
Abstract
Despite improvements in outcome, 15-25% of newly diagnosed multiple myeloma (MM) patients have treatment resistant high-risk (HR) disease with a poor survival. The lack of a genetic basis for HR has focused attention on the role played by epigenetic changes. Aberrant expression and somatic mutations affecting genes involved in the regulation of tri-methylation of the lysine (K) 27 on histone 3 H3 (H3K27me3) are common in cancer. H3K27me3 is catalyzed by EZH2, the catalytic subunit of the Polycomb Repressive Complex 2 (PRC2). The deregulation of H3K27me3 has been shown to be involved in oncogenic transformation and tumor progression in a variety of hematological malignancies including MM. Recently we have shown that aberrant overexpression of the PRC2 subunit PHD Finger Protein 19 (PHF19) is the most significant overall contributor to HR status further focusing attention on the role played by epigenetic change in MM. By modulating both the PRC2/EZH2 catalytic activity and recruitment, PHF19 regulates the expression of key genes involved in cell growth and differentiation. Here we review the expression, regulation and function of PHF19 both in normal and the pathological contexts of solid cancers and MM. We present evidence that strongly implicates PHF19 in the regulation of genes important in cell cycle and the genetic stability of MM cells making it highly relevant to HR MM behavior. A detailed understanding of the normal and pathological functions of PHF19 will allow us to design therapeutic strategies able to target aggressive subsets of MM.
Similar content being viewed by others

Background
Multiple myeloma (MM) is a hematopoietic malignancy of terminally differentiated plasma cells (PC) [1]. Multiple myeloma comprises 1.5% of all malignant diseases and accounts for 10% of all hematologic malignancies [2]. Despite the advent of novel therapies, MM remains difficult to treat and contributes to 20% of the deaths from hematologic malignancy. Recurrent relapses and increasingly aggressive drug resistant disease that grows outside the marrow cavity are typical features of MM progression [1]. The clinical stage of MM is preceded by a premalignant expansion of clonal plasma cells, recognized clinically as monoclonal gammopathy of undetermined significance (MGUS) and/or smoldering MM (SMM) and that have been shown to transform into MM, plasma cell leukemia (PCL) or extramedullary disease over time [3]. On average, 1% of cases of MGUS and 5-7% of SMM transform to MM annually; rates that are greater for higher-risk subgroups. At presentation, 15-25% of MM cases have high-risk (HR) disease that is associated with early relapse and high mortality rates. The percentage of patients with HR features increases at each relapse constituting a major therapeutic challenge. Although genetic features associated with HR have been identified none explain the majority of disease and epigenetic modifications including DNA methylation, chromatin accessibility, and histone modifications seem to play an important role and may also be therapeutically tractable [4,5,6].
Epigenetic variables include post transcriptional modification of histones that play an important role in the regulation of genes expression by recruiting transcription factors (TFs) and affecting chromosome structure and function [7, 8]. These modifications include phosphorylation on serine or threonine residues, methylation on lysine or arginine together with other modifications such as acetylation, ubiquitylation or sumoylation. Histone methylation is mediated by specific methyltransferases that add methyl groups to lysine (mono-, di-, and trimethyl), glutamate, or arginine residue. The tri-methylation of the lysine (K) 27 on histone 3 H3 (H3K27) is a critical determinant of chromatin accessibility required for gene expression and is regulated by the proteins of the polycomb group (PcG) [7, 8]. These polycomb proteins are an evolutionarily conserved group that regulate gene expression through histone modification brought about by their interaction to form chromatin-associated repressive complexes: Polycomb-repressive complex 1 (PRC1) is responsible for the ubiquitylation of the lysine 119 on histone 2A H2AK119ub1, and PRC2, catalyzes the methylation of H3K27.
Aberrant expression and somatic mutations affecting genes involved in the regulation of H3K27me3 deposition/removal are common in cancer. The deregulation of H3K27me3 has been shown to be involved in oncogenic transformation and tumor progression in a variety of hematological malignancies including MM [9,10,11]. In both MGUS and MM, PRC2 target genes, identified by their H3K27me3 marks, have been shown to decrease their level of expression as disease progresses [5, 6]. PRC2-mediated gene silencing as a mechanism of gene repression during MM progression has also been highlighted by genome-wide profiling of H3K27me3 marks combined with RNA-Seq [12]. This analysis identified increased silencing of H3K27me3 targets in MM patients at advanced stages of the disease and the expression of H3K27me3-marked genes correlated with poor patient survival [12]. Pharmacological inhibition of Enhancer of Zeste Homolog (EZH) 2 has anti-myeloma effects in both MM cell lines and CD138+ MM patient cells [1, 12]. These studies suggest that the mechanisms that control PRC2/EZH2 activity might serve as a novel therapeutic target for MM.
Polycomb repressive complex 2 (PRC2) is a multi-subunit epigenetic protein complex that regulates gene expression by catalyzing mono-, di- and tri-methylation of histone H3 on lysine 27 (H3K27me1, H3K27me2, and H3K27me3, respectively) and plays a role in safeguarding cellular identity by ensuring gene silencing appropriate for the cellular function at that stage of development [7, 8, 13]. PRC2 is comprised of the core subunits EZH1 or EZH2, Embryonic ectoderm development (EED) and Suppressor of zeste 12 (SUZ12), and RBBP4/7 [7, 8]. EZH1 and EZH2 are the catalytic subunits of PRC2 and are mutually exclusive within the complex retaining distinct enzymatic properties in-vitro, with EZH2 having higher methyltransferase efficiency under the same reaction conditions [14]. PRC2 activity can be modulated by different accessory subunits including PHF1, MTF2, PHF19, PALI1, EPOP, JARID2 and AEBP2 [7, 8]. Some of these accessory subunits display cell type–specific expression patterns, and some exist within a subset of the PRC2 complex. They are not essential for the basal activity of PRC2 but may direct PRC2 recruitment or modulate PRC2 activity under specific conditions [7, 8]. Recently, we have shown that aberrant overexpression of the PRC2 subunit PHF19 is the most significant overall contributor to HR status focusing attention on the role played by epigenetic change and PRC2 behavior in aggressive clinical states of MM [15, 16]. In this review, we discuss the current knowledge on the regulation of PRC2/EZH2 activity by PHF19, its biological impact in MM and the potential for anti-PHF19 targeted therapy.
Structure and function of PHF19 protein
The core of the PRC2 complex can interact with three paralogues of the Drosophila polycomb-like gene, Plant Homeodomain (PHD) Finger Protein (PHF)1/PCL1, MTF2/PCL2 and PHF19/PCL3 that modulate PRC2 enzymatic activities and recruitment to target genes important for development and differentiation [17,18,19,20,21].
The human PHF19 locus is located in chromosome 9q33.2 and was initially identified near a retroviral integration site in an immortalized human fibroblast cell line [22]. It has been suggested that the gene is expressed as multiple, alternatively spliced mRNAs with at least three different 5′ ends and that two types of mRNA are derived from the PHF19 gene encoding short (PHF19S, 207 aa) and long (PHF19L, 580 aa) isoforms that share 155 amino acids at their N-termini [22] (Fig. 1A). The Ensembl database reports 14 transcripts, three retained-intron transcripts, three processed transcripts and eight protein coding transcripts (Supplementary Table 1). The NCBI database reports 18 predicted transcripts produced either by NCBI’s genome annotation pipeline or copied from computationally annotated submissions to the International Nucleotide Sequence Database Collaboration (INSDC). However, subsequently curated RefSeq NCBI records report five protein coding transcripts (Supplementary Table 1). Human PHF19S mRNA is one out of 23 human gene mRNA that contain a ‘readthrough’ stop codon (these mRNAs have a UGA stop codon immediately followed by CUAG) [25]. This means that the ribosomes translating the PHF19S mRNA decode its UGA stop codon as a sense codon, thus extending PHF19S at its C terminus by an additional 156 amino acids to generate a protein of approximately 40 kDa. The readthrough efficiency for PHF19S mRNA was estimated to be around 2% in HEK293 cell line [25] but no data are available in cancers. The Human Protein Atlas database reports eight PHF19 isoforms (Supplementary Table 1). Cellular localization analysis of PHF19L and PHF19S isoforms showed that the long isoform is exclusively nuclear and that the short isoform is localized in both cytosol and the nucleus [22, 26]. Two putative nuclear localization signals are predicted to be located at amino acids 387–397 and 473–489 of the long isoform [22, 26].

Human PHF19 isoforms and Top 20 TFs and chromatin regulators that regulate PHF19 expression in cancer. A Schematic representation of PHF19 locus, gene and isoforms. B The Cistrome DB Toolkit (http://dbtoolkit.cistrome.org/) was used to identify what TF likely regulates PHF19 expression spanning a region of ∼10 kb upstream the transcription start site (TSS). The Y axis represents the regulatory potential score which were calculated by Cistrome DB Toolkit [23, 24]. The x-axis represents the different TFs
PHF19 contains an amino terminal Tudor domain and two PHD domains, one EH/WH (Extended Homology/Winged-Helix) domain and a C-terminal RC/CL (Reversed Chromo/Chromo-Like) domain [20, 21]. PHF19S lacks one of the second PHD-domains and the EH/WH domain and contains a specific C-terminal region (Fig. 1A). Of note, the murine Phf19 locus encodes only a long isoform (Phf19L, 578aa) with 90% homology with the human PHF19L protein sequence and with conserved Tudor, PHDs, EH/WH and RC/CL domains. In human embryonic kidney cells (HEK293T), both PHF19S and PHF19L have been shown to associate with the PRC2 components EZH2 and EED but not within the same complex [26]. The EH/WH domain of PHF19 interacts with unmethylated CpG DNA islands at promotors, enhancing the chromatin association of PRC2 [27]. The RC/CL domain of PHF19L is necessary for the interaction with SUZ12 and to stabilize the dimeric state of PRC2 promoting chromatin binding, possibly through an avidity effect [28]. The PHF19 Tudor domain has been shown to bind to H3K36me3 suggesting that it targets the PRC2 complex to the chromatin of actively transcribed genes [20, 29, 30]. H3K36me3 marks have been shown to inhibit the ability of PRC2 to methylate H3K27 [31]. PHF19 facilitates the removal of H3K36me3 through the recruitment of an H3K36me3 demethylase, NO66 or KDM2B [20, 29]. PHF19 has been shown to physically interact with NO66 [29] and to co-localize on the chromatin with the H3K36me2 demethylase KDM2B [20]. However, ChIP-Seq profiling data obtained from mouse embryonic stem cells (mESCs) showed a moderate genome-wide association between Phf19 and H3K36me3 [21, 30]. PHF19 may also be able to bind to H3K27me3, although with a lower affinity than it binds to H3K36me3 [29, 32]. A subset of PRC2 target genes with bivalent H3K36me3 and H3K27me3 colocalized with Phf19 in mESCs and F9 embryonic carcinoma cells [30]. Interestingly, Phf19 has been shown to localize at bivalent promotor marked by both H3K4me3 and H3K27me3 in mESCs [21, 29] suggesting that it may target PRC2 to genes involved in cell differentiation or cell cycle.
