
Abstract
Background
Intrahepatic cholangiocarcinoma (ICCA) is a heterogeneous group of malignant tumors characterized by high recurrence rate and poor prognosis. Heterochromatin Protein 1α (HP1α) is one of the most important nonhistone chromosomal proteins involved in transcriptional silencing via heterochromatin formation and structural maintenance. The effect of HP1α on the progression of ICCA remained unclear.
Methods
The effect on the proliferation of ICCA was detected by experiments in two cell lines and two ICCA mouse models. The interaction between HP1α and Histone Deacetylase 1 (HDAC1) was determined using Electrospray Ionization Mass Spectrometry (ESI-MS) and the binding mechanism was studied using immunoprecipitation assays (co-IP). The target gene was screened out by RNA sequencing (RNA-seq). The occupation of DNA binding proteins and histone modifications were predicted by bioinformatic methods and evaluated by Cleavage Under Targets and Tagmentation (CUT & Tag) and Chromatin immunoprecipitation (ChIP).
Results
HP1α was upregulated in intrahepatic cholangiocarcinoma (ICCA) tissues and regulated the proliferation of ICCA cells by inhibiting the interferon pathway in a Signal Transducer and Activator of Transcription 1 (STAT1)-dependent manner. Mechanistically, STAT1 is transcriptionally regulated by the HP1α-HDAC1 complex directly and epigenetically via promoter binding and changes in different histone modifications, as validated by high-throughput sequencing. Broad-spectrum HDAC inhibitor (HDACi) activates the interferon pathway and inhibits the proliferation of ICCA cells by downregulating HP1α and targeting the heterodimer. Broad-spectrum HDACi plus interferon preparation regimen was found to improve the antiproliferative effects and delay ICCA development in vivo and in vitro, which took advantage of basal activation as well as direct activation of the interferon pathway. HP1α participates in mediating the cellular resistance to both agents.
Conclusions
HP1α-HDAC1 complex influences interferon pathway activation by directly and epigenetically regulating STAT1 in transcriptional level. The broad-spectrum HDACi plus interferon preparation regimen inhibits ICCA development, providing feasible strategies for ICCA treatment. Targeting the HP1α-HDAC1-STAT1 axis is a possible strategy for treating ICCA, especially HP1α-positive cases.
Similar content being viewed by others

Introduction
Based on the anatomical location, cholangiocarcinoma is divided into four types: intrahepatic, perihilar, and extrahepatic cholangiocarcinoma (ICCA, PCCA and ECCA) and gallbladder cancer (GBC). ICCA is a heterogeneous group of malignant tumors comprising approximately 20% of all cholangiocarcinoma and is characterized by a high degree of malignancy and atypical early symptoms. The incidence of ICCA is relatively high in certain endemic areas with a higher incidence of inflammatory biliary diseases, and the incidence rate is increasing yearly [1]. Owing to the late-stage presentation, resistance to comprehensive treatments, and extremely high rate of postsurgical recurrence, the 5-year overall survival rate of ICCA is lower than 10% [2]. Therefore, the identification of novel molecular targets is critical.
Heterochromatin Protein 1α (HP1α, encoded by the CBX5 gene) is one of the most important HP1 family members and is a non-histone chromosomal protein involved in transcriptional silencing via heterochromatin formation and structural maintenance [3]. Many studies have shown that the distribution of HP1α on polytene chromosomes is not restricted to the chromocenters or telomeres. HP1α binds to chromatin mainly through direct interactions with modified histones, especially trimethylated H3K9 (H3K9me3), through the Chromo domain, and by interacting with other proteins through the Chromo shadow domain [4]. In cancerous lesions, HP1α is involved in the regulation of malignant behaviors, such as cell proliferation and cell cycle progression. Downregulation of HP1α has been demonstrated to inhibit the malignant biological behaviors of lung cancer, cervical cancer, and prostate cancer cells, for example, by impairing proliferation and inducing apoptosis [5,6,7]. Previously, we found that downregulation of Dicer and CyclinD1, which are binding proteins of HP1α, inhibited the proliferation of ICCA cells [8, 19,20]. Detailed information is provided in Table S1. The expression dataset of CCA in The Cancer Genome Atlas (TCGA) was downloaded from the UCSC Xena database. In this study, the fold-change values were obtained by logarithmic (log2FC) transformation. False-positive results were avoided by calculating of adjusted P values (adj. P) values using the Benjamini-Hochberg procedure. The differentially expressed genes (DEGs) were defined with the following cutoff values: adj. P < 0.05 and |log2FC| > 0.8. The tools and databases used for the bioinformatics analysis are summarized in Table S2 [21,22,23,24,25,26,27,28,29].
