
Abstract
Background
Variants that disrupt mRNA splicing account for a sizable fraction of the pathogenic burden in many genetic disorders, but identifying splice-disruptive variants (SDVs) beyond the essential splice site dinucleotides remains difficult. Computational predictors are often discordant, compounding the challenge of variant interpretation. Because they are primarily validated using clinical variant sets heavily biased to known canonical splice site mutations, it remains unclear how well their performance generalizes.
Results
We benchmark eight widely used splicing effect prediction algorithms, leveraging massively parallel splicing assays (MPSAs) as a source of experimentally determined ground-truth. MPSAs simultaneously assay many variants to nominate candidate SDVs. We compare experimentally measured splicing outcomes with bioinformatic predictions for 3,616 variants in five genes. Algorithms’ concordance with MPSA measurements, and with each other, is lower for exonic than intronic variants, underscoring the difficulty of identifying missense or synonymous SDVs. Deep learning-based predictors trained on gene model annotations achieve the best overall performance at distinguishing disruptive and neutral variants, and controlling for overall call rate genome-wide, SpliceAI and Pangolin have superior sensitivity. Finally, our results highlight two practical considerations when scoring variants genome-wide: finding an optimal score cutoff, and the substantial variability introduced by differences in gene model annotation, and we suggest strategies for optimal splice effect prediction in the face of these issues.
Conclusion
SpliceAI and Pangolin show the best overall performance among predictors tested, however, improvements in splice effect prediction are still needed especially within exons.
Similar content being viewed by others


Background
Splicing is the process by which introns are removed during mRNA maturation using sequence information encoded in the primary transcript. Sequence variants which disrupt splicing contribute to the allelic spectrum of many human genetic disorders, and it is estimated that overall, as many as 1 in 3 disease-associated single-nucleotide variants are splice-disruptive [1,2,3,4,5,6,7]. Splice-disruptive variants (SDVs) are most readily recognized at the essential splice site dinucleotides (GU/AG for U2-type introns) with many examples across Mendelian disorders [8,9,10,11,12]. SDVs can also occur at several so-called flanking noncanonical positions [13], and by some estimates, these outnumber essential splice mutations by several-fold [5, 14].
Variants beyond the splice-site motifs may be similarly disruptive but are more challenging to recognize [15]. Some of these disrupt splicing enhancers or silencers which are short motifs bound by splicing factors to stimulate or suppress nearby splice sites, to confer additional specificity, and to provide for regulated alternative splicing [16, 17]. These splicing regulatory motifs are widespread [18] and maintained by purifying selection [19], though they often feature partial redundancy and can tolerate some mutations. Some of the best-characterized cases come from genetic disorders, of which an archetypal example is spinal muscular atrophy caused by the loss of SMN1. Its nearly identical paralog SMN2 cannot functionally complement its loss, due to a fixed difference that eliminates an exonic splice enhancer (ESE). The resulting exon 7 skip** [20, 21] can be targeted by antisense oligonucleotides [22] to boost SMN2 protein expression sufficiently to provide therapeutic benefit. Other cases include synonymous variants, which as a class may be overlooked, and can disrupt existing splice regulatory elements or introduce new ones, as in the case of ATP6PA2-associated X-linked parkinsonism [23]. Systematically delineating splice-regulatory elements and their cognate factors, and defining the grammar or “splicing code” through which they act to shape splicing patterns has been a long-standing challenge for molecular and computational biology [24].
RNA analysis from patient specimens can provide strong evidence for splice-disruptive variants, and its inclusion in clinical genetic testing can improve diagnostic yield [5, 25,26,27,28]. However, advance knowledge of the affected gene is necessary for targeted RT-PCR analysis, while RNA-seq-based tests are not yet widespread [29, 30], and both rely upon sufficient expression in the blood or other clinically accessible tissues for detection. Therefore, a need remains for reliable in silico prediction of SDVs during genetic testing, and a diverse array of algorithms have been developed to this end. For instance, S-Cap [31] and SQUIRLS [32] implement classifiers that use features, such as motif models of splice sites, kmer scores for splice regulatory elements, and evolutionary sequence conservation, and are trained on sets of benign and pathogenic clinical variants. Numerous recent algorithms use deep learning to predict splice site likelihoods directly from the primary sequence; SDVs can then be detected by comparing predictions for wild-type and mutant sequence. Rather than training with clinical variant sets, SpliceAI [33] and Pangolin [34] use gene model annotations to label each genomic position as true/false based on whether it appears as an acceptor or donor in a known transcript. SPANR [6, 14, 51, 52], while saturation screens focus on individual exons [53,54,55,56,57,58] or motifs [36, 59] and measure the effects of every possible point variant within each target. Two broad MPSA datasets, Vex-seq [51] and MaPSy [6], were recently used to benchmark splicing effect predictors as part of the Critical Assessment of Genome Interpretation (CAGI) competition [60], and another, MFASS [14], has been used to validate a recent meta-predictor [43]. However, a limitation of benchmarking with broad MPSAs is that they may reflect an exon’s overall properties while lacking the finer resolution to assess different variants within it. For instance, an algorithm could perform well by predicting SDVs within exons with weak splice sites, or with evolutionarily conserved sequence, while failing to distinguish between truly disruptive and neutral variants within each.