Phf19 function has been characterized mainly in mESCs were it is dispensable for their self-renewal, it potentiates their differentiation and is required for the silencing of embryonic stem cell genes. In mESCs, Phf19 has been shown to localize with about 60-80% of SUZ12 binding sites [20, 21, 29, 30]. Phf19 colocalizes with another PCL protein MTF2 at a subset of PRC2 target genes [27]. shRNA-mediated Phf19 knockdown (KD) in mESCs leads to de-repression of a subset of PRC2 target genes involved in self-renewal, pluripotency and differentiation [21, 29]. These functions also correlate with a decrease in H3K27me3 at a subset of these genes [20, 21, 29, 30]. Mutation of conserved residues within the Phf19 Tudor domain, a domain implicated in recognizing H3K36me3, led to similar results [21, 30]. Overexpressing Phf19 in mESCs leads to enhanced global H3K27me3 levels [21]. Overall, these data suggest that in mESCs, Phf19 plays a role in PRC2 stabilization and in the spreading of PRC2/H3K27me3 to adjacent H3K36me3/2-positive nucleosomes after initial recruitment of PRC2 to the promotor by other PRC2 cofactors including Eed, Jarid2 or non-coding RNA [20, 21, 29, 30]. However, one should note that in mESCs, Phf19 is expressed approximately tenfold lower relative to Mtf2 and stoichiometry analysis of the different PRC2 complexes shows the number of PRC2 complexes containing Phf19 is minimal compared with Mtf2-containing complexes [33]. Furthermore, unlike in human cells, mouse cells lack the PHF19 short isoform that contains both the Tudor domain and the first PHD domain and is present in both cytoplasm and the nucleus of cells. These features suggest that the role of the PHF19L in recruiting PRC2 to target sites may be conserved between mice and human, however, the presence of the PHF19 short isoform may interfere or cooperate with the function of the PHF19L isoform leading to different roles and molecular mechanisms.
In normal tissue, analysis of PHF19 mRNAs by Northern blot showed that it is most abundant expression is in the thymus and heart, whereas they were barely detected in the lung and kidney [22]. RNA-Seq data shows that PHF19 expression is high in the artery, bone marrow and lymphoid tissues and the lowest in the pancreas and the liver (The Human Protein Atlas, https://www.proteinatlas.org/ENSG00000119403-PHF19/tissue). The ratio of PHF19S to PHF19L mRNAs varies among different tissues [22]. Placenta, skeletal muscle, and kidney express predominantly PHF19L mRNA, whereas liver and peripheral blood leukocytes contain more PHF19S mRNA [22]. In the mouse, Phf19 is expressed at low levels in all hematopoietic cells; however, differences in expression levels between stem, progenitor and mature progenitors are observed. Phf19 expression is modulated during activation and differentiation of mature blood cells such as B-cells and T-cells (see below). In normal mouse T-cells, the AKT pathway was shown to regulate the expression of Phf19 [34]. Increased Akt phosphorylation and activity through the downregulation of the Akt phosphatase Ship1 by miR-155 induces Phf19 transcription [34]. Further, the pharmacological inhibition of Akt in T-cells led to decreased expression of Phf19 [34].
Alterations of PHF19 expression in cancer
PHF19 is overexpressed in a number of human cancers including prostate cancer [35], glioblastoma, [36, 37], hepatocellular carcinoma [30, 38,39,PHF19 regulates CD8+ T-cell effector function The role of Phf19 was investigated in mouse CD8+ T-cells differentiation and function [34]. Phf19 was shown to orchestrate a transcriptional program able to restrain T cell senescence and to sustain CD8+ T-cell antitumor and immune responses [34]. Phf19 prevents terminal differentiation of T-cells through epigenetic silencing of the pro-effector and pro-exhaustion TFs including Eomes, Id2, Maf and Prdm1 [34]. In an infection model with recombinant vaccinia virus expressing the human glycoprotein 100 (gp100), Phf19 expression in CD8+ T-cells, similar to that of Ezh2, was strongly induced at the early stages (first 2 days) of the acute immune response, sharply downregulated at peak effector response and maintained at low levels throughout transition to memory phase [34]. In a mouse model of antitumor function, Phf19 overexpressing T-cells displayed limited senescence and sustained cytokine production and anti-tumor activity resulting in increased mice survival compared to controls [34]. This phenotype was dependent on the ability of Phf19 to modulate PRC2/EZH2 activity as Phf19 with mutated Tudor domain or it is overexpression in Ezh2-deficient T-cells abolished the phenotype [34]. These data have implication for cancer immunotherapy through epigenetic reprogramming of CD8+ T cell fate where Phf19 expression promotes effector function of these cells. These data suggest that in mature specialized blood cells (ex. CD8+ T-cells), Phf19 regulate PRC2 activity at genes involved in specialized function of these cells.
PHF19 in B-cell and plasma cells differentiation
In contrast to EZH2, the role of PHF19 in B-cell and plasma cell development and differentiation is not explored. Gene expression data shows that PHF19 is expressed at low levels in quiescent B-cells; however, its expression is upregulated in activated, proliferating, and fate-committed cells. In mouse B-cells, Phf19 expression is the highest in cycling pre-B (Fraction. Cprime, Fr.C) and germinal center (GC) B-cells (Fig. 2C). In human mature B-cells, PHF19 expression is highest in GC B-cells (centroblasts) and preplasmablasts (Fig. 2D). PHF19S expression is significantly higher than PHF19L expression in all B-cell and PC subsets (Fig. 2D).
PRC2/EZH2 in germinal center and PC differentiation
The germinal center reaction plays a critical role in myeloma pathogenesis. The PRC2 complex is an important mediator of the germinal center reaction and normal plasma cell differentiation; therefore, as a PRC2 activity regulator, PHF19 potentially plays an important role in these processes. Germinal centers are micro-structures that develop in secondary lymphoid organs as a result of B-cell activation by a specific antigen and the interaction with antigen-specific T-cells. This interaction allows the amplification and proliferation of B-cells, ensures high affinity antibody creation and the generation of memory B-cells or long-lived antibody secreting plasma cells. In mice, Ezh2, is required for GC formation in-vivo and promotes the proliferation of GC B-cells ex-vivo and its depletion leads to impaired PC production [62,63,64]. Ezh2 physically interacts with Bcl6 and represses PC differentiation via the suppression of Blimp1 and Irf4 transcription [65]. Ezh2 promotes the proliferation of GC B-cells through the suppression of the cell cycle inhibitors, Cdkn1a, Cdkn2a and Cdkn1b [66]. However, how Ezh2 is recruited to its target genes in GC B-cells and whether Phf19 play a role in this is unknown. EZH2 is actively involved in the de-novo establishment of bivalency at monovalent H3K4me3 promoters of GC exit genes [65]. This bivalent state at the gene promotors leads to a transient repressive state from which they can either become activated or stably repressed, depending on their differentiation commitment [65]. EZH2 gain-of-function mutations are among the most common genetic alterations identified in GC-derived B-cell lymphoma [62, 65]. Mutated EZH2 in B-cell lymphoma enables persistent epigenetic silencing of genes involved in PC differentiation and negative regulation of cell cycle [62, 66]. In MM, EZH2 is overexpressed and associated with adverse prognosis but is generally not mutated [4, 5].
