. The strains with equal biomass were transferred to the rice straw agar medium to observe the growth and biomass. A volume of 1 mL of T. guizhouense spores was inoculated for SSF, and all treatments were performed at 28 °C and 37 °C for seven days, with samples collected between day 2 and day 7. Each sampling point had three biological replicates.
Assays of enzyme activities and CO2 evolution rate
The endoglucanase activity and filter paper activity (FPA) were measured using CMC-Na (Sigma, USA) and filter paper (Whatman No.1) as substrates, following the method described by Xue et al. [56]. Xylanase activity was assayed with oat spelts xylan (Sigma, USA) as the substrates [57]. The assays for these three enzymes were conducted at 50 °C for 10 min in a 0.1 M acetate buffer (pH 4.8). Subsequently, the released reducing sugars were measured using the DNS method [58]. The cellobiohydrolase activity was determined in 0.1 M acetate buffer at 50 °C for 30 min with pNPC (Sigma, USA) as substrates according to Liu et al. [59]. One enzyme activity unit was defined as the number of enzymes required to liberate one μmol glucose or pNP per minute under the assayed conditions. The respiratory rate of T. guizhouense under SSF was evaluated by tracking the release of CO2. Briefly, the triangular flasks for SSF were sealed with a parafilm for 2 h before sampling, and then 20 mL of gas was extracted with a syringe. The collected gas was injected into the gas sampling bag (E-SWITCH, China), and the contents of CO2 were determined by gas chromatography (Agilent 7890A) equipped with a Porapak Q column and a flame ionization detector (FID) according to Zhang et al. [60].
Protein extraction and SWATH analysis
Protein extraction was performed after seven days' SSF as follows: 150 mL deionized ddH2O was added into the triangular flasks and then shaken at 180 rpm for 1 h, and the supernatants were collected by centrifugation at 12,000 rpm for 10 min and filtration through a 0.22 μm millipore filter membrane (Merck Millipore, German) for removing the impurities. The precipitates of the former step were used to extract the mycelial intracellular proteins using the NoviPure® Soil Protein Extraction Kit (MOBIO, USA) according to the manufacturer's protocol. The extracellular and intracellular proteins, both extracted from the same amount of fermentation substrates (mg·g−1 DW, protein/substrates), were directly mixed and concentrated by lyophilization (Powerdry LL3000, HETO, Denmark). The protein samples were stored at −80 °C for the subsequent experiment.
The SWATH (sequential window acquisition of all theoretical spectra) analysis of the proteomes was commissioned by a specialized company (GeneCreate, Wuhan, China). The protocols were described briefly as follows: 100 μg of proteins in different treatments was digested by trypsin (Promega, USA) at 37 °C overnight and then separated by the StrataX C18 pillar (Phenomenex, Torrance, CA, USA) and dried using a vacuum concentration meter. LC–ESI–MS/MS analysis was performed on an ABSCIEX nanoLC-MS/MS (Triple TOF 5600 plus) system. Data-dependent acquisition (DDA) was followed by SWATH acquisition, where all samples were mixed and detected in DDA, and the resulting data were used as a library for the analysis of each sample by SWATH. Spectral library generation and SWATH data processing were performed using skyline version 3.5 software, complying with the rules raised by Röst HL et al. [61]. Following the above instructions, each ion's XIC was automatically extracted with a retention time width of 5 min, and the area under the XIC curve (AUC) for individual ions was calculated. Fragment ions' areas that belong to one peptide were summed to get a peptide's abundance, and a summed abundance of peptides for a given protein was conducted to get the protein's abundance. To eliminate the random errors and sample bias, we normalized all the data among samples using a median normalization method [62]. Skyline employed the mProphet algorithm toward each extracted peak to assess the data confidence and control the false discovery rate.
Generation of T. guizhouense targeted gene deletion and overexpression mutants
Gene deletion and overexpression mutants were obtained through a gene replacement strategy using the polyethylene glycol (PEG)-mediated protoplast transformation procedure as described in Zhang et al. [63], with a hygromycin resistance cassette (HygB) in wt strain, or a Tgura3 compensation on Tgura3-deleted strain (ΔTgura3) to screen mutants without uridine. The detailed protocols were described in Additional file 1: Fig. S4, and the transformants were screened by PCR with a Phire® Plant Direct PCR Kit (Thermo Scientific, USA) and subjected to three rounds of single‐spore purification. For reverse complementation of ΔTgparp, a copy of the gene with its promoter and terminator region from the wt strain fused with the HygB gene at the C-terminus was amplified and inserted into ΔTgparp that was constructed by introducing Tgura3. The primers involved in the vector construction process are shown in Additional file 1: Table S2.