Cell culture
Human ICCA cell lines (HUCCT1, HCCC-9810, RBE, HUH28, and SSP-25) were maintained in our laboratory and cultured in RPMI-1640 medium. The kidney cell line, HEK-293T, was maintained in our laboratory and cultured in DMEM. All cell culture media were supplemented with 10% fetal bovine serum (FBS), 100 U/mL penicillin, and 100 µg/mL streptomycin at 37 °C in a humidified incubator with 5% CO2. The mycoplasma was detected by colorimetric method with Mycolor One-Step Mycoplasma Detector (Vazyme, China) according to the manufacturer’s instructions.
Tissue microarrays, immunohistochemical (IHC) analysis and HE staining
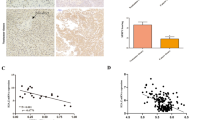
The difference in HP1α expression between CCA and para-cancerous tissues was evaluated using a tissue microarray purchased from Outdo Biotech, China, containing 36 CCA samples and nine para-cancerous tissue samples. Additionally, 40 surgical ICCA and 40 paired para-cancerous tissues used for IHC and 90 surgical ICCA and 90 paired para-cancerous tissues used for real-time quantitative PCR (RT-qPCR) were collected from the Department of Biliary-Pancreatic Surgery, Tongji Hospital of Huazhong University of Science and Technology, Wuhan, China. The follow-up of these 90 ICCA patients was performed after surgery, and the date of death or last follow-up was recorded. All the research was conducted in accordance with both the Declarations of Helsinki and Istanbul. The experimental protocols were approved by the Ethics Committee of Tongji Hospital of Huazhong University of Science and Technology (#TJ-IRB20230927). All patients signed informed consent forms.
The tissue samples were fixed with 4% polyoxymethylene and subjected to embedding and sectioning at 5 μm thickness. For HE staining, the sections were sequentially stained with hematoxylin and eosin. For IHC analysis, samples were deparaffinized, rehydrated and subjected to antigen retrieval. Then, the samples were blocked with 5% bovine serum albumin (BSA) prior to overnight incubation with primary antibodies at 4 °C (listed in Table S3). The next day, tissue samples were incubated with horseradish peroxidase (HRP)-conjugated secondary antibodies prior to DAB and hematoxylin staining. The intensity of staining (0, negative; 1, weak; 2, moderate; and 3, strong) and the percentage of positive cells (0, 0%; 1, 1–25%; 2, 26-50%; 3, 51–75%; and 4, 76-100%) were evaluated in a blinded manner. Finally, the IHC score was calculated (IHC score = staining intensity × percentage of positive cells).
Reagents and siRNA transfection
The inhibitors used in this study were as follows: Trichostatin A (TSA; HY-15144, 0.5 µM, 24 h), PF-06700841 (HY-112708, 50 nM, 24 h), decitabine (HY-A0004, 5 µM, 48 h), and GSK-J1 (HY-15648, 10 µM, 48 h) were purchased from MedChemExpress, China. Valproic acid (S1168, 1 µM, 48 h), Santacruzamate A (S7595, 20 µM, 48 h) and ACY-775 (S0864, 5 µM, 48 h) were purchased from Selleck, China. Human IFN-α2b (CYT-205, 60 ng/mL, 24 h) was purchased from Prospec (Israel). ICCA cells were transfected with siRNA (50 nM) and nonsense siRNA (50 nM) with Lipofectamine 2000 (Thermo Fisher Scientific, USA) following the manufacturer’s instructions. The siRNA constructs were synthesized by Sangon Biotech (China), and all the sequences are listed in Table S4.