During terminal differentiation into mature non-proliferating PCs, the GC B-cells or activated memory B-cells differentiate into plasmablasts that retain a proliferative capability in the context of a PC phenotype. A precursor population for plasmablast called a pre-plasmablast was first identified during the generation of plasma cells from human memory B cells in vitro [67, 68]. An in-vivo counterpart of the in vitro generated pre-plasmablasts was identified in human lymph nodes and tonsils [68, 69]. Interestingly, pre-plasmablasts co-express B and plasma cell TFs at low levels (BCL6, PAX5, IRF4, Blimp-1, XBP1) [68]. In this cell type, EZH2 seems to directly regulate the expression of genes involved in cell cycle (E2F7, CCNA2, E2F1, or AURKB) and DNA replication (including CDT1, POLD1, CDC45, MCM2, and MCM5) as these gene loci are enriched for EZH2, however, genes involved in memory B-cell phenotype maintenance seems to be an indirect target for EZH2 [70]. Interestingly, in this study EZH2 was found to be recruited to H3K27me3-free promoters of transcriptionally active genes known to regulate cell proliferation including AURKA, MCM5, CCND2 and RAD51AP1 [70]. The absence of H3K27me3 at the promotor of these genes raises question about the PRC2-independent activity of EZH2 but also about the regulation of PRC2/EZH2 activity at such promoters in the context of transient differentiating and proliferating cell states. PRC2-independent EZH2-mediated transcriptional activation was previously reported in prostate cancer and B-cell lymphoma through collaboration with transcriptional activators like E2F1 [71, 72]. It is plausible that PRC2/EZH2 activity at these transitionally expressed genes is inhibited by other factors as the PRC2 core subunit SUZ12 still colocalizes with EZH2 at some of these H3K27me3-free genes. In this context, an inhibitory role for the PCL subunits, PHF1 and MTF2 was demonstrated [73, 74]. MTF2 was shown to have both active and inhibitory roles in PRC2 function, however in a different cellular context. In ESCs, knockdown of Mtf2 causes reduced H3K27me3 at specific PRC2 targets, like Hox genes and pluripotency TFs, yet global H3K27me3 levels increase compared to control [74, 75]. In contrast, in embryonic fibroblasts Mtf2 was shown activate the expression of Cdkn2a by suppressing the catalytic activity of PRC2 [74]. The knockdown of Mtf2 showed increased H3K27me3 levels at the Cdkn2a locus, suggesting that Mtf2 can restrain PRC2 in certain contexts [74]. PHF1 was also shown to have PRC2 inhibitory function and one study showed that recognition of H3K36me3 by the PHF1 Tudor domain negatively regulates the enzymatic activity of PRC2 and lead to reduced levels of H3K27me3 in vitro and in HEK293T and K562 cells [73]. In a similar way in HSCs and prostate cancer cells, PHF19 KD was associated with increased levels of H3K27me3, and with a repression of a subset PRC2 target genes suggesting that in some context PHF19 may have PRC2 inhibitory functions.
PHF19 in germinal center and PC differentiation
Similar to EZH2 in the B-cell lineage, the expression of PHF19 is low in mature naïve cells; reaches high levels in GC B-cells and then returns to low level in PCs and memory B-cells (Fig. 2C and D). Specifically, PHF19 reaches its highest level of expression in centroblasts and pre-plasmablasts, two highly proliferative cell subsets (Fig. 2D). PHF19 is also highly expressed by centrocytes and plasmablasts. In-vitro, PHF19 is significantly upregulated after activation of memory B-cells by a combination of stimuli including CD40L, CpG oligodeoxynucleotide, IL2, IL10 and IL15 (Activated B-cells, act.BC) and by pre-plasmablast generated in-vitro (Fig. 2E). No study has comprehensively investigated the role of PHF19 (neither PHF1 nor MTF2) in B-cell development or plasma cell. However, using a bone marrow transplantation model of retrovirally transduced cells with PHF19 or an empty vector, one study showed that PHF19 overexpression does not induce any difference in the percentages of mature B-cells or T-cells in the bone morrow or the spleen between mice overexpressing PHF19 and controls [12]. Notably, in germinal center B cells, EZH2 establish bivalency at genes involved in PC differentiation and negative regulation of the cell cycle promoting cell proliferation [62, 66]. Interestingly, upregulated genes after pharmacological inhibition of EZH2 showed overlap only with bivalent genes but not with H3K27me3-only enriched genes suggesting that bivalent genes might play an important role in MM pathogenesis and could be amenable to therapeutic targeting.
In MM cell lines, pharmacological inhibition of EZH2 decreased the global H3K27 methylation and had anti-myeloma effects both in vitro and in vivo [1, 12, 96,97,98]. Inhibition of EZH2 in MM leads to the downregulation of a c-MYC signature, the upregulation of cell cycle control genes including the CDK inhibitors CDKN1A/p21 and CDKN2B/p15 leading to cell cycle arrest and apoptosis [1, 98]. New evidence shows that in MM cell lines that the sensitivity to EZH2 inhibitor correlated with distinct metabolic signatures resulting from a dysregulation of genes involved in methionine cycling [99].
The t(4;14) subset of MM is notable for a global reduction of H3K27me3 levels. t(4;14) MM cells exhibit an increased expression of the H3K36 methyltransferase NSD2 resulting in increased levels of H3K36me2 and an imbalance in the distribution of the H3K36me2 and H3K27me3 marks [97]. H3K36me2 has been shown to inhibit both PRC2 binding to nucleosomes and in the methylation of histones [31, 100]. However, t(4;14) MM cells exhibit enhanced recruitment of EZH2 and enrichment of H3K27me3 at specific genomic loci involved in normal germinal center B-cells differentiation and in a subset of c-MYC targets genes in B-cells suggesting that EZH2-mediated repression of these genes may be important for NSD2-induced oncogenesis [97]. Accordingly, t(4;14)+ NSD2 overexpressing MM cell lines are more sensitive to EZH2 inhibition compared to cell lines with NSD2 low expression [97].
H3K27me3 marks are removed by the histone demethylases ubiquitously transcribed tetratricopeptide repeat X chromosome (UTX; also known as KDM6A) and jumonji domain-containing protein 3 (JMJD3; also known as KDM6B) [101]. In MM, UTX/ KDM6A is mutated or deleted in about 5% of primary MM cases [81]. In an isogenic cell line system, loss of UTX leads to a failure in the activation of the expression of some genes involved in cell growth, adhesion, survival and movement, and promotes proliferation, clonogenicity, adhesion, and tumorigenicity of MM cells in-vitro and in-vivo [102]. Interestingly, treatment of UTX null cells with EZH2 inhibitors reactivated the expression of nearly half of the genes repressed by UTX loss [102]. Furthermore, UTX-deficient MM cells showed increased sensitivity to EZH2 inhibition compared to MM cells expressing wild-type UTX [102]. These data from t(4;14)+ NSD2 overexpressing MM and from UTX-deficient MM suggest that EZH2 may have context-dependent oncogenic activities in MM.
Transcriptional regulation of PHF19 expression in Myeloma
In MM, the transcriptional regulation of PHF19 gene is largely unexplored to date. While PHF19 is located on chromosome 9q33.2, a chromosome frequently gained in MM, we (unpublished) and others [103] have shown only a negative correlation between trisomy Chr9 and PHF19 expression indicating that high levels of PHF19 are not induced by DNA copy number gain of Chr9.
In the MM1S cell line, our analysis of ChIP-seq data from the Cistrome databases showed that PHF19 locus is decorated with active chromatin marks H3K27ac and H3K4me3 and POL2RA (Fig. 3A). ATAC-seq signals showed that the chromatin is accessible at the PHF19 locus. Interestingly, BRD4, which bookmarks transcribed genes and active genes during mitosis is also identified at the PHF19 locus (Fig. 3A). The Mediator of RNA polymerase II transcription subunit 1 (MED1) is also present at the PHF19 locus. ChIP-seq analysis of transcription factors show that the TSS of the PHF19 gene is bound by E2F1, IRF4 and MYC (Fig. 3A). Further the PHF19 gene was one of the 681 genes identified as associated with a cis-located super-enhancer region (coordinate on hg38 genome version chr9:120868122-120905040) [105] (Fig. 3A). This region is enriched for enhancer’s marks H3K27ac, H3K4me1 and accessible chromatin (ATAC-Seq) in the KMS11 MM cell line (not shown). Analysis of chromatin accessibility and TF binding at this Enhancer region showed the presence of IRF4, MYC and BRD4 at open chromatin spots (Fig. 3A). Notably, this enhancer was not identified in glioblastoma cell line U-87 MG or in small cell lung cancer cell line H2171 suggesting that it might be specific to plasma cells. Furthermore, this enhancer is different from the trans-located enhancer identified in Ewing sarcoma (See above, [47]). This epigenetic landscape in MM1S cell line, indicates that the PHF19 locus is transcriptionally active and this correlates with the level of mRNA expression of PHF19 (CCLE and Keats lab datasets) (Fig. 3B). Microarray RNA expression data of PHF19S and PHF19L from the CCLE datasets, shows that PHF19S is expressed at a higher level than PHF19L in MM cell lines (Fig. 3C). In our hands, this pattern of mRNA expression of PHF19S and PHF19L was also observed at the protein level by western blot in all the MM cell lines tested (Fig. 3C) (data not shown).

Chromatin marks and transcription factors at the PHF19 locus and PHF19 expression in human MM cell lines. A Snapshot from UCSC genome browser showing binding events within the PHF19 locus in the MM1S cell line. B PHF19 expression as reported using RNA-seq in the Cancer Cell Line Encyclopedia (CCLE) database [104] (https://depmap.org/portal/download/, accessed on 25 July 2021) (upper) and as reported in Keats lab dataset (https://www.keatslab.org/data-repository) (Lower). C PHF19 expression as reported using microarray in the CCLE database
As IRF4 was one of the TF present at the enhancer and TSS of PHF19, we checked if PHF19 expression is affected by the CRISPR mediated ablation of IRF4 in MM cell lines [106]. In this context, PHF19 was not identified among the differentially expressed genes in MM1S, OPM2 or H929 cells suggesting that IRF4 is not directly involved in PHF19 expression regulation. The analysis of published RNA-seq data from the use of the BRD4 inhibitor (AZD5153) vs control in MM1S cells identified PHF19 as one of the downregulated genes [107]. The use of this BRD4 inhibitor also downregulated PHF19 in other MM cell lines including IM9, MOLP8, OPM2 and RPMI8226 [107]. PHF19 has been identified as a potential MYC target by ChIP-seq experiments [108], however, as far as we know there is no direct evidence that can link MYC to the regulation of PHF19 expression (For example knockdown or pharmacologic inhibition of MYC followed by evaluation of PHF19 expression levels). Both BRD4 inhibition [107] and IRF4-depletion lead to significant downregulation of MYC [106, 109]. However, BRD4 inhibition but not IRF4-depletion led to significant downregulation of PHF19 expression. The pharmacological inhibition of H3K4me3 Lysine demethylase 5A (KDM5A), which also leads to a significant downregulation of MYC and MYC-targets, did not lead to significant changes in PHF19 expression [110]. These data suggest that PHF19 is not a target for MYC, however, the involvement of MYC in PHF19 expression needs to be further resolved in MM.