Quantitative RT-PCR and biomass assays of T. guizhouense
For mycelial qRT-PCR, total RNA extraction was performed using the RNeasy® Plant Mini Kit (Qiagen, Germany), and cDNA synthesis was completed using the PrimeScript RT Reagent Kit (RR036A, Takara, Dalian, China) according to the manufacturer's instructions. The qRT-PCR was performed using SYBR Premix Ex Taq II (RR820A, Takara, Dalian, China) and the CFX Connect Real-Time System (Bio-Rad, Hercules, USA).
For biomass assay, four-day-old T. guizhouense mycelia growing at 37 °C under SSF were collected and mixed evenly, and the total DNA was extracted with the EZNATM Soil DNA kit (Omega Bio-Tek, Inc., Norcross, GA, USA). The number of T. guizhouense copies was determined with SYBR Premix Ex Taq II (RR820A, Takara, Dalian, China), and the amplification of the fragment using the extracted DNA as a model was performed on a CFX Connect Real-Time System (Bio-Rad, Hercules, USA).
ATP content and MMP assays
ATP content was determined using the ATP Content Kit (Beyotime, China) according to the manufacturer's instructions. Briefly, fresh mycelia were collected from the rice straw plate covered with cellophane at 72 h, weighed, homogenized, and then centrifuged at 12,000 rpm for 5 min at 4 °C. The supernatants were mixed with ATP detection working dilution in a white 96-well plate. Luminance (RLU) was measured using a SpectraMax® i3x microplate reader (Molecular Devices, Sunnyvale, CA, USA). Standard curves were also generated, and the ATP content of each treatment was calculated. Total ATP levels were expressed as nmol·g−1 mycelia. As for the determination of MMP, the fresh mycelium tissues were measured using the JC-10 Assay Kit (Solarbio, Shanghai, China). Fresh mycelial tissues were incubated with 0.5 mL JC-10 working solution at 37 °C for 20 min. Subsequently, the samples were washed at least three times with a washing buffer. Cleaned mycelial tissues were directly captured using a confocal fluorescence microscope (TCS SP8, Leica, Germany) coupled with a digital camera. Simultaneously, the fluorescence intensity of stained mycelial tissues was measured using the SpectraMax® i3x microplate reader (Molecular Devices, Sunnyvale, California, USA).
Construction of the ATP sensor and lignocellulases-GFP fractions
FRET-based ATP sensor contained an ε subunit to connect the variants of CFP (mseCFP) and YFP (cp173-mVenus) and could monitor the ATP levels in the cytosol or mitochondria independently [38]. Here, a new FRET-based ATP sensor based on the ε subunit from the bacterial F0F1-ATP synthase was constructed to track the real-time content of ATP in different mutants (wt-ATP, ΔTgparp-ATP). The pDR-GW AT1.03YEMK plasmid (http://n2t.net/addgene:28003; RRID: Addgene_28003) contains the functional fragment of the ATP sensor, which was obtained via PCR. Subsequently, additional functional fragments were PCR amplified from the T. guizhouense genome, including the upstream region of ura3, the ORF fragment of ura3, the downstream region of ura3, and a strong promoter fragment. These fragments were then fused together using fusion PCR to generate a long fragment designated as ura3-HygB-ura3 upstream-promoter-ATP sensor-ura3 downstream, and it was transformed into the protoplasts of different mutants through the polyethylene glycol (PEG)-mediated protoplast transformation procedure [63]. Due to the presence of ura3 in the upstream region of the inserted fragment, there were repetitive sequences between the inserted fragment and the genome, inducing DNA repair and resulting in the loss of the segment between the repetitive sequences. Exploiting this feature, we were able to simultaneously insert the ATP sensor gene into the genome while knocking out the ura3 gene. Consequently, the HygB gene was lost along with it, yielding a ura3-deficient FRET sensor strain for subsequent genetic editing. The ura3 gene encodes orotidine-5'-phosphate decarboxylase, converting 5-fluoroorotic acid to a toxic form (5-fluorouracil). Thus, the ura3-deficient transformation was selected using 5-fluoroorotic acid [64].
For the generation of lignocellulases-GFP fusions proteins, the DNA fractions of the representative lignocellulase genes, including EGL (endo-1,4-β-glucanase, OPB37031), CBH (cellobiohydrolase, OPB45635), BGL (β-glucosidase, KKP01743), and XYL (endo-1,4-β-xylanase, OPB45659) were amplified with T. guizhouense DNA as template, and the GFP fragment was amplified using plasmid pEGFP-N1 as template (Clontech, USA). The DNA fractions of different lignocellulase genes were ligated with the GFP fragment using the overlap**-PCR technique based on the instructions of CloneAmp HiFi PCR Premix (Clontech, USA). Mutants of egl-gfp, cbh-gfp, bgl-gfp, and xyl-gfp were obtained through homologous recombination based on wt and ΔTgparp.
