
Abstract
Background
Nanopore-based DNA sequencing relies on basecalling the electric current signal. Basecalling requires neural networks to achieve competitive accuracies. To improve sequencing accuracy further, new models are continuously proposed with new architectures. However, benchmarking is currently not standardized, and evaluation metrics and datasets used are defined on a per publication basis, impeding progress in the field. This makes it impossible to distinguish data from model driven improvements.
Results
To standardize the process of benchmarking, we unified existing benchmarking datasets and defined a rigorous set of evaluation metrics. We benchmarked the latest seven basecaller models by recreating and analyzing their neural network architectures. Our results show that overall Bonito’s architecture is the best for basecalling. We find, however, that species bias in training can have a large impact on performance. Our comprehensive evaluation of 90 novel architectures demonstrates that different models excel at reducing different types of errors and using recurrent neural networks (long short-term memory) and a conditional random field decoder are the main drivers of high performing models.
Conclusions
We believe that our work can facilitate the benchmarking of new basecaller tools and that the community can further expand on this work.
Similar content being viewed by others
Background
Sequencing of DNA (or RNA) can be achieved by translocating nucleic acids through a protein nanopore. By passing an electric current through the nanopore, a signal is measured that is representative for the chemical nature of the different nucleotides inside the pore. Therefore, capturing this current yields a signal that can be translated into a DNA sequence. In 2014, Oxford Nanopore Technologies (ONT) released the first commercial sequencing devices based on this principle.
Basecalling is the process of translating the raw current signal to a DNA sequence [1]. It is a fundamental step because almost all downstream applications depend on it [2]. Basecalling is a challenging task due to several reasons. First of all, the current signal level does not correspond to a single base but is most dominantly influenced by the several nucleotides that reside inside the pore at any given time. Secondly, DNA molecules do not translocate at a constant speed. Therefore, the number of signal measurements is not a good estimate of sequence length. Instead, the detection of signal changes is required to determine that the next base has entered the pore.
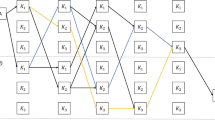
To address the basecalling challenge, a wide array of basecallers has been developed both by ONT and the wider scientific community. Basecallers evolved from statistical tests, to hidden Markov models (HMMs) and finally to the use of neural networks [3, 4]. Wick et al. (2019) benchmarked Chiron [5] and four other ONT basecallers that were being developed at the time: Albacore, Guppy, Scrappie, and Flappie. Chiron used developments in the speech to text translation field as it applied a convolutional neural network (CNN) to extract the features from the raw signal, a recurrent neural network (RNN) to relate such features in a temporal manner, and a connectionist temporal classification (CTC) decoder [6] to avoid having to segment the signal. Since then, many other basecallers have been developed and published by the community (Fig. 1a): Mincall [Full size image
Basecalling failure is a major determinant of reported performance
We first noted that the number of reads that failed basecalling varies substantially. Distinctively, Causacall, Halcyon, and Mincall only managed to properly basecall 66%, 79%, and 87% of the reads respectively, while the rest of the models managed > 90% (Fig. 2a). It is therefore critical to include such a metric in model evaluation since models could be skip** difficult to basecall reads, which would skew results towards a false higher performance.
Different methods prevail at different measures
We evaluated the performance of the different architectures using the alignment event rates (Fig. 2b). Bonito performed best in three out of the four metrics. It has the highest median match rate (90%) and the lowest median mismatch (2.5%) and deletion (4.3%) rates. Causalcall achieved the lowest median insertion rate (1.7%); however, it performed worst in the other three metrics with the lowest median match rate (77.6%) and highest mismatch (6%) and deletion (14.4%) rates. Halcyon shows the highest variation in performance rates, demonstrating that in addition to the median, the distribution across the reads is important to consider while comparing basecallers. It is therefore critical to not only report the error rates but also their distributions.
Homopolymer error rates are correlated with alignment event rates
Homopolymers are especially difficult to basecall because, for long stretches of the same base, the signal does not change, and since the DNA translocation speed is not constant, the number of measurements is not a good indicator of the length of the homopolymer. For such stretches of DNA, Bonito performed best with the lowest median error rate (14.9% averaged across all four bases) and the lowest median error rate for each base individually. Causalcall performed significantly worse than the rest of the models with the highest median error rate (44.5% averaged across bases) (Fig. 2c). We observed a performance correlation between alignment event rates and homopolymer error rates; however, the latter have a significantly higher error rate likely due to the inherent difficulty of basecalling such stretches.