Clinical and biological consequences of PHF19 overexpression in Multiple Myeloma
PHF19 is overexpressed in MM and is higher in malignant PC compared to normal bone marrow PC [15, 52, 103]. The expression of PHF19 increases with increasing disease stage and during the progression from the premalignant stages MGUS and smoldering MM into MM [15, 16, 52, 84, 103]. The highest PHF19 expression is observed in the most aggressive variant, PCL. PHF19 expression has been identified as the best individual predictor of risk in a MM DREAM challenge [15]. We note, however, that PHF19 was not identified among the prognostic gene expression profiles of MM including the GEP70 [111], EMC92 [112] and the IFM15 [113]. In a recent study aiming at establishing a transcriptional regulatory network in order to predict disease progression, PHF19 was identified as a member of a genetic program (Program 68) that predicts high-risk behavior and early relapse [114]. Along with other genes in this program (e.g., CKS1B, PCNA, E2F1, FOXM1), PHF19 expression was associated with cell proliferation and was among the best genes predictors of high-risk status [114]. In a recent preprint study analyzing single cell RNA-seq of FACS-sorted CD138+ MM cells using Seurat, showed that MM cells cluster into five major subtypes (clusters) with the cell subtype-2 expressing high level of PHF19 compared to the others subtypes [115]. This observation suggests that only a subpopulation of MM tumor cells express high levels of PHF19 bringing the association between PHF19 and MM aggressiveness to a cellular level. The presence of the PHF19high myeloma cell subtype-2 is showed to be associated with myeloma progression using a new deep learning tools called DEGAS [115]. Interestingly, this PHF19 high myeloma cell subtype also exhibited high expression of genes involved in cell cycle including HELLS, EZH2, TYMS, ZWINT, and MKI67 [115].
Multiple lines of evidence suggest PHF19 is a mediator of adverse prognosis because of its potential to control the expression of genes associated with drug resistance and proliferation [15, 22, 30, 35, 36, 52, 84, 103]. Our team identified PHF19 and EZH2 among the deregulated genes between SMM and MM in the seven major subtypes of MM (D1-HRD, D2, CCND1-11q13, CCND3-6p21, MMSET, MAF and MAFB) [84]. In a global challenge to identify biomarkers of high-risk behavior in MM, bioinformatic teams from around the world competed to identify expression patterns that correlated with poor outcome from eight data sets totaling 2447 patients [15]. This effort identified PHF19 as the most significant contributor, more so even than NSD2 (MMSET), CKS1B and MAF expression, known mediators of MM risk [15]. Incorporation of PHF19 and MMSET expression with age and ISS identified a simple model of high-risk MM [15] and PHF19 high expression is associated with poor prognosis [15, 16, 52, 103]. Higher PHF19 expression is significantly associated with multiple high-risk genetic factors such as IGH translocation groups, non-hyperdiploidy, TP53 mutations and the overall number of drivers per sample [15]. Furthermore, well known proliferation genes including EZH2, MCM4, TYMS, AURKB, CHEK1, MCM2, ZWINT, CCNA2 and BIRC5 were highly correlated with PHF19 expression [15].
Using an shRNA loss-of-function approach, several study have shown that MM cell lines with different genetic background (ARP1, OCI-My5, JJN3, MM1S, L363, RPMI8226, H929 and KMS11) are dependent on PHF19 for their proliferation [15, 16, 52, 103]; however, the mechanism by which this is mediated remains undefined. The knockdown of both PHF19L and PHF19S or only PHF19L has been shown to diminish the cell growth and colony formation of several MM cell lines [16, 52, 103], however, only the ectopic rescue of PHF19L restored normal cell growth and colony formation [103]. Mice xenografted with MM cell line knocked down for PHF19L showed suppressed tumor growth and prolonged survival compared with controls [16, 52, 103]. In the study by Yu T et al. [52], the ectopic overexpression of PHF19L in ARP1 and OCI-My5 MM cells, increased cell growth and induced drug resistance to bortezomib, epirubicin, or melphalan and promoted tumor growth in vivo [52]. Cell cycle analysis showed that knockdown of PHF19 lead to cell cycle defect characterized by the accumulation of the cell in G0/G1 of the cell cycle [15, 103].
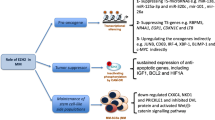
Importantly, mechanistic investigation of PHF19 function in MM has given conflicting results, (Fig. 4). On one hand, Ren et al. [103] showed that PHF19 is critical for the maintenance of the H3K27me3 landscape across the chromatin, with the exception of CpG rich promoters and that it represses cell cycle inhibitor genes e.g. CDKN1A/C and JAK-STAT pathway genes. In contrast, Yu et al. [52] reported that PHF19 depletion promotes the phosphorylation of EZH2 leading to EZH2 inactivation via the PI3K/AKT pathway, thus causing a decrease in H3K27me3 and H3K27me2 marks thus promoting the expression of genes involved in MM cell survival, proliferation and conferring drug resistance via JUN, KLF, RELB, HIF1α, BCLXL and MCL1. Schinke et al. showed that PHF19 KD leads to the downregulation of major cancer players such as BCL2, MYC and EGR1 in ARP1 and JJN3 cell lines and to the downregulation of genes within the JAK/STAT pathway in JJN3 cell line only suggesting that PHF19 is somehow involved in the upregulation of these genes [16]. Despite these differences, the three studies show that PHF19 overexpression is associated with more aggressive proliferation and drug resistance in MM cell lines making it critical to resolve the key mechanism by which this is mediated.

Schema showing suggested mechanism of action of PHF19 overexpression in MM cell lines. Ren Z et al. showed that PHF19 promotes PRC2 activity and represses cell cycle inhibitor genes. Yu T et al. showed that PHF19 impedes PRC2 activity by promoting the phosphorylation of EZH2 via AKT pathway leading to EZH2 inactivation leading to an increase in expression of genes that play an important role in MM. Schinke C et al. showed that PHF19 repressed the expression of tumor suppressor protein (TSP) and upregulated the expression of pro-survival and proliferation genes
In MM, inactivation of EZH2 by phosphorylation was shown to lead to cell-adhesion mediated drug resistance to doxorubicin and the alkylating agent 4-OHCY [95]. Adhesion and cell-cell contact signals activated the PI3K/Akt pathway to induce the phosphorylation of EZH2 on the Ser-21, leading to H3K27 hypomethylation resulting in the activation of antiapoptotic genes including IGF1 [95]. Pharmacological and genetic inhibition of the IGF1R-PI3K-AKT pathway reverses CAM-DR by promoting EZH2 dephosphorylation and H3K27 hypermethylation both in vitro and in refractory murine MM models [95]. EZH2 is also phosphorylated by CDK1 on Thr-345 and Thr-487 that promotes EZH2 ubiquitination and subsequent degradation by the proteasome [116] or disrupted EZH2 binding with the other PRC2 components SUZ12 and EED, and thereby inhibited EZH2 methyltransferase activity [117]. AKT pathway is one of the upstream regulators of PHF19 expression [34, 43] and PHF19 promotes AKT pathway signaling [45] and CDK1 seems to be one of the downstream target of PHF19 activity in MM (See below). These observations suggest that PHF19 might regulate PRC2/EZH2 activity via an effect on mediating the expression of CDK1 and the promotion of AKT activity.
PHF19 is involved in the regulation of genes important in cell cycle and the genetic stability of MM cells
In order to define the transcriptional program and biological pathways promoted by PHF19, we analyzed the gene expression data from the Multiple Myeloma Research Foundation (MMRF) CoMMpass study of 683 newly diagnosed MM and 35 relapse patients, (Fig. 5). PHF19 is expressed in all MM subtypes with higher expression levels seen in the non-hyperdiploid subgroups (χ2=38, p=5.7.10-10) and HR patients defined by GEP70 (Fig. 5A and B). Using the logrank test we identified a PHF19 expression level (of 9.65) as an optimal cut point for overall survival (OS) splitting the population into high and low PHF19 expressing groups. The cases with elevated PHF19 expression (35%, 241/683) were associated with inferior OS (Fig. 5C). PHF19 expression was significantly higher at relapse (Fig. 5D). These data confirm and extend the results of previous reports using different MM series [15, 16, 52, 103, 118].