Confocal imaging and fluorescence intensity analysis of the protoplasts
The protoplast extraction of the lignocellulases-GFP fusion strains was performed according to the method reported by Li et al. [65]. Briefly, GFP fusion strains were cultured in the rice straw medium at 37 °C for 19 h to obtain the fresh mycelia, resuspended with 5 mL of lysing enzymes solution. A 100 mL lysing enzymes solution system contains 750 mg lysing enzyme (L1412, Sigma-Aldrich, USA), 21.86 g sorbitol, and 1.36 g KH2PO4, and the pH was adjusted to 5.50. The mixture was incubated at 37 °C at 100 rpm for 2 h. The release of protoplasts during the incubation was monitored on a confocal fluorescence microscope (TCS SP8, Leica, Germany) with a 60 × water immersion objective, and GFP was excited at 488 nm and emitted at 510 nm. The fluorescence of the protoplast samples was detected on flow cytometry (BD FACSAria SORP, Becton–Dickinson, USA). Based on the forward scatter characteristics of the protoplasts, an initial scatter gate was applied to collect data from individual cells. GFP protein was excited using a 488 nm laser, and fluorescence signals were detected using a 528/29 bandpass filter. Flow cytometry analyzed 20,000 cells per sample, and gates were set based on differences in fluorescence values to define the positive control expressing GFP [66]. The data were collected and analyzed using the FlowJo software (Version 7.6.1, FlowJo LLC, Ashland, Oregon).
Confocal imaging and fluorescence intensity analysis of ATP sensor and lignocellulases-GFP
Four-day-old mycelia were collected from the rice straw medium at 37 °C. For ER staining, fresh hyphae were incubated with ER-Tracker™ Red (ThermoFisher, Cat.M7512, Ex/Em = 587 nm/615 nm) at a concentration of 100 nM at room temperature for 5 min in darkness, and immediately washed three times with PBS buffer (pH 7.2). Confocal imaging of the ATP sensor was recorded on a confocal fluorescence microscope (TCS SP8, Leica, Germany) using a 60 × oil immersion objective. Excitation was provided at 435 nm, and emissions were at 475 nm and 527 nm, respectively. Data were presented as the ratio of signal intensities with a ratio of 527 nm to 475 nm. For confocal imaging of lignocellulases-GFP, mycelia were detected on a confocal fluorescence microscope (TCS SP8, Leica, Germany) with a 60 × water immersion objective, and GFP was excited at 488 nm and emitted at 510 nm.
All fluorescences were measured in ImageJ software. Briefly, image colors were extracted through Image-Color-Split Channels, and the RGB format was divided into 8-bit. Auto-threshold was applied to adjust the threshold to avoid exceeding limits, and the intensity analysis was performed by plot profile as previously described by Zhao et al. [41]. Data were completed as at least three independent biological replicates. Statistical data are expressed as means ± standard errors (SE) from all valid pixels.
Western blot
Different lignocellulases-GFP fusion strains were inoculated into the rice straw medium. After 72 h, the fungal mycelia were collected, and ER-enriched fractions were separated and isolated by Endoplasmic Reticulum Isolation Kit according to the manufacturer's instructions (ER0100, Sigma). SDS-PAGE sample buffer was employed to elute proteins, and together with the supernatant from the fourth day of SSF was used for GFP immunoblot analysis. For GFP-tagged protein detection, the concentrations of ER proteins and secreted proteins were adjusted to the same level (1 mg·mL−1) by the BCA protein assay kit (P0009, Beyotime, China), and the samples were separated by 12% SDS-PAGE and transferred to the PVDF membrane (Millipore). GFP monoclonal antibody (GF28R) (MA5-15256, Thermo Fisher) and Goat anti-Mouse IgG (H + L) Highly Cross-Adsorbed Secondary Antibody Alexa Fluor Plus 488 (A32723, Thermo Fisher) were used for immunoblot detection. GFP monoclonal antibody served as the primary antibody (1:2000), and anti-mouse IgG served as the secondary antibody (1:20,000). The Odyssey Infrared Imaging System (version 2.1) was used to detect and analyze the immunoblot signals.
Data availability
All data supporting the findings of this study are available within the paper and its additional materials. Source data that support the findings of this study are available from the corresponding author on reasonable request.
References
Druzhinina IS, Seidl-Seiboth V, Herrera-Estrella A, Horwitz BA, Kenerley CM, Monte E, Mukherjee PK, Zeilinger S, Grigoriev IV, Kubicek CP. Trichoderma: the genomics of opportunistic success. Nat Rev Microbiol. 2011;9:749–59.
Puyam A. Advent of Trichoderma as a bio-control agent-a review. Journal of Applied and Natural Science. 2016;8:1100–9.
Harman GE, Howell CR, Viterbo A, Chet I, Lorito M. Trichoderma species—opportunistic, avirulent plant symbionts. Nat Rev Microbiol. 2004;2:43–56.
Strakowska J, Błaszczyk L, Chełkowski J. The significance of cellulolytic enzymes produced by Trichoderma in opportunistic lifestyle of this fungus. J Basic Microbiol. 2014;54:S2–13.