Plasmid construction and lentiviral transduction
Full-length and truncated sequences of Tripartite Motif Containing 28 (TRIM28), HDAC1 and CBX5, namely, full-length HDAC1 (Flag tagged, NM_004964.3), full-length CBX5 (HA tagged, NM_001127322.1), full-length TRIM28 (Flag tagged, NM_005762.3), truncated HDAC1 (lacking the histone deacetylase or disordered region) and truncated CBX5 (lacking the chromo domain or shadow domain), were generated by PCR and were inserted separately into the plasmid vector pHAGE-puro. Gene-specific small hairpin RNAs were synthesized by Sangon Biotech and were inserted into the pLKO.1-puro vector. HEK293T cells were transfected with the vectors mentioned above, along with the envelope plasmid pMD2.G and the packaging plasmid psPAX. The supernatant was filtered and collected after three days. ICCA cells were infected with the specific lentiviruses in the presence of HiTransG (1:25, GeneChem, China) for 24 h and then treated with 1 µg/ml puromycin to establish stable cell lines.
RNA extraction, RT‒qPCR and RNA sequencing (RNA-seq)
Total RNA from cell and tissue samples was extracted with RNA Extraction Reagent (Vazyme) and reverse transcribed into cDNA using HiScript III RT SuperMix for qPCR (+ gDNA Wiper) (Vazyme) according to the manufacturer’s instructions. RT-qPCR was performed using Hieff® qPCR SYBR Green Master Mix (No ROX, Yeasen, China) and specific primers (Table S4) in an iQ5™ quantitative PCR system (Bio-Rad, USA). Expression levels were calculated using the 2 − ΔΔCt method. Primers were synthesized by Sangon Biotech.
Total RNA samples were used for RNA-seq performed by HaploX (China). Quality was evaluated using a 4200 TapeStation system (Agilent, USA) and quantified using a Life Invitrogen Qubit 3.0 fluorometer (Thermo Fisher Scientific). The samples were used for library preparation, followed by RNA-seq by Illumina PE150 sequencing (Illumina, USA). The raw data were used for further bioinformatics analysis.
Western blot
Fresh cell samples were lysed with RIPA buffer in the presence of proteinase inhibitor cocktail and PhosSTOP phosphatase inhibitor (Roche, Switzerland). Total protein was extracted, mixed with 5× loading buffer, and denatured at 95 °C for 5 min. Proteins in individual lysate samples (30 µg per lane) were separated by sodium dodecyl sulfate‒polyacrylamide gel electrophoresis and transferred to nitrocellulose membranes (Millipore, USA). Then, the membranes were blocked with 5% BSA and incubated with primary antibodies overnight at 4 °C. The next day, the membranes were incubated with secondary antibodies, and the signals were visualized using enhanced chemiluminescence (Thermo Fisher Scientific). The results were analyzed using Image Lab software (Bio-Rad).
Purification and isolation of nuclear and cytoplasmic proteins
Nuclear and cytoplasmic proteins were purified and isolated with Nuclear and Cytoplasmic Protein Extraction Kit (Beyotime, China). Briefly, ICCA cells were mixed with cytoplasmic extraction buffer A followed by vigorous vortexing and incubation in an ice bath. Cytoplasmic extraction buffer B was then added, followed by vigorous vortexing and centrifugation. The supernatant was collected as a cytoplasmic sample. The precipitate was mixed with nuclear extraction buffer, followed by vigorous vortexing, incubation in an ice bath, and centrifugation. The supernatant was collected as a nuclear sample for further analysis.
Coimmunoprecipitation (co-IP) and Electrospray Ionization Mass Spectrometry (ESI-MS)
For the exogenous co-IP assay, HEK293T cells were transfected with expression vectors carrying full-length or truncated coding sequences. For the endogenous co-IP assay, wild-type ICCA cells were cultured without any specific treatment. The cells were harvested and lysed with IP buffer (20 mM Tris-HCl [pH 7.4], 150 mM NaCl, 1 mM EDTA, and 1% NP-40) in the presence of proteinase inhibitor cocktail and PhosSTOP. The samples were subjected to ultrasonication and centrifugation. 5% of the supernatant was collected as the input sample, and the remaining supernatant was incubated with primary antibodies and protein A + G agarose beads (Med Chem Express). The next day, the agarose beads were washed with IP buffer and heated to 95 °C in 2× loading buffer as the IP sample. All the protein samples were used for western blotting analysis. The IP samples of HEK293T cells transfected with the HP1α expression vectors and control vectors were used for ESI-MS analysis. The analysis was performed by Applied Protein Technology (China).