Utility of PhredQ scores varies across methods
To evaluate the relationship between the predicted bases and their PhredQ scores. We first consider the distributions of the scores of the correct and incorrect bases (Fig. 2d). For all model architectures, correct bases have higher scores than incorrect bases; Causalcall has the smallest overlap between the two distributions (0.7%), followed by the rest of the models with similar overlaps (6–8%) except Halcyon (12%) and Bonito (32%). Secondly, we calculated AUCs by sorting the reads based on their average quality scores and determining the area under the normalized cumulative score (Fig. 2e). All architectures showed a correlation between read quality and average match rate. Not surprisingly, Bonito performed best with an AUC of 0.91. CATCaller and SACAll are, however, close contenders both with an AUC of 0.886. Importantly, each model has its own PhredQ score offset that determines how their quality scores are calibrated. As a result, quality scores across models, even when compared in a standardized benchmark, are not directly numerically comparable.
The signatures of basecalling errors
Finally, we evaluated the different types of mistakes in the context of the two neighboring bases in the basecalls (Additional file 1: Fig. S2). In general, these “error signatures” reveal that there are differences between the accuracy of the models depending on the predicted base context. Across all models, cytosine has low error rates (\(\approx\)10%) when predicted in CCT or TCT context; however, it can have significantly higher error rates (> 30%) when predicted in the context of the 3-mers TCC or TCG. We noticed that many of the contexts with higher error rates contain a CG motif, suggesting the increased error might be due to the potential methylation status of cytosine. To evaluate if specific models have particular error biases we performed hierarchical clustering on the pairwise Jensen-Shannon divergences between the error signatures (Additional file 1: Fig. S3). This revealed that Causalcall and Halcyon are the two most different models in terms of “error signatures”. The rest of the models have similar error profiles (lowest Pearson correlation coefficient between them: 0.95). We can conclude that basecalling errors are biased since they are not uniformly distributed across the 3-mer contexts. However, the error profiles are very similar between basecallers, suggesting that training data may play a stronger role in defining these error biases than the architecture of the model itself.
Architecture analysis
The benchmarking setup also allows straightforward investigation of which components of the neural networks provide the main performance gains. To this end, we created novel architectures by combining the convolution and encoder and decoder modules from existing basecallers, as well as some additional modules. We again used the human task to evaluate the different models. In total, ninety different models were evaluated and ranked based on the sum of rankings across all metrics (Fig. 3a, Additional file 1: Fig. S4). Out of the original models, Bonito again performs best but reached 9th place in the overall ranking. Consequently, our grid search reveals eight new model architectures that perform better in general. However, improvements in performance made by these models are small; for example, in comparison to the Bonito model, the alignment event rate improvements and homopolymer error rates are smaller than 1%, suggesting that we may be reaching the performance limits obtainable given the training data used.

Benchmark of architecture components. a Top 25 best performing model combinations. b Comparison of CTC to CRF decoder. c Comparison of simple (Bonito, CATCAller, SACall) to complex (Causalcall, Mincall, URNano) convolutions. d Comparison of bidirection LSTM depth (1, 3, or 5 layers). e Comparison of RNN to Transformer encoders
CRF decoder is vastly superior to CTC
We observed that most of the high performing models used the CRF decoder module. We therefore compared the change in performance between pairs of models whose only difference was the decoder (Fig. 3b, Additional file 1: Fig. S5). We see that for almost all models, using a CRF decoder leads to a general improvement of performance with a mean increase in match rate of 4% and mean decreases of mismatch, insertion, and deletion rates of 1%, 1%, and 2% respectively. Some exceptions are models that used the Mincall or URNano convolution modules, which have a mean increase in insertion rates of 1%, although their other alignment rate metrics still improve significantly. This is in concordance with our previous results, where Causalcall and URNano demonstrated the lowest insertion rates of all the models, showcasing that it is their convolutional architectures that boosts performance for this type of metric. Notably, a decrease in homopolymer error rates is also observed for the models with Mincall or URNano convolution modules that include a CRF decoder. However, results for the other models are more varied and depend on the base. Consistently with these improvements, we observe an average improvement of 3% in the AUCs. However, the PhredQ overlap between correct and incorrect predictions worsened with a median increase of 30%.