Here we leverage saturation MPSAs as a complementary, high-resolution source of benchmarking data to evaluate eight recent and widely used splice predictors. Algorithms using deep learning to model splicing impacts using extensive flanking sequence contexts, SpliceAI and Pangolin, consistently showed the highest agreement with measured splicing effects, while other tools performed well on specific exons or variant types. Even for the best performing tools, predictions were less concordant with measured effects for exonic variants versus intronic ones, indicating a key area of improvement for future algorithms.
Results
A validation set of variants and splice effects
We aggregated splicing effect measurements for 2230 variants from four massively parallel splice assay (MPSA) studies, focusing on saturation screens targeting all single nucleotide variants (SNVs) in and around selected exons [53, 54, 57, 58] (Fig. 1A). We also included 1386 variants in BRCA1 from a recent saturation genome editing (SGE) study, in which mutations were introduced to the endogenous locus by CRISPR/Cas9-mediated genome editing, with splicing outcomes similarly measured by RNA sequencing [61]. Splice-disruptive variants (SDVs) and, conversely, neutral variants, were defined as specified by the respective studies, to account for their differences in gene target and methodology. For contrast with these saturation-scale datasets, we also prepared a more conventional, gene-focused benchmarking dataset by manually curating a set of 296 variants in the tumor suppressor gene MLH1 from clinical variant databases and literature reports. In sum, this benchmarking dataset contained 3912 SNVs across 33 exons spanning six genes (Additional file 1: Fig. S1–S5; Additional file 2: Table S1).

Variants used for splice effect predictor benchmarking. A Validation sets can be drawn from pathogenic clinical variants and, conversely, common polymorphisms in frequently screened disease genes (top panel), broadly targeted massively parallel splice assays (MPSAs) interrogating a few variants across many exons (middle panel), and saturation MPSAs in which all possible variants are created for a few target exons (bottom panel). B Variant classes defined by exon/intron region and proximity to splice sites (upper), with the percent coverage of the possible SNVs within each variant class (denoted by color) for each dataset in the benchmark set (for BRCA1, missense, and stop-gain variants were excluded and not counted in the denominator)
As expected, MPSAs measured most of the possible single-nucleotide variants at each target (93.3% of SNVs) with relatively uniform coverage by exon/intron region (Fig. 1B). From the BRCA1 SGE study, we retained only intronic or synonymous variants because missense variants’ effects could be mediated via protein alteration, splicing impacts, or both. Targeted exons varied in their robustness to splicing disruption, from POU1F1 exon 2 (10.2% SDV), to MST1R (also known as RON) exon 11 (68.4% SDV; Additional file 1: Fig. S6), reflecting both intrinsic differences between exons as well as different procedures for calling SDVs across MPSA studies. In contrast to the high coverage of the mutational space from MPSA and SGE datasets, reported clinical variants only sparsely covered the mutational space (1.6% of the possible SNVs in MLH1 exons +/− 100 bp) and were heavily biased towards splice sites (59.5% of reported variants within +/− 10 bp of a splice site; Additional file 1: Fig. S7). Larger clinical variant sets used to train classifiers showed a similar skew: 94.6% of the SQUIRLS training variants [32] and 88.9% of the pathogenic S-Cap training set [31] were within +/− 10 bp of splice sites. Thus, MPSAs offer high coverage without the variant class biases present among clinical variant sets.