Seiboth B, Ivanova C, Seidl-Seiboth V. Trichoderma reesei: a fungal enzyme producer for cellulosic biofuels. Biofuel Production-Recent Developments And Prospects. 2011;1:309–40.
Gusakov AV. Alternatives to Trichoderma reesei in biofuel production. Trends Biotechnol. 2011;29:419–25.
Huberman LB, Liu J, Qin L, Glass NL. Regulation of the lignocellulolytic response in filamentous fungi. Fungal Biol Rev. 2016;30:101–11.
Foreman PK, Brown D, Dankmeyer L, Dean R, Diener S, Dunn-Coleman NS, Goedegebuur F, Houfek TD, England GJ, Kelley AS. Transcriptional regulation of biomass-degrading enzymes in the filamentous fungus Trichoderma reesei. J Biol Chem. 2003;278:31988–97.
Meenu K, Singh G, Vishwakarma R: Molecular mechanism of cellulase production systems in Trichoderma. In Biotechnology and Biology of Trichoderma. Elsevier; 2014: 319–324.
Benz JP, Chau BH, Zheng D, Bauer S, Glass NL, Somerville CR. A comparative systems analysis of polysaccharide-elicited responses in N eurospora crassa reveals carbon source-specific cellular adaptations. Mol Microbiol. 2014;91:275–99.
Tanaka M, Shintani T, Gomi K. Unfolded protein response is required for Aspergillus oryzae growth under conditions inducing secretory hydrolytic enzyme production. Fungal Genet Biol. 2015;85:1–6.
Kaufman RJ. Stress signaling from the lumen of the endoplasmic reticulum: coordination of gene transcriptional and translational controls. Genes Dev. 1999;13:1211–33.
Walker GM, White NA. Introduction to fungal physiology. Fungi. 2017;1:1–35.
Bhabhra R, Askew D. Thermotolerance and virulence of Aspergillus fumigatus: role of the fungal nucleolus. Med Mycol. 2005;43:87–93.
Zhang W, Parker K, Luo Y, Wan S, Wallace L, Hu S. Soil microbial responses to experimental warming and clip** in a tallgrass prairie. Glob Change Biol. 2005;11:266–77.
**a Y, Wang J, Guo C, Xu H, Wang W, Yang M, Shen Q, Zhang R, Miao Y. Exploring the multi-level regulation of lignocellulases in the filamentous fungus Trichoderma guizhouense NJAU4742 from an omics perspective. Microb Cell Fact. 2022;21:1–14.
Tao C, Wang Z, Liu S, Lv N, Deng X, **ong W, Shen Z, Zhang N, Geisen S, Li R. Additive fungal interactions drive biocontrol of Fusarium wilt disease. New Phytol. 2023;238:1198–214.
Mota TM, Oshiquiri LH, Lopes ÉCV, Barbosa Filho JR, Ulhoa CJ, Georg RC. Hsp genes are differentially expressed during Trichoderma asperellum self-recognition, mycoparasitism and thermal stress. Microbiol Res. 2019;227: 126296.
Heimel K. Unfolded protein response in filamentous fungi—implications in biotechnology. Appl Microbiol Biotechnol. 2015;99:121–32.
Wi SG, Cho EJ, Lee D-S, Lee SJ, Lee YJ, Bae H-J. Lignocellulose conversion for biofuel: a new pretreatment greatly improves downstream biocatalytic hydrolysis of various lignocellulosic materials. Biotechnol Biofuels. 2015;8:1–11.
Hiscox J, Savoury M, Vaughan IP, Müller CT, Boddy L. Antagonistic fungal interactions influence carbon dioxide evolution from decomposing wood. Fungal Ecol. 2015;14:24–32.
Chubukov V, Gerosa L, Kochanowski K, Sauer U. Coordination of microbial metabolism. Nat Rev Microbiol. 2014;12:327–40.
Wang T-Y, He F, Hu Q-W, Zhang Z. A predicted protein–protein interaction network of the filamentous fungus Neurospora crassa. Mol BioSyst. 2011;7:2278–85.
Zhang K, Li Y, Li T, Li Z-G, Hsiang T, Zhang Z, Sun W. Pathogenicity genes in Ustilaginoidea virens revealed by a predicted protein–protein interaction network. J Proteome Res. 2017;16:1193–206.
Rissel D, Heym PP, Thor K, Brandt W, Wessjohann LA, Peiter E. No silver bullet–Canonical Poly (ADP-Ribose) Polymerases (PARPs) are no universal factors of abiotic and biotic stress resistance of Arabidopsis thaliana. Front Plant Sci. 2017;8:59.
Ogawa T, Muramoto K, Takada R, Nakagawa S, Shigeoka S, Yoshimura K. Modulation of NADH levels by Arabidopsis Nudix hydrolases, AtNUDX6 and 7, and the respective proteins themselves play distinct roles in the regulation of various cellular responses involved in biotic/abiotic stresses. Plant Cell Physiol. 2016;57:1295–308.