High PHF19 level is associated with high-risk and PHF19 transcriptional signature in MM suggests that it might regulate cell cycle progression. A and B PHF19 is expressed in all MM subtypes with higher expression levels seen in the non-hyperdiploid subgroups (X2=38, p=5.7e-10) and HR patients defined by GEP70. Interestingly, PHF19 expression is significantly higher at relapse (t=2.8, df=34, p=0.006). C Using a logrank test we identify a PHF19 expression level of 9.65 as an optimal cut point for overall survival (OS) splitting the population into high and low PHF19 expressing groups. The OS of patients with elevated PHF19 expression is significantly shorter than patients with lower PHF19 expression (HR=2.98 (2.2-4), p=3.68e-13. D PHF19 expression is significantly higher at relapse (t=2.8, df=34, p=0.006). E PHF19high and low groups were defined using an elbow test. F Volcano plot showing genes differentially expressed between PHF19high and PHF19low MM samples. Analysis identified 835 differentially expressed genes (DEG) (Fold change >2, FDR < 0.05), with 547 (65%) upregulated and 288 (35%) downregulated genes. G Gene set enrichment analysis (GSEA) analysis of the differentially expressed genes between PHF19 high and low MM samples (Gene ontology (GO)). H Venn diagram showing the overlap of: 1- DEG between PHF19high and PHF19low MM samples, 2- Downregulated genes between PHF19-KD and PHF19-WT in MM1S cell line (i.e. genes upregulated by PHF19), 3- Upregulated genes between PHF19-overexpression (rescue) and PHF19-KD in MM1S cell line (i.e. genes upregulated by PHF19) and 4- upregulated genes between PHF19high and PHF19low MM samples. I Venn diagram showing the overlap of: 1- the 294 genes defining PHF19 signature in MM, 2- DEG between centroblasts (CB) and bone marrow plasma cells (BMPC) and 3- DEG between preplasmablasts (PrePB) and BMPC
PHF19 high and low groups were also defined using an elbow test (Fig. 5E). Using this cutpoint to define the transcriptional signature associated with high PHF19 expression we compared the gene expression profile of high vs low PHF19 samples and identified 835 differentially expressed genes (DEG) (Fold change >2, FDR < 0.05), with 547 (65%) upregulated and 288 (35%) downregulated genes (Fig. 5F). GSEA showed that these genes are enriched for cell division, DNA replication and chromosome segregation processes (Fig. 5G). To directly link the regulation of expression of these genes/pathways to PHF19, we analyzed RNA-seq data from the MM1S cell line in which PHF19 was knocked down by shRNA and identified differentially expressed genes between PHF19-KD cells and PHF19-WT [103]. Overlap** the DEG from this analysis in MM1S cell lines and from patients’ samples, we identified 294 common genes and defined them as the PHF19 transcriptional signature of MM (Supplementary Table 2). Interestingly, 224 of these genes (82%) were upregulated in PHF19-high vs low MM samples, are downregulated after PHF19 depletion in the MM1S cells and are upregulated after restoring the expression of PHF19 in depleted cells (Fig. 5H). These data suggest that PHF19 is involved in the transcriptional activation of these genes.
We analyzed the RNA-seq data of HSCs from a PHF19 knockout mouse model [56]. PHF19 in HSCs was shown to control the balance between quiescence and proliferation as well as the balance between self-renewal, differentiation and maintenance [56]. In total 52 out of the 294 genes identified in our MM analysis were also found to be DE between Phf19-WT and Phf19-KO mouse HSCs. Interestingly, 43 out of the 52 genes, were downregulated after PHF19-depletion in MM cells and in mouse HSCs suggesting that PHF19 positively regulate the transcription of these genes. These 43 genes are mostly involved in cell cycle progression and include genes like TYMS, AURKB, CCNA2 and BIRC5 (Supplementary Table 3) that significantly correlated with high PHF19 expression in MM [15]. This observation suggests that PHF19 activity may stimulate the expression of cell cycle associated genes in different cellular context including malignant PCs affecting their proliferation and maintenance and leading to therapeutic resistance and HR behavior.
In normal physiology, PHF19 may be involved in regulating late-stage B-cell development so its aberrant expression could be relevant to the pathology of MM. To address this idea we explored the relevance of the 294 genes in GC B-cell and plasma cells (PC) differentiation program by analyzing their expression in normal human GC B-cells (centroblasts) and PCs. PHF19 expression was the lowest in quiescent mature naïve B-cell and PC and the highest in proliferating centroblast and pre-plasmablast (Fig. 2D). We show that 175 genes and 180 genes (about 61%) of these 294 genes are shared with DEG between centroblasts or pre-plasmablast and mature bone marrow PC, respectively (Fig. 5L). The majority of these genes are involved in cell cycle regulation.
Analysis of published single cell RNA-seq data [119, 120] showed that PHF19 is upregulated by cycling GC B-cells and plasmablasts compared to non-cycling GC, memory B-cells and naïve B-cells (Fig. 6A). Specifically, in GC B-cells the expression of PHF19 peaks in the G2-M phase of the cell cycle and this is followed by the modulation of several genes of the PHF19 transcriptional signature that we identified in MM and that are involved in cell cycle regulation including PLK4, BUB1, CENPE, NUF2 and AURKB (Fig. 6B). This pattern of expression suggests that PHF19 could play a role in the transcriptional regulation of genes specific to the G2-M checkpoint, the mitotic cell cycle transition by which a cell in G2 commits to M phase and where the cell must maintain the integrity and the proper segregation of the recently duplicated chromosomes. The deregulation of the expression of these genes by PHF19 overexpression may lead to abnormal cell cycle, increased cellular proliferation and genomic instability. The G2M checkpoint plays a critical role in cell cycle and its deregulation can lead to cancer. Cancers with high activity of G2M pathway genes tend to be more aggressive. These data suggest that PHF19 overexpression in PC may re-induce the expression of genes involved in proliferation and cell cycle, a situation that would be biologically relevant to the malignant behavior of a cell type otherwise destined for quiescence or apoptosis when the initial immune response is no longer required. Furthermore, single cell RNA-seq analysis identified that only a subpopulation of MM malignant cells overexpress PHF19 [115]. Interestingly, this PHF19high myeloma cell subpopulation seems to be the cell cycling fraction as they also exhibited high expression of genes involved in cell cycling including HELLS, EZH2, TYMS, ZWINT, and MKI67 [115].

Single cell RNA-seq data from germinal centers links PHF19 expression modulation to cell cycle. A PHF19 expression is the highest in cycling GC B-cells and preplasmablasts and plasmablasts. Upper panel shows a UMAP projection and cluster identification from single cell RNAseq profiles of human B cells maturation in tonsils [120]. Lower panel shows UMAP plot showing expression of PHF19 in the different B cells clusters as shown in the upper panel (www.tonsilimmune.org/, accessed on 16 June 2021). B Heatmap displaying the relative expression fold change (log2) of PHF19 and selected genes in clusters of dark zone B cells representing different stages of the cell cycle (data are from [119]. Ten clusters were identified by PhenoGraph in dark zone GC B cells based on the expression of genes associated with the S-G2-M stages of the cell cycle: three clusters of cells transitioning from G1 to S phase (C1, C2 and C3), two clusters in the S-phase (C4 and C5), three clusters of cells transitioning from G2 to M phase (C6, C7 and C8), one cluster in the M-phase (C9) and one cluster of cells transitioning from M to G1 phase (C10). The differential expression analysis is performed by comparing each cluster to all the others. PHF19 expression peaks in the G2-M phase of the cell cycle and this is followed by the modulation of several genes of the PHF19 transcriptional signature
Molecular classification of MM by unsupervised hierarchic clustering of mRNA expression profiles in CD138-enriched plasma cells from 414 newly diagnosed patients identified seven disease subtypes influenced by known genetic lesions [121]. These seven subgroups are MAF (c-MAF and MAFB), Cyclin D-1 (CCND1), Cyclin D-2 (CCND1 and CCND3), MMSET-activating translocations, hyperdiploidy (HD), low bone disease and proliferation subgroup (PR). PHF19 expression is the highest in the PR subgroup (Fig. 7A). Using Genomicscape [55] platform we performed coexpression analysis and found that 209 genes positively correlated with PHF19 expression (Pearson correlation coefficient ≥ 0.5 and p value ≤ 0.05). The heatmap shows the top 30 coexpressed genes with PHF19 in the seven molecular subgroups (Fig. 7B). GSEA analysis (Biological Process ontology, GO:BP) of these 209 genes showed an enrichment for cell cycle regulation, the integrity of mitosis and chromosome segregation processes (Fig. 7C). Overlap** the genes from this analysis and the 294 genes identified as the PHF19 transcriptional signature of MM (see above) showed that 128 of the 209 genes (61%) are common (Fig. 7D).

PHF19 expression is associated with cell proliferation in MM. A PHF19 expression in molecular subtype of MM, proliferation subgroup (PR, n=47), low bone disease (LBD, n=58), MMSET (n=68), hyperdiploidy (HD, n=116), Cyclin D-1 (CD-1, n=28), Cyclin D-2 (CD-2, n=60) and MAF (n=37). B Coexpression analysis of PHF19 using GenomicScape database [55] (http://genomicscape.com/microarray/expression.php, accessed on 03 September 2021). Heat maps showing the top 30 genes positively correlated with PHF19 in a UAMS cohort [121]. C Gene set enrichment analysis (GSEA) analysis was performed using g:Profiler web tool (Biological Process ontology, GO:BP, https://biit.cs.ut.ee/gprofiler/gost). D Venn diagram showing the overlap between the 294 genes identified as the PHF19 transcriptional signature of MM and the 209 genes that positively correlate with expression of PHF19
Overall, these data strongly implicate PHF19 in the regulation of genes important in the proliferation, genetic stability and cell cycle of cells making it highly relevant to the basis of HR behavior. Thus, if we are to understand how HR behavior is mediated it is essential to understand the cellular and epigenetic mechanisms by which PHF19 promotes aggressive disease behavior; understanding these mechanisms will allow us to design of therapeutic strategies able to target aggressive disease.
Conclusions and perspectives
Overall, data from different cell-context and experimental design strongly implicate PHF19 in the regulation of genes important in cell cycle regulation and the genetic stability of cancer cells making it highly relevant to the basis of HR MM behavior. In MM, the control mechanism for the abnormal H3K27me3 patterns is elusive and the role played by PHF19 as a regulator of PRC2 activity in this process is still not clear. Until recently the PRC complex had been described as an epigenetic repressor, however, emerging evidence suggests a scenario in which PRC proteins can also have a dynamic effect in mediating gene expression levels. Thus, while PHF19 is known to recruit and increase the transcriptional repressive activity of PRC2 through an increased deposition of the repressive mark H3K27me3, new studies in MM, prostate cancer and mouse hematopoietic stem cells have shown that PHF19-depletion is associated with a focal increase of H3K27me3 mark and that PHF19 may also play a role in the activation of gene expression. Experimentally the conflicting data highlight the limitations of MM cell lines for the assessment of the mode of action of epigenetically active genes and suggest the need to investigate PHF19 mechanisms in a more appropriate biological model. The current data indicate that PHF19 either plays a role in context-dependent regulation of PRC2 activity or alternatively that it may have activity independent of PRC2. To differentiate these two possibilities, it is crucial to identify the genes that are regulated by PHF19 and then to investigate how these genes are epigenetically controlled in a relevant model system i.e germinal center B-cells, plasmablasts or plasma cells. Furthermore, the role of PHF19 short isoform still to be clarified in MM as this form contains both the Tudor domain and the first PHD domain and is present in the nucleus of cells meaning it still can interact with H3K36me3 and have an impact on the long isoform function. The roles and molecular mechanism that may be established by both PHF19 isoform should be more clearly investigated.