Comparing bioinformatic predictions with MPSA measured effects
We selected eight recent and widely used predictors to evaluate: HAL [36], S-Cap [31], MMSplice [37], SQUIRLS [32], SPANR [Full size image
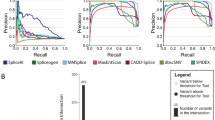
To systematically benchmark each predictor, we treated the splicing status from the experimental assays and curated clinical variant set as ground truth. We quantified the ability of each predictor to distinguish between the splice disruptive (n=1,060) and neutral (n=2852) variants in the benchmark set by taking the area under the precision-recall curve (prAUC) per classifier/gene (Fig. 3A). We next asked if classifiers’ performance differed by variant type and location. Algorithms consistently performed better for intronic than for exonic variants (median prAUC for introns: 0.773; for exons: 0.419; Fig. 3B), despite a similar proportion of SDVs in exons and introns (28.4% and 25.9% SDV, respectively). This difference persisted even when removing canonical splice dinucleotide variants (Additional file 1: Fig. S10). More finely subdividing the benchmark variant set by regions (defined as in Fig. 1B) demonstrated that performance suffers farther from splice sites where the overall load of SDVs is lower (Additional file 1: Fig. S11). To summarize overall performance, we counted the number of instances in which each predictor either had the highest prAUC or was within the 95% confidence interval of the winning tool’s prAUC (Fig. 3C). Every tool scored well for at least one dataset or variant class, but Pangolin and SpliceAI had the best performance most frequently (7 and 3 datasets/variant classes, respectively).

Splice effect predictors’ classification performance on benchmark variants. A Precision-recall curves showing algorithms’ performance distinguishing SDVs and splicing-neutral variants in each dataset. B Precision-recall curves of tools’ performance differentiating SDVs and splice neutral variants in exons (left) and introns (right). C Top panel: tally, for each algorithm, of the number of individual datasets and variant classes (defined as in Fig. 1B) for which that algorithm had the highest prAUC or was within the 95% confidence interval of the best performing tool. Bottom panel: signed difference between the best performing tool’s prAUC and a given tool’s prAUC; each dot corresponds to an individual dataset or variant class
Benchmarking in the context of genome-wide prediction
In practice, a splicing effect predictor must sensitively identify SDVs while maintaining a low false positive rate across many thousands of variants identified in an individual genome. We therefore evaluated each tool’s sensitivity for SDVs within our benchmark set, as a function of its genome-wide SDV call rate. We used a background set of 500,000 simulated SNVs drawn at random from in or near (+/− 100 bp) internal protein-coding exons (Additional file 1: Fig. S12; Additional file 2: Table S1). We scored these background SNVs with each tool and computed the fraction of the background set called as SDV as a function of the tool-specific score threshold. Although the true splice-disruptive fraction of these background variants is unknown, we normalized algorithms to each other by taking, for each algorithm, the score threshold at which it called an equal fraction (e.g., 10%) of the genomic background set as SDV. We then computed the sensitivity across the benchmark-set SDVs using this score threshold and termed this the ‘transcriptome-normalized sensitivity’. Taking SpliceAI as an example, at a threshold of deltaMax ≥0.06, 10% of the background set is called as SDV. Applying the same threshold (deltaMax≥0.06) to BRCA1 SGE variants in the benchmark set, SpliceAI reaches 98.2% sensitivity and 80.7% specificity (Fig. 4A).

Transcriptome-normalized sensitivity. A Example shown for SpliceAI. Upper panel shows SpliceAI scores for the 500,000 background set variants (teal histogram) and the cumulative fraction (black line) of variants above a given score threshold (10% of background set variants with deltaMax ≥0.06). Below, histograms of SpliceAI scores for BRCA1 SGE benchmark variants, either SDVs (middle) or splicing-neutral variants (bottom), and the resulting transcriptome-normalized sensitivity and specificity at a deltaMax cutoff of 0.06. B Transcriptome-normalized sensitivity (at 10% background set SDV) versus within-benchmark variant set prAUC by benchmarked dataset. C Transcriptome-normalized sensitivity on benchmark exonic and intronic variants plotted as a function of the percent of the background variant set called SDV
We repeated this process, using for each algorithm the score threshold at which 10% of the background set was called as SDV, and applying this threshold to the benchmark set (Additional file 3: Table S2). Transcriptome-normalized sensitivity varied widely between algorithms, but SpliceAI, ConSpliceML, and Pangolin emerged as consistent leaders (median across datasets of 87.3%, 85.8%, and 79.9%, respectively). Mirroring the results seen entirely within the benchmarking variant set (Fig. 3), median transcriptome-normalized sensitivity was lower for exonic vs intronic variants for all tools examined by an average of 36.9%, and the same pattern remained after removing intronic variants at essential splice sites. These results were not specific to the transcriptome-wide threshold of 10%: the same three algorithms scored highly for thresholds at which 5% or 20% of the background set scored as SDV. Performance also varied by exon target (Fig. 4B); for example, many of the SDVs in FAS exon 6 and MST1R exon 11 were not detected by any algorithm at a threshold which would classify 10% of the background set as SDV. The effects measured by MPSAs in these specific exons may be particularly subtle, posing difficult targets for prediction, and suggesting that existing tools may need scoring thresholds tuned to specific exons or variant regions. Finally, to explore the tradeoff between SDV recall and overall call rate, we quantified the transcriptome-normalized sensitivity for SDVs in the benchmark set, as a function of percent of the background set called SDV and took the area under the resulting curve, analogous to the prAUC statistic. Again, performance was consistently lower within exons than introns, across algorithms and datasets (Fig. 4C).