Feng XZ, Krishnan K, Richie DL, Aimanianda V, Hartl L, Grahl N, Powers-Fletcher MV, Zhang ML, Fuller KK, Nierman WC, et al. HacA-Independent Functions of the ER Stress Sensor IreA Synergize with the Canonical UPR to Influence Virulence Traits in Aspergillus fumigatus. Plos Pathogens. 2011;7:1.
Kadam A, Jubin T, Roychowdhury R, Garg A, Parmar N, Palit SP, Begum R. Insights into the functional aspects of poly (ADP-ribose) polymerase-1 (PARP-1) in mitochondrial homeostasis in Dictyostelium discoideum. Biol Cell. 2020;112:222–37.
Ogawa T, Ishikawa K, Harada K, Fukusaki E, Yoshimura K, Shigeoka S. Overexpression of an ADP-ribose pyrophosphatase, AtNUDX2, confers enhanced tolerance to oxidative stress in Arabidopsis plants. Plant J. 2009;57:289–301.
Tapodi A, Debreceni B, Hanto K, Bognar Z, Wittmann I, Gallyas F, Varbiro G, Sumegi B. Pivotal role of Akt activation in mitochondrial protection and cell survival by poly (ADP-ribose) polymerase-1 inhibition in oxidative stress. J Biol Chem. 2005;280:35767–75.
Zorova LD, Popkov VA, Plotnikov EY, Silachev DN, Pevzner IB, Jankauskas SS, Babenko VA, Zorov SD, Balakireva AV, Juhaszova M. Mitochondrial membrane potential. Anal Biochem. 2018;552:50–9.
**ao Y, Liu L, Zhang T, Zhou R, Ren Y, Li X, Shu H, Ye W, Zheng X, Zhang Z. Transcription factor MoMsn2 targets the putative 3-methylglutaconyl-CoA hydratase-encoding gene MoAUH1 to govern infectious growth via mitochondrial fusion/fission balance in Magnaporthe oryzae. Environ Microbiol. 2021;23:774–90.
Shekhova E, Kniemeyer O, Brakhage AA. Induction of mitochondrial reactive oxygen species production by itraconazole, terbinafine, and amphotericin B as a mode of action against Aspergillus fumigatus. Antimicrobial Agents Chemother. 2017. https://doi.org/10.1128/aac.00978-00917.
Zhu G, Chen S, Zhang Y, Lu L. Mitochondrial membrane-associated protein mba1 confers antifungal resistance by affecting the production of reactive oxygen species in aspergillus fumigatus. Antimicrob Agents Chemother. 2023;67:e00225-e223.
Back SH, Schröder M, Lee K, Zhang K, Kaufman RJ. ER stress signaling by regulated splicing: IRE1/HAC1/XBP1. Methods. 2005;35:395–416.
Hartl FU, Bracher A, Hayer-Hartl M. Molecular chaperones in protein folding and proteostasis. Nature. 2011;475:324–32.
Persson LB, Ambati VS, Brandman O. Cellular control of viscosity counters changes in temperature and energy availability. Cell. 2020;183(1572–1585): e1516.
Imamura H, Huynh Nhat KP, Togawa H, Saito K, Iino R, Kato-Yamada Y, Nagai T, Noji H. Visualization of ATP levels inside single living cells with fluorescence resonance energy transfer-based genetically encoded indicators. Proc Natl Acad Sci. 2009;106:15651–6.
Buchberger A, Bukau B, Sommer T. Protein quality control in the cytosol and the endoplasmic reticulum: brothers in arms. Mol Cell. 2010;40:238–52.
Tyedmers J, Mogk A, Bukau B. Cellular strategies for controlling protein aggregation. Nat Rev Mol Cell Biol. 2010;11:777–88.
Zhao X, Fang Y, Yang Y, Qin Y, Wu P, Wang T, Lai H, Meng L, Wang D, Zheng Z. Elaiophylin, a novel autophagy inhibitor, exerts antitumor activity as a single agent in ovarian cancer cells. Autophagy. 2015;11:1849–63.
Bloom AL, Panepinto JC. RNA biology and the adaptation of Cryptococcus neoformans to host temperature and stress. Wiley Interdisciplinary Reviews: RNA. 2014;5:393–406.
Tiwari S, Thakur R, Shankar J. Role of heat-shock proteins in cellular function and in the biology of fungi. Biotechnol Res Int. 2015;2015:1.
Depaoli MR, Hay JC, Graier WF, Malli R. The enigmatic ATP supply of the endoplasmic reticulum. Biol Rev. 2019;94:610–28.
Patel A, Malinovska L, Saha S, Wang J, Alberti S, Krishnan Y, Hyman AA. ATP as a biological hydrotrope. Science. 2017;356:753–6.
Balchin D, Hayer-Hartl M, Hartl FU. In vivo aspects of protein folding and quality control. Science. 2016;353:aac4354.