Availability of data and materials
The material supporting the conclusion of this review has been included within the article.
The datasets analyzed during the current study are available in the following repository:
http://dbtoolkit.cistrome.org/
http://servers.binf.ku.dk/bloodspot/
http://genomicscape.com/microarray/expression.php
https://depmap.org/portal/download/
Abbreviations
- act.BC:
-
Activated B-cells
- DEG:
-
Differentially expressed genes
- EED:
-
Embryonic ectoderm development
- EZH:
-
Enhancer of Zeste Homolog
- GC:
-
Germinal center
- GSEA:
-
Gene Set enrichment analysis
- HD:
-
Hyperdiploidy
- H3K27me3:
-
Tri-methylation of the lysine (K) 27 on histone 3 H3
- HR:
-
High-risk
- HSC:
-
Hematopoitic stem cell
- KD:
-
Knockdown
- mESCs:
-
Mouse embryonic stem cells
- MGUS:
-
Monoclonal gammopathy of undetermined significance
- MM:
-
Multiple myeloma
- MMRF:
-
Multiple Myeloma Research Foundation
- mRNA:
-
Messenger ribonucleic acid
- PC:
-
Plasma cells
- PCL:
-
Plasma cell leukemia
- PHF:
-
Plant Homeodomain Finger Protein
- PRC2:
-
Polycomb Repressive Complex 2
- SMM:
-
Smoldering multiple myeloma
- SRBC:
-
Sheep red blood cells
- SUZ12:
-
Suppressor of zeste 12
- TFs:
-
Transcription factors
References
Pawlyn C, Bright MD, Buros AF, Stein CK, Walters Z, Aronson LI, et al. Overexpression of EZH2 in multiple myeloma is associated with poor prognosis and dysregulation of cell cycle control. Blood Cancer J. 2017;7(3):e549.
Rajkumar SV. Multiple myeloma: 2018 update on diagnosis, risk-stratification, and management. Am J Hematol. 2018;93(8):981–1114.
Landgren O, Kyle RA, Pfeiffer RM, Katzmann JA, Caporaso NE, Hayes RB, et al. Monoclonal gammopathy of undetermined significance (MGUS) consistently precedes multiple myeloma: a prospective study. Blood. 2009;113(22):5412–7.
Walker BA, Wardell CP, Chiecchio L, Smith EM, Boyd KD, Neri A, et al. Aberrant global methylation patterns affect the molecular pathogenesis and prognosis of multiple myeloma. Blood. 2011;117(2):553–62.
Pawlyn C, Kaiser MF, Davies FE, Morgan GJ. Current and potential epigenetic targets in multiple myeloma. Epigenomics. 2014;6(2):215–28.
Dupere-Richer D, Licht JD. Epigenetic regulatory mutations and epigenetic therapy for multiple myeloma. Curr Opin Hematol. 2017;24(4):336–44.
van Mierlo G, Veenstra GJC, Vermeulen M, Marks H. The complexity of PRC2 subcomplexes. Trends Cell Biol. 2019;29(8):660–71.
Morgan MAJ, Shilatifard A. Reevaluating the roles of histone-modifying enzymes and their associated chromatin modifications in transcriptional regulation. Nat Genet. 2020;52(12):1271–81.
Di Carlo V, Mocavini I, Di Croce L. Polycomb complexes in normal and malignant hematopoiesis. J Cell Biol. 2019;218(1):55–69.
Radulovic V, de Haan G, Klauke K. Polycomb-group proteins in hematopoietic stem cell regulation and hematopoietic neoplasms. Leukemia. 2013;27(3):523–33.
Varlet E, Ovejero S, Martinez AM, Cavalli G, Moreaux J. Role of polycomb complexes in normal and malignant plasma cells. Int J Mol Sci. 2020;21(21).
Agarwal P, Alzrigat M, Parraga AA, Enroth S, Singh U, Ungerstedt J, et al. Genome-wide profiling of histone H3 lysine 27 and lysine 4 trimethylation in multiple myeloma reveals the importance of Polycomb gene targeting and highlights EZH2 as a potential therapeutic target. Oncotarget. 2016;7(6):6809–23.
Lavarone E, Barbieri CM, Pasini D. Dissecting the role of H3K27 acetylation and methylation in PRC2 mediated control of cellular identity. Nat Commun. 2019;10(1):1679.
Margueron R, Li G, Sarma K, Blais A, Zavadil J, Woodcock CL, et al. Ezh1 and Ezh2 maintain repressive chromatin through different mechanisms. Mol Cell. 2008;32(4):503–18.
Mason MJ, Schinke C, Eng CLP, Towfic F, Gruber F, Dervan A, et al. Multiple myeloma DREAM challenge reveals epigenetic regulator PHF19 as marker of aggressive disease. Leukemia. 2020;34(7):1866–74.
Schinke CD, Bird JT, Qu P, Yaccoby S, Lyzogubov VV, Shelton R, et al. PHF19 inhibition as a therapeutic target in multiple myeloma. Curr Res Transl Med. 2021;69(3):103290.
Lonie A, D'Andrea R, Paro R, Saint R. Molecular characterisation of the Polycomblike gene of Drosophila melanogaster, a trans-acting negative regulator of homeotic gene expression. Development. 1994;120(9):2629–36.
Cao R, Wang H, He J, Erdjument-Bromage H, Tempst P, Zhang Y. Role of hPHF1 in H3K27 methylation and Hox gene silencing. Mol Cell Biol. 2008;28(5):1862–72.
Casanova M, Preissner T, Cerase A, Poot R, Yamada D, Li X, et al. Polycomblike 2 facilitates the recruitment of PRC2 polycomb group complexes to the inactive X chromosome and to target loci in embryonic stem cells. Development. 2011;138(8):1471–82.
Ballare C, Lange M, Lapinaite A, Martin GM, Morey L, Pascual G, et al. Phf19 links methylated Lys36 of histone H3 to regulation of polycomb activity. Nat Struct Mol Biol. 2012;19(12):1257–65.
Hunkapiller J, Shen Y, Diaz A, Cagney G, McCleary D, Ramalho-Santos M, et al. Polycomb-like 3 promotes polycomb repressive complex 2 binding to CpG islands and embryonic stem cell self-renewal. PLoS Genet. 2012;8(3):e1002576.
Wang S, Robertson GP, Zhu J. A novel human homologue of Drosophila polycomblike gene is up-regulated in multiple cancers. Gene. 2004;343(1):69–78.
Zheng R, Wan C, Mei S, Qin Q, Wu Q, Sun H, et al. Cistrome data browser: expanded datasets and new tools for gene regulatory analysis. Nucleic Acids Res. 2019;47(D1):D729–D35.
Mei S, Qin Q, Wu Q, Sun H, Zheng R, Zang C, et al. Cistrome data browser: a data portal for ChIP-Seq and chromatin accessibility data in human and mouse. Nucleic Acids Res. 2017;45(D1):D658–D62.
Loughran G, Jungreis I, Tzani I, Power M, Dmitriev RI, Ivanov IP, et al. Stop codon readthrough generates a C-terminally extended variant of the human vitamin D receptor with reduced calcitriol response. J Biol Chem. 2018;293(12):4434–44.
Boulay G, Rosnoblet C, Guerardel C, Angrand PO, Leprince D. Functional characterization of human Polycomb-like 3 isoforms identifies them as components of distinct EZH2 protein complexes. Biochem J. 2011;434(2):333–42.
Li H, Liefke R, Jiang J, Kurland JV, Tian W, Deng P, et al. Polycomb-like proteins link the PRC2 complex to CpG islands. Nature. 2017;549(7671):287–91.
Chen S, Jiao L, Liu X, Yang X, Liu X. A dimeric structural scaffold for PRC2-PCL targeting to CpG Island Chromatin. Mol Cell. 2020;77(6):1265–78 e7.
Brien GL, Gambero G, O'Connell DJ, Jerman E, Turner SA, Egan CM, et al. Polycomb PHF19 binds H3K36me3 and recruits PRC2 and demethylase NO66 to embryonic stem cell genes during differentiation. Nat Struct Mol Biol. 2012;19(12):1273–81.
Cai Z, Qian ZY, Jiang H, Ma N, Li Z, Liu LY, et al. hPCL3s promotes hepatocellular carcinoma metastasis by activating beta-catenin signaling. Cancer Res. 2018;78(10):2536–49.
Schmitges FW, Prusty AB, Faty M, Stutzer A, Lingaraju GM, Aiwazian J, et al. Histone methylation by PRC2 is inhibited by active chromatin marks. Mol Cell. 2011;42(3):330–41.
Dong C, Nakagawa R, Oyama K, Yamamoto Y, Zhang W, Dong A, et al. Structural basis for histone variant H3tK27me3 recognition by PHF1 and PHF19. Elife. 2020;9.
Kloet SL, Makowski MM, Baymaz HI, van Voorthuijsen L, Karemaker ID, Santanach A, et al. The dynamic interactome and genomic targets of Polycomb complexes during stem-cell differentiation. Nat Struct Mol Biol. 2016;23(7):682–90.
Ji Y, Fioravanti J, Zhu W, Wang H, Wu T, Hu J, et al. miR-155 harnesses Phf19 to potentiate cancer immunotherapy through epigenetic reprogramming of CD8(+) T cell fate. Nat Commun. 2019;10(1):2157.
Jain P, Ballare C, Blanco E, Vizan P, Di Croce L. PHF19 mediated regulation of proliferation and invasiveness in prostate cancer cells. Elife. 2020;9.