Determining optimal score cutoffs
Integrating splice effect predictors into variant interpretation pipelines requires a pre-determined score threshold beyond which variants are deemed disruptive. We explored whether our benchmarking efforts could inform this by identifying the score threshold that maximized Youden’s J statistic (J=sensitivity+specificity-1; Additional file 4: Table S3). For each algorithm, we first identified optimal score thresholds on each dataset individually to explore differences across genes and exons. For most tools we evaluated, ideal thresholds varied considerably across exons, regions, and variant classes, such that a threshold derived from one was suboptimal for others (Fig. 5). For some tools, including HAL and ConSpliceML, thresholds optimized on individual datasets spanned nearly the tools’ entire range of scores, while for others such as SQUIRLS, SpliceAI, and Pangolin, the optimal thresholds were less variable. For the tools with consistently high classification performance and transcriptome-normalized sensitivity — SpliceAI, Pangolin, and ConSpliceML (Figs. 3 and 4) — we found that the optimal thresholds were usually lower than the threshold recommended by the tools’ authors, largely consistent with conclusions of other previous benchmarking efforts [44, 45, 47, 48]. Optimal thresholds also differed by variant class, suggesting that tuning cutoffs by variants’ annotated effects, like those implemented in S-Cap, may offer some improvement for classification accuracy on variants genome-wide.

Substantial variability of score thresholds by dataset and variant type. For each algorithm (panel), the score thresholds (y-axis) that maximized Youden’s J is shown, across each benchmark variant dataset (blue points), by variant type (red points), and compared to previous reports (green points). Dashed gray line shows tool developers’ recommended thresholds. Solid lines indicate medians
Variant effects at alternative splice sites
Alternative splicing can present challenges for variant effect prediction. Several of the tools tested here require gene model annotation, and their scores may be influenced by the inclusion or exclusion of nearby alternative isoforms in these annotations. In particular, SpliceAI and Pangolin use these annotations by default to apply a “mask” which suppresses scores from variants that either strengthen known splice sites or weaken unannotated splice sites, under the assumption that neither would be deleterious. Masking reduces the number of high-scoring variants genome-wide: among the background set, nearly one in every four splice-disruptive variants (deltaMax ≥0.2) identified without masking were suppressed (called neutral) by enabling it (Fig. 6A). Even without masking, annotation differences can introduce more subtle changes: among background set variants called by Pangolin as splice disruptive (absolute score ≥ 0.1), ~0.7% of variants called SDV with one annotation set (GENCODE) were called neutral by another (MANE Select), and vice versa (Fig. 6B). Therefore, while masking may be a necessary filter to reduce the number of variants for follow-up, it requires the provided annotation to be complete and further assumes there is no functional sensitivity to the relative balance among alternative splice forms.