Enzyme linked Immunosorbent Assay (ELISA)
The supernatants of ICCA cells were collected without dilution. The concentrations of IFN-α and IFN-γ were evaluated using a Human IFNA1 ELISA Kit (KE00044, Proteintech, China) and a Human IFN-gamma ELISA Kit (RK00015, ABclonal, China) according to the manufacturer’s instructions. The supernatant and the reaction mixture were incubated at 37 °C, and the absorbance at 450 nm was measured using a MULTISKAN FC microplate reader (Bio-Rad).
Cleavage under targets and tagmentation (CUT & tag)
A Hyperactive Universal CUT&Tag Assay Kit for Illumina (Vazyme) was used to evaluate the genomic occupancy of DNA binding proteins. Briefly, 1 × 105 ICCA cells were collected and the nuclei were extracted. Specific DNA fragments were obtained as previously described [Full size image
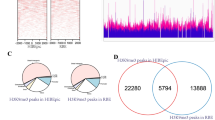
We failed to detect binding between HP1α and STAT1 (Figure S3C); therefore, we explored the interaction between HP1α and the STAT1 promoter region. We failed to detect any difference in luciferase activity upon HP1α overexpression, although insertion of the promoter region (1000 bp to the TSS) significantly enhanced transcription (Figure S3D). Similarly, TSA application failed to influence the transcriptional activity mediated by the STAT1 promoter, which is inconsistent with the observations in ICCA cells (Figure S3E), indicating that indirect binding to chromatin, for example, interactions via histone modifications, should be examined. Although an interaction between HP1α and H3K9me3 was observed, no binding to H3K27ac or H3K9ac was observed (Fig. 4F and S3F). To explore the distribution in local chromatin, we calculated the number of sites in each section. Most of the sites were located in the 700–1000 bp region (Figure S3G). CUT & Tag was performed to evaluate the occupancy of HP1α and histone markers (Fig. 5G, S3H). The binding sequences of the different markers are summarized in Table S8. The sequence of the HP1α binding sites was similar to that of H3K9me3, rather than acetylated histones. Most peaks in the promoters were located upstream of the 1000 bp. Except for H3K27me3, all targets exhibited peaks in the promoter region of STAT1, which was similar to the results obtained in the GEO database (Figure S3I, S3J). To extend this conclusion, we screened for all genes that contained four peaks of histone marks in the same regions (9063 genes, Figure S3K, S3L). Among these 9063 genes, 5871 contained HP1α peaks, and some genes were involved in cell cycle regulation and viral infection (Figure S3M). Among these 5871 genes, 1157 genes were significantly regulated by TSA and were highly correlated with cell cycle regulation (Figure S3N).
HP1α was found to be significantly enriched in the 700–1000 bp region of the STAT1 promoter compared to other proximal sites (Fig. 5H). The binding of HP1α to this region was inhibited by deletion of the Chromo domain or by treatment with TSA (Fig. 5I). Similarly, HDAC1 could bind to this region, and this binding was inhibited by deletion of the Histone Deacetylase domain or treatment with TSA (Fig. 5J). Additionally, only increases in H3K27ac and decreases in H3K9me3 were observed in this region after HP1α knockdown (Fig. 5K). Both H3K27ac and H3K9me3 were found to be enriched in the promoters of certain genes involved in infection and cell cycle (Figure S4A, S4B). ChIP was performed to evaluate the effect of the HP1α-HDAC1 complex on the local distribution of the two histone markers. Once either HP1α or HDAC1 was knocked down, we observed higher levels of H3K27ac and lower levels of H3K9me3, which could be partly rescued by overexpression of the other parts (Fig. 5L and M). Considering this evidence, we suggest that the HP1α-HDAC1 complex regulates the basal activation of IFN signaling by transcriptionally downregulating STAT1.
The proliferation of ICCA cells was inhibited by directly activating IFN signaling pathway
To evaluate the effect of the IFN signaling pathway, IFN-α2b, a cytokine preparation that can directly and potently activate IFN signaling, was used. We attempted to determine the optimal concentration, and 60 ng/mL was selected because higher concentrations had limited inhibitory effects (Fig. 6A). HUCCT1 cells were treated and RNA-seq was performed. We found that certain ISGs were upregulated (Fig. 6B). Among the 5871 genes mentioned above, 161 were regulated by IFN-α2b, some of which were involved in IFN signaling and translation initiation (Figure S3O).