Deng Q, Hou J, Feng L, Lv A, Ke X, Liang H, et al. PHF19 promotes the proliferation, migration, and chemosensitivity of glioblastoma to doxorubicin through modulation of the SIAH1/beta-catenin axis. Cell Death Dis. 2018;9(11):1049.
Li G, Warden C, Zou Z, Neman J, Krueger JS, Jain A, et al. Altered expression of polycomb group genes in glioblastoma multiforme. PLoS One. 2013;8(11):e80970.
Chen S, Fu Z, Wen S, Yang X, Yu C, Zhou W, et al. Expression and diagnostic value of miR-497 and miR-1246 in hepatocellular carcinoma. Front Genet. 2021;12:666306.
Xu H, Hu YW, Zhao JY, Hu XM, Li SF, Wang YC, et al. MicroRNA-195-5p acts as an anti-oncogene by targeting PHF19 in hepatocellular carcinoma. Oncol Rep. 2015;34(1):175–82.
**aoyun S, Yuyuan Z, Jie X, Yingjie N, Qing X, Yuezhen D, et al. PHF19 activates hedgehog signaling and promotes tumorigenesis in hepatocellular carcinoma. Exp Cell Res. 2021;406(1):112690.
Ruan S, Zhang H, Tian X, Zhang Z, Huang H, Shi C, et al. PHD finger protein 19 enhances the resistance of ovarian cancer cells to compound fuling granule by protecting cell growth, invasion, migration, and stemness. Front Pharmacol. 2020;11:150.
Tao F, Tian X, Ruan S, Shen M, Zhang Z. miR-211 sponges lncRNA MALAT1 to suppress tumor growth and progression through inhibiting PHF19 in ovarian carcinoma. FASEB J. 2018:fj201800495RR.
Ghislin S, Deshayes F, Middendorp S, Boggetto N, Alcaide-Loridan C. PHF19 and Akt control the switch between proliferative and invasive states in melanoma. Cell Cycle. 2012;11(8):1634–45.
Murakami H, Ito S, Tanaka H, Kondo E, Kodera Y, Nakanishi H. Establishment of new intraperitoneal paclitaxel-resistant gastric cancer cell lines and comprehensive gene expression analysis. Anticancer Res. 2013;33(10):4299–307.
Wang H, Xu P, Sun G, Lv J, Cao J, Xu Z. Downregulation of PHF19 inhibits cell growth and migration in gastric cancer. Scand J Gastroenterol. 2020;55(6):687–93.
Zhang J, Lv W, Liu Y, Fu W, Chen B, Ma Q, et al. LINC_00355 promotes gastric cancer progression by upregulating PHF19 expression through sponging miR-15a-5p. BMC Cancer. 2021;21(1):657.
Gollavilli PN, Pawar A, Wilder-Romans K, Natesan R, Engelke CG, Dommeti VL, et al. EWS/ETS-driven ewing sarcoma requires BET bromodomain proteins. Cancer Res. 2018;78(16):4760–73.
Li P, Sun J, Ruan Y, Song L. High PHD Finger Protein 19 (PHF19) expression predicts poor prognosis in colorectal cancer: a retrospective study. PeerJ. 2021;9:e11551.
Abdelfettah S, Boulay G, Dubuissez M, Spruyt N, Garcia SP, Rengarajan S, et al. hPCL3S promotes proliferation and migration of androgen-independent prostate cancer cells. Oncotarget. 2020;11(12):1051–74.
Garcia-Montolio M, Ballare C, Blanco E, Gutierrez A, Aranda S, Gomez A, et al. Polycomb factor PHF19 controls cell growth and differentiation toward erythroid pathway in chronic myeloid leukemia cells. Front Cell Dev Biol. 2021;9:655201.
Pan MR, Hsu MC, Chen LT, Hung WC. G9a orchestrates PCL3 and KDM7A to promote histone H3K27 methylation. Sci Rep. 2015;5:18709.
Yu T, Du C, Ma X, Sui W, Yu Z, Liu L, et al. Polycomb-like protein 3 induces proliferation and drug resistance in multiple myeloma and is regulated by miRNA-15a. Mol Cancer Res. 2020;18(7):1063–73.
Li Z, Liu L, Du C, Yu Z, Yang Y, Xu J, et al. Therapeutic effects of oligo-single-stranded DNA mimicking of hsa-miR-15a-5p on multiple myeloma. Cancer Gene Ther. 2020;27(12):869–77.
Bagger FO, Kinalis S, Rapin N. BloodSpot: a database of healthy and malignant haematopoiesis updated with purified and single cell mRNA sequencing profiles. Nucleic Acids Res. 2019;47(D1):D881–D5.
Kassambara A, Reme T, Jourdan M, Fest T, Hose D, Tarte K, et al. GenomicScape: an easy-to-use web tool for gene expression data analysis. Application to investigate the molecular events in the differentiation of B cells into plasma cells. PLoS Comput Biol. 2015;11(1):e1004077.
Vizan P, Gutierrez A, Espejo I, Garcia-Montolio M, Lange M, Carretero A, et al. The Polycomb-associated factor PHF19 controls hematopoietic stem cell state and differentiation. Sci Adv. 2020;6(32):eabb2745.
Majewski IJ, Ritchie ME, Phipson B, Corbin J, Pakusch M, Ebert A, et al. Opposing roles of polycomb repressive complexes in hematopoietic stem and progenitor cells. Blood. 2010;116(5):731–9.
Lee SC, Miller S, Hyland C, Kauppi M, Lebois M, Di Rago L, et al. Polycomb repressive complex 2 component Suz12 is required for hematopoietic stem cell function and lymphopoiesis. Blood. 2015;126(2):167–75.
Yu W, Zhang F, Wang S, Fu Y, Chen J, Liang X, et al. Depletion of polycomb repressive complex 2 core component EED impairs fetal hematopoiesis. Cell Death Dis. 2017;8(4):e2744.
Kinkel SA, Galeev R, Flensburg C, Keniry A, Breslin K, Gilan O, et al. Jarid2 regulates hematopoietic stem cell function by acting with polycomb repressive complex 2. Blood. 2015;125(12):1890–900.
Rothberg JLM, Maganti HB, Jrade H, Porter CJ, Palidwor GA, Cafariello C, et al. Mtf2-PRC2 control of canonical Wnt signaling is required for definitive erythropoiesis. Cell Discov. 2018;4:21.
Beguelin W, Popovic R, Teater M, Jiang Y, Bunting KL, Rosen M, et al. EZH2 is required for germinal center formation and somatic EZH2 mutations promote lymphoid transformation. Cancer Cell. 2013;23(5):677–92.
Guo M, Price MJ, Patterson DG, Barwick BG, Haines RR, Kania AK, et al. EZH2 represses the B cell transcriptional program and regulates antibody-secreting cell metabolism and antibody production. J Immunol. 2018;200(3):1039–52.
Caganova M, Carrisi C, Varano G, Mainoldi F, Zanardi F, Germain PL, et al. Germinal center dysregulation by histone methyltransferase EZH2 promotes lymphomagenesis. J Clin Invest. 2013;123(12):5009–22.
Beguelin W, Teater M, Gearhart MD, Calvo Fernandez MT, Goldstein RL, Cardenas MG, et al. EZH2 and BCL6 cooperate to assemble CBX8-BCOR complex to repress bivalent promoters, mediate germinal center formation and lymphomagenesis. Cancer Cell. 2016;30(2):197–213.
Beguelin W, Rivas MA, Calvo Fernandez MT, Teater M, Purwada A, Redmond D, et al. EZH2 enables germinal centre formation through epigenetic silencing of CDKN1A and an Rb-E2F1 feedback loop. Nat Commun. 2017;8(1):877.
Jego G, Bataille R, Pellat-Deceunynck C. Interleukin-6 is a growth factor for nonmalignant human plasmablasts. Blood. 2001;97(6):1817–22.
Jourdan M, Caraux A, Caron G, Robert N, Fiol G, Reme T, et al. Characterization of a transitional preplasmablast population in the process of human B cell to plasma cell differentiation. J Immunol. 2011;187(8):3931–41.
Jourdan M, Caraux A, De Vos J, Fiol G, Larroque M, Cognot C, et al. An in vitro model of differentiation of memory B cells into plasmablasts and plasma cells including detailed phenotypic and molecular characterization. Blood. 2009;114(25):5173–81.
Herviou L, Jourdan M, Martinez AM, Cavalli G, Moreaux J. EZH2 is overexpressed in transitional preplasmablasts and is involved in human plasma cell differentiation. Leukemia. 2019;33(8):2047–60.
Xu K, Wu ZJ, Groner AC, He HH, Cai C, Lis RT, et al. EZH2 oncogenic activity in castration-resistant prostate cancer cells is Polycomb-independent. Science. 2012;338(6113):1465–9.
Xu H, Xu K, He HH, Zang C, Chen CH, Chen Y, et al. Integrative analysis reveals the transcriptional collaboration between EZH2 and E2F1 in the regulation of cancer-related gene expression. Mol Cancer Res. 2016;14(2):163–72.
Musselman CA, Avvakumov N, Watanabe R, Abraham CG, Lalonde ME, Hong Z, et al. Molecular basis for H3K36me3 recognition by the tudor domain of PHF1. Nat Struct Mol Biol. 2012;19(12):1266–72.
Li X, Isono K, Yamada D, Endo TA, Endoh M, Shinga J, et al. Mammalian polycomb-like Pcl2/Mtf2 is a novel regulatory component of PRC2 that can differentially modulate polycomb activity both at the Hox gene cluster and at Cdkn2a genes. Mol Cell Biol. 2011;31(2):351–64.
Walker E, Chang WY, Hunkapiller J, Cagney G, Garcha K, Torchia J, et al. Polycomb-like 2 associates with PRC2 and regulates transcriptional networks during mouse embryonic stem cell self-renewal and differentiation. Cell Stem Cell. 2010;6(2):153–66.