Palazzo L, Leidecker O, Prokhorova E, Dauben H, Matic I, Ahel I. Serine is the major residue for ADP-ribosylation upon DNA damage. Elife. 2018;7: e34334.
Pascal JM. The comings and goings of PARP-1 in response to DNA damage. DNA Repair. 2018;71:177–82.
Müller-Ohldach M, Brust D, Hamann A, Osiewacz HD. Overexpression of PaParp encoding the poly (ADP-ribose) polymerase of Podospora anserina affects organismal aging. Mech Ageing Dev. 2011;132:33–42.
De Murcia JM, Ricoul M, Tartier L, Niedergang C, Huber A, Dantzer F, Schreiber V, Amé J-C, Dierich A, LeMeur M. Functional interaction between PARP-1 and PARP-2 in chromosome stability and embryonic development in mouse. EMBO J. 2003;22:2255–63.
Semighini CP, Savoldi M, Goldman GH, Harris SD. Functional characterization of the putative Aspergillus nidulans poly (ADP-ribose) polymerase homolog PrpA. Genetics. 2006;173:87–98.
Kothe GO, Kitamura M, Masutani M, Selker EU, Inoue H. PARP is involved in replicative aging in Neurospora crassa. Fungal Genet Biol. 2010;47:297–309.
Chiarugi A, Moskowitz MA. PARP-1–a perpetrator of apoptotic cell death? Science. 2002;297:200–1.
Luo M, Singh RK, Tanner JJ. Structural determinants of oligomerization of Δ1-pyrroline-5-carboxylate dehydrogenase: Identification of a hexamerization hot spot. J Mol Biol. 2013;425:3106–20.
Csordás G, Weaver D, Hajnóczky G. Endoplasmic reticulum–mitochondrial contactology: structure and signaling functions. Trends Cell Biol. 2018;28:523–40.
Xue D, Lin D, Gong C, Peng C, Yao S. Expression of a bifunctional cellulase with exoglucanase and endoglucanase activities to enhance the hydrolysis ability of cellulase from a marine Aspergillus niger. Process Biochem. 2017;52:115–22.
Miao Y, Kong Y, Li P, Li G, Liu D, Shen Q, Zhang R. Effect of CBM1 and linker region on enzymatic properties of a novel thermostable dimeric GH10 xylanase (Xyn10A) from filamentous fungus Aspergillus fumigatus Z5. AMB Express. 2018;8:1–10.
Sinegani AAS, Emtiazi G. The relative effects of some elements on the DNS method in cellulase assay. J Appl Sci Environ Manag. 2006;10:93–6.
Liu D, Li J, Zhao S, Zhang R, Wang M, Miao Y, Shen Y, Shen Q. Secretome diversity and quantitative analysis of cellulolytic Aspergillus fumigatus Z5 in the presence of different carbon sources. Biotechnol Biofuels. 2013;6:1–16.
Zhang L, Wang S, Liu S, Liu X, Zou J, Siemann E. Perennial forb invasions alter greenhouse gas balance between ecosystem and atmosphere in an annual grassland in China. Sci Total Environ. 2018;642:781–8.
Röst HL, Rosenberger G, Navarro P, Gillet L, Miladinović SM, Schubert OT, Wolski W, Collins BC, Malmström J, Malmström L. OpenSWATH enables automated, targeted analysis of data-independent acquisition MS data. Nat Biotechnol. 2014;32:219–23.
Oberg AL, Mahoney DW. Statistical methods for quantitative mass spectrometry proteomic experiments with labeling. BMC Bioinformatics. 2012;13:1–18.
Zhang J, Miao Y, Rahimi MJ, Zhu H, Steindorff A, Schiessler S, Cai F, Pang G, Chenthamara K, Xu Y. Guttation capsules containing hydrogen peroxide: an evolutionarily conserved NADPH oxidase gains a role in wars between related fungi. Environ Microbiol. 2019;21:2644–58.
Liu Y, Li T, Zhu H, Zhou Y, Shen Q, Liu D. Cysteine facilitates the lignocellulolytic response of Trichoderma guizhouense NJAU4742 by indirectly up-regulating membrane sugar transporters. Biotechnol Biofuels Bioproducts. 2023;16:159.
Li C, Pang A-P, Yang H, Lv R, Zhou Z, Wu F-G, Lin F. Tracking localization and secretion of cellulase spatiotemporally and directly in living Trichoderma reesei. Biotechnol Biofuels. 2019;12:1–12.
Wang G, Jia W, Chen N, Zhang K, Wang L, Lv P, He R, Wang M, Zhang D. A GFP-fusion coupling FACS platform for advancing the metabolic engineering of filamentous fungi. Biotechnol Biofuels. 2018;11:1–12.
Acknowledgements
This research was financially supported by National Natural Science Foundation of China (32172680, 31972513 and 32302679), Natural Science Foundation of Jiangsu Province (BK20230991), the Fundamental Research Funds for the Central Universities (KYPT2023006), and the National Postdoctoral Research Fellowship Program (GZC20231126).