The proliferation of ICCA cells is inhibited by directly activating IFN signaling pathway. A. The effect of various concentrations of IFN-α2b on the proliferation of ICCA cells evaluated by CCK-8 assay. B. The heatmap of the RNA-seq data of IFN-α2b-treated ICCA cells. The gradual change from red to blue represents changes in gene expression from high to low. C. Western blot analysis of HP1α, IFN signaling participants and proliferation and cell cycle-promoting factors upon IFN-α2b treatment and HP1α overexpression. D. RT-qPCR analysis of the expression of HP1α, STAT1 and ISGs upon IFN-α2b treatment and HP1α overexpression. E. Analysis of the expression level of intranuclear STAT1 and p-STAT1 upon IFN-α2b treatment and HP1α overexpression by Western blot. F. The effect of IFN-α2b treatment and HP1α overexpression on the distribution of cell cycle of ICCA cells. G-I. The result of proliferation assay. G. Colony formation assay. H. CCK-8 assay over 24 h. I. CCK-8 assay within 1 h. J. RT-qPCR analysis of the expression of HP1α and IFN signaling participants at different point of time. K. Western blot analysis of the expression and the activation level of STAT1 at different point of time. L, M. Effect of drug treatment on the distribution of H3K9me3 and H3K27ac in the STAT1 promoter. L. TSA treatment. M. IFN-α2b treatment. *P < 0.05; **P < 0.01
IFN-α2b significantly induced the expression of STAT1, p-STAT1, ISG15, SAMHD1, and MX1 and downregulated the protein factors (Fig. 6C and D). HP1α is not regulated by IFN-α2b. IFN-α2b treatment induced synchronous upregulation of intranuclear STAT1 and p-STAT1 (Fig. 6E). IFN-α2b inhibited proliferation and induced S-phase arrest (Fig. 6F and H, S3P). HP1α overexpression partially reversed these effects. Notably, an inhibitory effect was observed as early as 24 h after treatment (Fig. 6H). In contrast, no similar effect was observed within 1 h (Fig. 6I). To investigate this possibility, we examined the expression levels of the related molecules at different time points. Although all of these factors, except for STAT1, were upregulated before the 1 h time point, their expression was upregulated more drastically at 24 h and gradually decreased thereafter (Fig. 6J). However, HP1α expression was not affected. The protein levels of p-STAT1, but not total STAT1, increased within 1 h. Up to 24 h, the levels of both total STAT1 and p-STAT1 were elevated (Fig. 6K). IFN-α2b and TSA induced a significant increase in H3K27ac levels in the STAT1 promoter. TSA also decreased the local H3K9me3 levels (Fig. 6L and M). It could be inferred that increased total STAT1, rather than increased p-STAT1, is the underlying cause of proliferation inhibition, which is consistent with the finding that the HP1α-STAT1 axis regulates ICCA cell proliferation (Fig. 3).
Broad-spectrum HDACi plus IFN preparation regimen improves the antiproliferation effects and inhibits the development of ICCA
Proliferation assays were performed to evaluate the effects of broad-spectrum HDACi and IFN preparations. Compared with IFN-α2b alone, TSA plus IFN-α2b further inhibited the proliferation of ICCA cells (Fig. 7A and B, S4C). When TSA was applied with IFN-α2b-treated cells, we observed G1/G0-phase arrest, which was similar to the recruitment effect of chemotherapy regimens (Fig. 7C). TSA plus IFN-α2b further downregulated proliferation and cell cycle factors (Fig. 7D).