Complex convolutions are most robust, but simple convolutions are still very competitive
Another main architectural difference is the complexity and depth of the convolutional layers: ranging from two or three simple convolutional layers like in Bonito, CATCaller, and SACall, to more elaborate convolutional modules like Causalcall, URNano, or Mincall. We find that the top four ranked models use the URNano or Causalcall architecture (Fig. 3a). However, the six following models all use one of the simpler CNNs. More complex convolutional architectures perform better in general, specifically Causalcall and URNano (Fig. 3c, Additional file 1: Fig. S6). Simple convolutional architectures can also perform as good or even better, however they are more dependent on the encoder architecture that follows.
RNNs are superior to transformers and are depth dependent
Transformer layers have gained popularity in other fields due to increased performance and speed [Models Model architectures were recreated using Pytorch (v1.9.0) based on their description in their publications and their github repositories. If their implementation was done using Pytorch, code was reused as much as possible (Additional file 1: Fig. S13-27). Non-recurrent models (all except Halcyon) were trained for 5 epochs with a batch size of 64. All models were trained on the same task data which was also given as input in the same order. Models inital random parameters were initialized via a uniform distribution with values ranging from − 0.08 to 0.08. Reads were sliced in non-overlap** chunks of 2000 data points. Models were trained using an Adam optimizer (initial learning rate = \(1e^{-3}\), \(\beta _1\) = 0.9, \(\beta _2\) = 0.999, weight decay = 0). Learning rate was initially increased linearly for 5000 training steps from 0 to the initial learning rate of the optimizer as a warm-up; the learning rate was then decreased using a cosine function until the last training step to a minimum of \(1e^{-5}\). To improve model stability, gradients were clipped between − 2 and 2. Halcyon was trained similarly to non-recurrent models with the following differences: models were trained first for 1 epoch with non-overlap** chunks of 400 data points, then for 2 epochs with chunks of 1000 data points and finally for 2 epochs with chunks of 2000 data points. This was necessary because training directly using 2000 data points chunks led to unstable model training. This phenomenon is also described in the original Halcyon publication [17], requiting this transfer learning approach to ameliorate the issue. Recurrent models were also trained without warm-up and with a 0.75 scheduled sampling. During training, 5% of the training data was used for validation from which accuracy and loss were calculated without gradients. Validation data was the same for all models. The state of the model was saved every 20,000 training steps. The model state was chosen based on the best validation accuracy during training. Models were evaluated on hold out test data from the task being evaluated. URNano used cross-entropy as its loss; however, since the objective of the benchmark was basecalling and not signal segmentation, we used a CTC decoder instead. All the other models were recreated as stated in their respective publications; when in doubt, their github implementations were used as reference. Since Causalcall and Halcyon performed worse than the rest of the models, we evaluated the original models Causalcall, Halcyon, and Guppy model published by the authors and compared them against our PyTorch implementations (SAdditional file 1: Fig. S28, Additional file 1: Table S5). When evaluating the original Halcyon, we were unable to completely basecall all 25k reads in the test set due to memory limitations; we therefore compared our recreation based only on the \(\approx\)13k reads that were basecalled by the original Halcyon model from the human task test set. We used Guppy (v5.0.11, latest version) for the comparison between original models and our PyTorch implementations. In terms of reads that we consider evaluable, we see small differences (less than 3%) between the original versions and our implementations of Causalcall and Guppy. However, we see some differences in the types of failed reads between the original Causalcall, which has 7% more reads that failed map**, whereas our recreation had 3% more reads with short alignments. Surprisingly, basecalls from the original Halcyon produce only 46% of reads suitable for evaluation, (30% less than our recreation). A significant 34% of reads failed map** (30% more than our recreation) to the reference. There is also an increase, although smaller, on the 19% of reads that have short alignments to the reference (5% more than our recreation) (Additional file 1: Fig. S28a). Regarding our implementation of Guppy, we find that differences are small, with at most a 2% difference. We then looked at the alignment event rates (Additional file 1: Fig. S28b). Differences between the two Guppy models were very small, with the largest being a 1.3% difference in increased match rate from the original version. The original Causalcall showed improved match performance, with increased match (2%) and a decreased deletion (6%) rates; however, it showed a slight increase in mismatch (1%) and insertion (1%) rates as well as higher variability across reads. Finally, the