Author information
Authors and Affiliations
Contributions
TL and DL designed the experiments. TL performed most of the experiments. YL, HZ, LC, and YZ assisted in the execution of a subset of experiments. TL wrote the manuscript. DL and QS assisted in the revision of the manuscript. All authors have read the manuscript and approved the submission to this journal.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Fig. S1
. Growth and straw utilization efficiency of T. guizhouense by using rice straw as sole carbon sources at different temperatures. (a) Growth of T. guizhouense on plates at different temperatures using rice straw as sole carbon sources. (b) Scanning electron micrographs of the substrates including CK (raw material), T28 and T37, respectively. Fig. S2. Quality control of SWATH analysis results. (a) Experimental design to compare the extracellular proteins extracted from T. guizhouense at different temperatures. (b) The distribution of proteins' unique peptides number achieved the data analysis quality. (c) The length of identified peptides. (d) Classification of the SWATH-quantified proteins according to the biological function. (e) Distribution of proteins ratio of each sample. (f) Correlation of the quantitative values for all the identified proteins between replicated injections. Fig. S3. Model of the critical proteins for the lignocellulose utilization based on the SWATH and PPI analysis. PPI analysis predicted five proteins were considered critical during the lignocellulose utilization process. (a) The PPI network was constructed based on the STRING database between different treatments. (b) Proposed model of T. guizhouense in the lignocellulose utilization at different temperature. By compared with T28, four downregulated proteins including Nonribosomal peptide synthetases (NRPS protein, OPB39084), delta-1-pyrroline-5-carboxylate dehydrogenase (P5CDH, OPB43669), aldehyde dehydrogenase (NAD+) (ALDH, KKP01310), and ADPRase (OPB41268) and one up-regulated protein (PARP, OPB37503) were deemed as pivotal in T37. NRPS was the modular protein that produced peptide antibiotics and siderophores. P5CDH preferentially used NAD+ as a coenzyme to transform glutamate semialdehyde into glutamate. ALDH, a nicotinamide adenine dinucleotide (phosphate) (NAD(P))-dependent enzyme, was involved in detoxification, biosynthesis, antioxidant functions, and structural and regulatory mechanisms. ADPRase and PARP were also involved in the metastasis of ADPR. Intracellular free ADP‐ribose was a highly reactive and potentially toxic molecule. Activation of ADPRase or inhibition of PARP could relieve ER stress by increasing cellular ATP levels. AMP and ribose 5‐phosphate (Ribose 5‐P) might be beneficial for nucleotide recycling, resulting in the suppression of the over-consumption of NAD+ and ATP. Fig. S4. Knockout or over-expression of different functional genes in T. guizhouense. (a) Schematic diagram for genes disruption through double crossover recombination. (b) Schematic diagram for gene over-expression. (c) Diagram of construction principle for preparing the mutants of lignocellulases-GFP fusion strains. HA1 and HA2 were used as two arms of homologous recombination and the HygB gene was used as the biomarker for screening. (d) Analysis of expression level of Tgadprase gene relative to Tef in wt and OE-Tgadprase strain determined by qPCR. Data were calculated from three biological replicates. Error bars represent ± SDs. *** P < 0.001. A P-value < 0.05 is regarded as statistically significant. The expression value was normalized to wt. (e–f) Verification of ΔTgparp (left) and ΔTgire (right) by PCR to verify homologous recombination and whether gene exists. (g) Growth of mutants (ΔTgire and ΔTgireΔTgparp) inoculated on PDA at 28 °C (top row) and rice straw medium at 37 °C (bottom row), respectively. (h) Verification of ΔTgireΔTgparp by PCR to verify homologous recombination and whether gene exists. (i) Verification of recombinant strains with GFP labeling by PCR using two relevant primer pairs of E-egl (E-cbh, E-blg, and E-xyl) and E-GFP to verify homologous recombination. Fig. S5. Southern blot analysis of gene Tgparp. The genomic DNA was digested with the restriction enzymes and hybridized with the probes amplified by primers listed in Table. S2. Lane M: DNA marker; Lane 1: wt-1; Lane 2: ΔTgparp-1; Lane 3: positive plasmid-1; Lane 4: wt-2; Lane 5: ΔTgparp-2; Lane 6: Positive plasmid-2. Fig. S6. The profile of carbon utilization using Biolog FF Microplates at 28 °C. Three independent biological experiments were performed and the data were collected. Error bars represent ± SDs. Fig. S7. Transcriptional level of representative lignocellulase genes in wt and ΔTgparp