Influence of masking and annotation choice. (A) Venn charts showing counts of background set variants (n=500,000) called as SDV only when masking is disabled (red), enabled (blue), or in both cases (overlap, purple) for SpliceAI and Pangolin. (B) Same for Pangolin, run with masking using two different annotation sets. C Tracks of SpliceAI scores (y-axis) for all SNVs in FGFR2 exon IIIc showing either score using masking and default annotation (upper panel) or scores with masking and only the FGFR2c isoform (lower panel) vs hg38 position (x-axis), formatted as in Fig. 2. Symbols denote known benign or pathogenic variants from ClinVar or published reports. A cluster of pathogenic exon IIIc acceptor disrupting variants is missed when annotation does not include exon IIIc (yellow region), and exon IIIc donor disrupting variants have intermediate scores (blue region). D SpliceAI masked scores perfectly separate known pathogenic and benign variants when exon IIIc is included but not using default annotations
We examined the effects of annotation choices and masking options at the two alternatively spliced exons in our benchmark variant set. In the first, POU1F1, two functionally distinct isoforms (alpha and beta) result from a pair of competing acceptors at exon 2. Alpha encodes a robust transactivator and normally accounts for ≥97% of POU1F1 expression in the human pituitary [62,63,64,65]. Beta exhibits dominant negative activity, and SDVs that increase its expression cause combined pituitary hormone deficiency [57, 66]. We focused on SpliceAI, in which the default annotation file includes only the alpha transcript. Predictions were broadly similar after updating annotations to include only the beta isoform or to include both: 13.8% (n=130/941) and 10.5% (n=99/941) of the variants, respectively, changed classifications compared to SpliceAI run with default annotations (each at an SDV cutoff of deltaMax≥0.08 which was optimal across that dataset; Additional file 1: Fig. S13). Among these were several pathogenic SDVs including c.143-5A>G which is associated with combined pituitary hormone deficiency (CPHD) [67], scored as highly disruptive by MPSA [57], and was validated in vivo by a mouse model [68]. With the default annotations (alpha isoform only) and when including both isoforms, SpliceAI scores c.143-5A>G as disruptive (deltaMax =0.21 and 0.16, respectively). However, when only the beta isoform is included, this variant is predicted neutral (deltaMax <0.001). A similar pattern emerged at a cluster of six pathogenic SDVs which disrupt a putative exonic splicing silencer which normally suppresses beta isoform expression [57]. Therefore, counterintuitively, pathogenic SDVs which act by increasing beta isoform usage go undetected when using annotation specific to that isoform.
The choice of canonical transcript may be less clear when alternative isoforms’ expression is more evenly balanced, as in the case of WT1, a key kidney and urogenital transcription factor gene [69] covered by our benchmarking set. Exon 9 of WT1 has two isoforms, KTS+ and KTS−, named for the additional three amino acids included when the downstream donor is used [70, 71]. In the healthy kidney, KTS+ and KTS− are expressed at a 2:1 ratio [72, 73]. Decreases in this ratio cause the rare glomerulopathy Frasier’s syndrome [72,73,74], while increases are associated with differences in sexual development (DSD) [75]. We ran SpliceAI using annotations including KTS+ alone (its default), KTS− alone, and with both isoforms (Additional file 1: Fig. S14). A cluster of variants, including one associated with DSD near the unannotated KTS− donor [75] (c.1437A>G), appear to weaken that donor but are masked because the KTS− donor is absent from the default annotations. Conversely, another variant (c.1447+3G>A) associated with DSD appears to increase the KTS+/KTS− ratio but is also masked because it strengthens the annotated KTS+ donor (deltaMax=0 with default annotation), and similarly scores as neutral when the annotation is updated to include both isoforms (deltaMax=0.02). That variant scores somewhat more highly (deltaMax=0.12) when only the KTS− annotation is used, but that in turn results in failure to capture several known Frasier’s Syndrome pathogenic variants near the KTS+ donor [58, 72, 73, 76,77,78,79]. This case illustrates that predictors can fail even when all functionally relevant isoforms are included, because masking may suppress SDVs for which the pathogenic effects result from strengthening of annotated splice sites and disrupting the balance between alternative isoforms. This challenge was not specific to SpliceAI; for instance, Pangolin also showed poor recovery of KTS− SDVs (only 25% correctly predicted) due to a similar masking operation.
POU1F1 and WT1 do not represent exceptional cases. Among RNA-seq junction usage data from the GTEx Consortium [65], we estimate 18.0% of all protein-coding genes (n=3571/19,817 genes) have at least one alternate splice site that is expressed and at least modestly used (≥20% PSI) in at least one tissue, yet is absent from SpliceAI default annotations (Additional file 1: Fig. S15). One of these is FGFR2, a tyrosine kinase gene with key roles in craniofacial development [80,81,82]. Mutually exclusive inclusion of its exons IIIb and IIIc results in two isoforms with different ligand specificities [80, 81, 83], and disruption of exon IIIc splicing causes Crouzon, Apert, and Pfeiffer Syndromes, which share overlap** features including craniosynostosis (premature cranial suture fusion) [84,85,86,87]. Pathogenic variants cluster near exon IIIc splice sites and at a synonymous site that activates cryptic donor usage within the exon [84, 86,87,88,89,90,91,92,93,94,95,96,97] (Fig. 6C). The default annotation excludes exon IIIc, causing all four pathogenic variants at its acceptor to be scored splice neutral, but when IIIc is included in the annotation, all four are predicted with high confidence (all ≥0.99; Fig. 6D, Additional file 2: Table S1). Disabling masking could capture cases such as this, but may not be a viable option in practice as reduces overall performance, and increases the number of high-scoring variants which must be reviewed [43].