original Halcyon performed worse in all metrics except deletion rate. However, its performances are less variable across reads. Homopolymer error rates show a similar trend (Additional file 1: Fig. S28c), the original Causalcall performs significantly better with a more similar to the other models average error rate (25.6%), while the other two models show very similar performances. We finally compared the models regarding their PhredQ scores: when comparing Bonito to Guppy, we saw a large difference in the scale of the scores (Additional file 1: Fig. S28d); however, Guppy still had an overlap between the distributions of 28%. On the other hand, the original Causalcall showed a significant increase in the overlap between distributions (48%). (Additional file 1: Fig. S28e). Correlating with the event rates results, the original versions of Causalcall and Guppy performed slightly better than our recreated counterparts with AUCs of 0.837 and 0.937 respectively. The original Halcyon does not report any PhredQ scores. With these results, we concluded that although there are some differences between the original models and our recreations, these are minor and could be attributed to training strategies and used data. Most models contain a convolutional module that later directly feeds into an encoder (recurrent/transformer) module. To be able to combine modules from different models without changing the original number of channels, we included a linear layer in between the convolution and encoder modules to up-scale or down-scale the number of channels. After this additional linear layer, we applied the last activation function of the preceding convolutional module. Contrary to the other models, the convolution modules from URNano and Causalcall do not reduce the amount of input timepoints. For those modules, we also included an extra convolution layer with the same configuration as the last convolution layer in Bonito (kernel size = 19, stride = 5, padding = 9). This layer had the same number of channels as the last convolutional layer of URNano or Causalcall. This convolution layer was necessary in order to both use transformer encoders and/or a CRF decoder due to memory requirements. We also included three non-used encoder architectures: either one, three or five RNN-LSTM bidirectional stacked layers with 256 channels each. Evaluation metrics are based on the alignment between the predicted sequence and the reference sequence. Alignment is done using Minimap2 (2.21) [29] with the ONT configuration for all metrics except accuracy. Accuracy is based on the Needleman-Wunsch global alignment algorithm implemented in Parasail (1.2.4) [30]. The global alignment is configured with a match score of 2, a mismatch penalty of 1, a gap opening penalty of 8 and a gap extension penalty of 4. Accuracy is used to evaluate the best performing state of the models during training based on the validation fraction of the data. During training, short sequences have to be aligned; however, during testing, complete reads have to be aligned, for which Minimap2 is necessary. Accuracy is defined as the number of matched bases in the alignment divided by the total number of bases in the reference sequence. Match, mismatch, insertion, and deletion rates are calculated as the number of events of each case divided by the length of the reference unless otherwise stated. Homopolymer regions are defined as consecutive sequences of the same base of length 5 or longer. Error rates on homopolymer regions are calculated by counting the number of homopolymers with errors (one or more mismatches, insertions, or deletions) and dividing it by the number of homopolymer bases. PhredQ scores are calculated using the fast_ctc_decode library from ONT. Average quality scores are calculated for all the correct and incorrect bases for each read. Differences between mean scores between correct and incorrect bases are reported. AUCs are calculated by sorting the basecalled reads according to their mean Phred quality score and calculating the average match rate for cumulative fraction of reads in steps of 50. Error profiles are calculated for all 3-mers by counting the number of events (mismatches for each base, insertions and deletions) in the context of the two neighboring bases of the event itself according to the basecalls. Rates for each event are calculated by dividing each event count by the total number of 3-mer occurrences in the read. Error profiles are also calculated for each base, independently of their context. Randomness of error is defined as the Jensen-Shannon divergence between each 3-mer error profile and their corresponding base error profile. Packages and their versions used for training and evaluation can be found on (Additional file 1: Table S6). All analysis were run on Python 3.7.8 and CUDA version 10.2. We used the following hardware requirements: 32 CPU cores and 64Gb of RAM (data processing and model performance evaluation); it is possible to reduce these requirements at the expense of longer compute time; 4 CPU cores, 128Gb of RAM, and 1 NVIDIA RTX6000 GPU (model training basecalling).Model training
Original model recreation and benchmark
Comparison of original models to model recreations
Architecture analysis
Evaluation metrics
Accuracy
Alignment rates
Homopolymer error rates
PhredQ scoring
Error profiles
Software and hardware requirements