at 37 °C. Data were calculated from three biological replicates. Error bars represent ± SDs. * P < 0.05, ** P < 0.01. A P-value < 0.05 was regarded as statistically significant. The expression values are normalized to wt. Endoglucanase genes: 1–5 (A1A106567.1, A1A101831.1, A1A107632.1, A1A104288.1, and A1A110611.1); cellobiohydrolase genes: 6–9 (A1A104298.1, A1A108865.1, A1A104556.1, and A1A102650.1); glucosidase genes: 10–13 (A1A110026.1, A1A105994.1, A1A107778.1, and A1A111035.1); hemicellulase and auxiliary hydrolyzing genes: 14–19 (A1A111258.1, A1A111547.1, A1A102817.1, A1A112191.1, A1A105029.1, and A1A100991.1). Fig. S8. The ATP level between wt and ΔTgparp with the gradual increase of tempeature (from 28 °C to 37 °C). Three independent biological experiments were performed and data was collected. Error bars represent ± SDs. ** P < 0.01, ** P < 0.001. A P-value < 0.05 was regarded as statistically significant. Fig. S9. Growth of T. guizhouense inoculated at 37 °C for 48 h when different concentrations of exogenous ATP-Na2 were added. Fig. S10. Construction and growth of strains with the ATP sensor. (a) Schematic of the ATP sensor. (b) Equally harvested biomass of strains wt-ATP and ΔTgparp-ATP were inoculated on PDA at 28 °C (left) and rice straw medium at 37 °C (right), respectively. (c) Verification of the strains wt-ATP and ΔTgparp-ATP through PCR by using two relevant primer pairs of E-ATP-F and E-ATP-R to verify the ATP sensor fragment, and E3 was used to verify the existence of gene Tgparp. Fig. S11. Flow cytometry dot plot of the protoplasts containing the lignocellulases-GFP at different treatments under heat stress. The lignocellulases-GFP fusion strains contained egl-gfp (a), cbh-gfp (b), bgl-gfp (c), and xyl-gfp (d); the dot plot of wt is represented in blue, while ΔTgparp are depicted in red. Fig. S12. Growth condition and lignocellulose utilization efficiency of RNAi-Tgparp under heat stress by knocking down of Tgparp through the RNA interference technology. (a) Schematic diagram for the generation of RNAi-Tgparp. MCS1: Sense strand of gene Tgparp, MCS2: Antisense strand of gene Tgparp. (b) Relative expression of gene Tgparp between wt, RNAi-Tgparp-1, and RNAi-Tgparp-2. The expression value is normalized to wt. (c) Biomass of wt, RNAi-Tgparp-1 and RNAi-Tgparp-2 inoculated on PDA (top row) and rice straw medium (bottom row), respectively. (d) Comparison of the lignocellulolytic activities including FPA, EG, CBH, and XYL between wt, RNAi-Tgparp-1, and RNAi-Tgparp-2 under heat stress. (e) Relative expression of genes related to DNA repair between wt, ΔTgparp and RNAi-Tgparp-1. Data were calculated from three biological replicates. Error bars represent ± SDs. ** P < 0.01, *** P < 0.001. A P-value < 0.05 is regarded as statistically significant. Fig. S13. Construction and growth of strains with NAD+ sensor. (a) Schematic of the ATP sensor. (b) The growth condition of wt with mCherry-cpYFP / mCherry-FiNad and ΔTgparp with mCherry-cpYFP / mCherry-FiNad inoculated on PDA at 28 °C (top row) and rice straw medium at 37 °C (bottom row), respectively. (c) Verification of the strains that successfully expressed the mCherry-cpYFP or mCherry-FiNad in wt and ΔTgparp. Fig. S14. Confocal imaging and fluorescence intensity analysis of NAD+ in T. guizhouense. (a-b) Confocal imaging analysis of hyphae from wt-mCherry-cpYFP (left) and ΔTgparp-mCherry-cpYFP (right) under heat stress, bar = 3 μm. (c-d) Confocal imaging analysis of hyphae from wt-mCherry-FiNad (left) and ΔTgparp-mCherry-FiNad (right) under heat stress, bar = 3 μm. (e–h) Fluorescence intensity of the two channels (mCherry and cpYFP) detected in different strains. (i-l) Green/Red ratios plotted for single pixels of the mycelium with the mCherry-cpYFP or mCherry-FiNad in wt and ΔTgparp. Blue numbers represent the average of all values. (m–n) Green/Red ratio of mCherry-cpYFP or mCherry-FiNad in wt and ΔTgparp from more mycelia repeats. The box plot means the median values of ratios. (o) Ratio of mCherry-FiNad/mCherry-cpYFP between wt and ΔTgparp according to the results in graphs m and n. (p) Biochemical analysis of cellular NAD+ content under heat stress between wt and ΔTgparp. Table S1. SWATH quantifies proteins of Trichoderma guizhouense NJAU4742 at different temperatures using straw as the only carbon source. Table S2. PCR primers used in this study.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Li, T., Liu, Y., Zhu, H. et al. Cellular ATP redistribution achieved by deleting Tgparp improves lignocellulose utilization of Trichoderma under heat stress. Biotechnol Biofuels 17, 54 (2024). https://doi.org/10.1186/s13068-024-02502-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13068-024-02502-8