Broad-spectrum HDACi plus IFN preparation regimen maximizes the antiproliferation effects on ICCA. A, B. Evaluation of the proliferation inhibition induced by the TSA plus IFN-α2b regimen. (A) CCK-8 assay. (B) Colony formation assay. Blank, solvent for IFN-α2b, and murine IFN-α2. C. The distribution of cell cycle of ICCA cells. D. Western blot analysis of HP1α and proliferation and cell cycle-promoting factors. E. RT-qPCR analysis of the effect of TSA plus IFN-α2b regimen on ISG expression. Red, upregulated by TSA. Blue, downregulated by TSA. Green, failed to be significantly regulated by TSA. F. Presentation of liver lesions induced by hydrodynamic tail vein injection in the different treatment groups. Solvent, the recommended solvent for TSA in animal experiments. G. Scheme of the animal experiment. (1) Blank + Solvent; (2) IFN-α2 + Solvent; (3) IFN-α2 + TSA. One week after the hydrodynamic tail vein injection, different treatments were applied for two weeks. The survival status of mice was then assessed. Blue arrow, Solvent. Orange arrow, TSA. Green arrow, Blank. Red arrow, murine IFN-α2. H-K. Evaluation of the severity of tumorigenesis and the effect of the regimen. H. Ratio of liver weight to total weight. I. Ascites volume. J. ICCA lesions areas. K. Ki-67 positivity rate in the liver lesions. L. Survival analysis of the mice in different treatment groups. M. Presentation of the subcutaneous tumors. N, O. Volume (N) and Ki-67 positive rate of subcutaneous tumors (O) after different treatments. P. Graphic illustration of the HP1α-HDAC1-STAT1 regulatory axis in ICCA. STAT1 is directly and epigenetically repressed by the HP1α-HDAC1 complex. Treatment with TSA plus IFN significantly inhibited the proliferation of ICCA cells by targeting the HP1α-HDAC1 complex, increasing the total STAT1 level, downregulating HP1α, increasing basal IFN signaling, and directly stimulating the IFN pathway. *P < 0.05. **P < 0.01
Given that both reagents could induce IFN signaling, we screened for common targets using RNA-seq data of both TSA and IFN-α2b, for example, ISGs (Figure S4D, Table S9). As previously reported [37], TSA abrogated IFN-α2b-mediated upregulation of some ISGs that were either downregulated or unaffected by TSA (Fig. 7E). STAT1 and three ISGs, including SAMHD1 and ISG15, were further upregulated by TSA, which was consistent with the RNA-seq results. The upregulation of some ISGs (including MX1) was not reversed by TSA treatment (Fig. 6E). CD274 and PDCD1LG2 (encoding PD-L1 and PD-L2, respectively) were used as the positive controls as both genes were upregulated upon IFN stimulations [38]. HP1α knockdown downregulated the mRNA level of CD274 and PDCD1LG2 (Figure S4E). IFN-α2b induced the transcription of both genes, which was slightly rescued by TSA treatment (Figure S4F). The mRNA level of CD274 was positively correlated with that of HP1α in ICCA samples (Figure S4G). When HP1α was knocked down, CD274 was then downregulated (Figure S4H). IFN-α2b upregulated the expression of PD-L1, while TSA downregulated PD-L1 whether IFN-α2b was applied or not (Figure S4H).
This combination has been validated in a mouse model. We established ICCA models and found that although IFN-α2 alone ameliorated the progression of ICCA, the additional application of TSA further inhibited this oncogenic process. This therapeutic effect was abrogated by Hp1α overexpression (Fig. 7F and G, and S4I). Neither the TSA plus IFN-α2 regimen nor IFN-α2 monotherapy led to morphological changes in the kidneys (Figure S4J). All ICCA lesions were confirmed to be of an epithelial origin (Figure S4K). The TSA plus IFN-α2 regimen further reduced the weight and area of hepatic lesions and the rate of Ki-67 positivity, yet the volume of ascites was not influenced. Hp1α-overexpressing tumors were more severe and insensitive to TSA and IFN-α2 (Fig. 7H and K, S4L). Murine Cd274 was significantly upregulated upon Hp1α overexpression, which was similar to the findings in ICCA samples (Figure S4M). In addition, the TSA plus IFN-α2 regimen further improved the prognosis of the mouse model, which was not observed in the Hp1α overexpression group (Fig. 7L). Additionally, TSA plus IFN-α2 further reduced the volume of subcutaneous tumors and the rate of Ki-67 positivity (Fig. 7M and O, S4N).
In conclusion, the following mechanism is proposed. Although minimal IFN signaling is maintained, STAT1 is repressed by the HP1α-HDAC1 complex in a direct and epigenetic manner. Treatment with broad-spectrum HDACi plus IFN significantly inhibited the proliferation of ICCA cells by increasing the total STAT1 level, downregulating HP1α, increasing basal IFN signaling, and directly stimulating the IFN pathway (Fig. 7P).