Ning F, Wang C, Niu S, Xu H, **a K, Wang N. Transcription factor Phf19 positively regulates germinal center reactions that underlies its role in rheumatoid arthritis. Am J Transl Res. 2018;10(1):200–11.
Buil A, Martinez-Perez A, Perera-Lluna A, Rib L, Caminal P, Soria JM. A new gene-based association test for genome-wide association studies. BMC Proc. 2009;3(Suppl 7):S130.
Chang M, Rowland CM, Garcia VE, Schrodi SJ, Catanese JJ, van der Helm-van Mil AH, et al. A large-scale rheumatoid arthritis genetic study identifies association at chromosome 9q33.2. PLoS Genet. 2008;4(6):e1000107.
Zhan F, Hardin J, Kordsmeier B, Bumm K, Zheng M, Tian E, et al. Global gene expression profiling of multiple myeloma, monoclonal gammopathy of undetermined significance, and normal bone marrow plasma cells. Blood. 2002;99(5):1745–57.
Zhan F, Tian E, Bumm K, Smith R, Barlogie B, Shaughnessy J Jr. Gene expression profiling of human plasma cell differentiation and classification of multiple myeloma based on similarities to distinct stages of late-stage B-cell development. Blood. 2003;101(3):1128–40.
Pawlyn C, Kaiser MF, Heuck C, Melchor L, Wardell CP, Murison A, et al. The spectrum and clinical impact of epigenetic modifier mutations in myeloma. Clin Cancer Res. 2016;22(23):5783–94.
Erokhin M, Chetverina O, Gyorffy B, Tatarskiy VV, Mogila V, Shtil AA, et al. Clinical correlations of polycomb repressive complex 2 in different tumor types. Cancers. 2021;13(13).
Herviou L, Kassambara A, Boireau S, Robert N, Requirand G, Muller-Tidow C, et al. PRC2 targeting is a therapeutic strategy for EZ score defined high-risk multiple myeloma patients and overcome resistance to IMiDs. Clin Epigenetics. 2018;10(1):121.
Boyle EM, Rosenthal A, Ghamlouch H, Wang Y, Farmer P, Rutherford M, et al. Plasma cells expression from smouldering myeloma to myeloma reveals the importance of the PRC2 complex, cell cycle progression, and the divergent evolutionary pathways within the different molecular subgroups. Leukemia. 2021.
Neo WH, Lim JF, Grumont R, Gerondakis S, Su IH. c-Rel regulates Ezh2 expression in activated lymphocytes and malignant lymphoid cells. J Biol Chem. 2014;289(46):31693–707.
Rizk M, Rizq O, Oshima M, Nakajima-Takagi Y, Koide S, Saraya A, et al. Akt inhibition synergizes with polycomb repressive complex 2 inhibition in the treatment of multiple myeloma. Cancer Sci. 2019;110(12):3695–707.
Croonquist PA, Van Ness B. The polycomb group protein enhancer of zeste homolog 2 (EZH 2) is an oncogene that influences myeloma cell growth and the mutant ras phenotype. Oncogene. 2005;24(41):6269–80.
Pan YM, Wang CG, Zhu M, **ng R, Cui JT, Li WM, et al. STAT3 signaling drives EZH2 transcriptional activation and mediates poor prognosis in gastric cancer. Mol Cancer. 2016;15(1):79.
Rizq O, Mimura N, Oshima M, Saraya A, Koide S, Kato Y, et al. Dual Inhibition of EZH2 and EZH1 sensitizes PRC2-dependent tumors to proteasome inhibition. Clin Cancer Res. 2017;23(16):4817–30.
Gonzalez ME, DuPrie ML, Krueger H, Merajver SD, Ventura AC, Toy KA, et al. Histone methyltransferase EZH2 induces Akt-dependent genomic instability and BRCA1 inhibition in breast cancer. Cancer Res. 2011;71(6):2360–70.
Kim J, Lee Y, Lu X, Song B, Fong KW, Cao Q, et al. Polycomb- and methylation-independent roles of EZH2 as a transcription activator. Cell Rep. 2018;25(10):2808–20 e4.
Yan J, Li B, Lin B, Lee PT, Chung TH, Tan J, et al. EZH2 phosphorylation by JAK3 mediates a switch to noncanonical function in natural killer/T-cell lymphoma. Blood. 2016;128(7):948–58.
Li Z, Li M, Wang D, Hou P, Chen X, Chu S, et al. Post-translational modifications of EZH2 in cancer. Cell Biosci. 2020;10(1):143.
Cha TL, Zhou BP, **a W, Wu Y, Yang CC, Chen CT, et al. Akt-mediated phosphorylation of EZH2 suppresses methylation of lysine 27 in histone H3. Science. 2005;310(5746):306–10.
Kikuchi J, Koyama D, Wada T, Izumi T, Hofgaard PO, Bogen B, et al. Phosphorylation-mediated EZH2 inactivation promotes drug resistance in multiple myeloma. J Clin Invest. 2015;125(12):4375–90.
Kalushkova A, Fryknas M, Lemaire M, Fristedt C, Agarwal P, Eriksson M, et al. Polycomb target genes are silenced in multiple myeloma. PLoS One. 2010;5(7):e11483.
Popovic R, Martinez-Garcia E, Giannopoulou EG, Zhang Q, Zhang Q, Ezponda T, et al. Histone methyltransferase MMSET/NSD2 alters EZH2 binding and reprograms the myeloma epigenome through global and focal changes in H3K36 and H3K27 methylation. PLoS Genet. 2014;10(9):e1004566.
Alzrigat M, Jernberg-Wiklund H, Licht JD. Targeting EZH2 in multiple myeloma-multifaceted anti-tumor activity. Epigenomes. 2018;2(3).
Nylund P, Atienza Parraga A, Haglof J, De Bruyne E, Menu E, Garrido-Zabala B, et al. A distinct metabolic response characterizes sensitivity to EZH2 inhibition in multiple myeloma. Cell Death Dis. 2021;12(2):167.
Yuan W, Xu M, Huang C, Liu N, Chen S, Zhu B. H3K36 methylation antagonizes PRC2-mediated H3K27 methylation. J Biol Chem. 2011;286(10):7983–9.
Agger K, Cloos PA, Christensen J, Pasini D, Rose S, Rappsilber J, et al. UTX and JMJD3 are histone H3K27 demethylases involved in HOX gene regulation and development. Nature. 2007;449(7163):731–4.
Ezponda T, Dupere-Richer D, Will CM, Small EC, Varghese N, Patel T, et al. UTX/KDM6A loss enhances the malignant phenotype of multiple myeloma and sensitizes Cells to EZH2 inhibition. Cell Rep. 2017;21(3):628–40.
Ren Z, Ahn JH, Liu H, Tsai YH, Bhanu NV, Koss B, et al. PHF19 promotes multiple myeloma tumorigenicity through PRC2 activation and broad H3K27me3 domain formation. Blood. 2019;134(14):1176–89.
Barretina J, Caponigro G, Stransky N, Venkatesan K, Margolin AA, Kim S, et al. The cancer cell line encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature. 2012;483(7391):603–7.
Loven J, Hoke HA, Lin CY, Lau A, Orlando DA, Vakoc CR, et al. Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell. 2013;153(2):320–34.
Fedele PL, Liao Y, Gong JN, Yao Y, van Delft MF, Low MSY, et al. The transcription factor IRF4 represses proapoptotic BMF and BIM to licence multiple myeloma survival. Leukemia. 2021;35(7):2114–8.
Rhyasen GW, Hattersley MM, Yao Y, Dulak A, Wang W, Petteruti P, et al. AZD5153: a novel bivalent BET bromodomain inhibitor highly active against hematologic malignancies. Mol Cancer Ther. 2016;15(11):2563–74.
Lin CY, Loven J, Rahl PB, Paranal RM, Burge CB, Bradner JE, et al. Transcriptional amplification in tumor cells with elevated c-Myc. Cell. 2012;151(1):56–67.
Shaffer AL, Emre NC, Lamy L, Ngo VN, Wright G, **ao W, et al. IRF4 addiction in multiple myeloma. Nature. 2008;454(7201):226–31.
Ohguchi H, Park PMC, Wang T, Gryder BE, Ogiya D, Kurata K, et al. Lysine demethylase 5A is required for MYC driven transcription in multiple myeloma. Blood Cancer Discov. 2021;2(4):370–87.
Shaughnessy JD Jr, Zhan F, Burington BE, Huang Y, Colla S, Hanamura I, et al. A validated gene expression model of high-risk multiple myeloma is defined by deregulated expression of genes map** to chromosome 1. Blood. 2007;109(6):2276–84.
Kuiper R, Broyl A, de Knegt Y, van Vliet MH, van Beers EH, van der Holt B, et al. A gene expression signature for high-risk multiple myeloma. Leukemia. 2012;26(11):2406–13.
Decaux O, Lode L, Magrangeas F, Charbonnel C, Gouraud W, Jezequel P, et al. Prediction of survival in multiple myeloma based on gene expression profiles reveals cell cycle and chromosomal instability signatures in high-risk patients and hyperdiploid signatures in low-risk patients: a study of the Intergroupe Francophone du Myelome. J Clin Oncol. 2008;26(29):4798–805.
Wall MA, Turkarslan S, Wu WJ, Danziger SA, Reiss DJ, Mason MJ, et al. Genetic program activity delineates risk, relapse, and therapy responsiveness in multiple myeloma. NPJ Precis Oncol. 2021;5(1):60.
Johnson T, Yu C, Huang Z, Xu S, Wang T, Dong C, et al. Diagnostic evidence GAuge of single cells (DEGAS): A flexible deep-transfer learning framework for prioritizing cells in relation to disease. bioRxiv. 2021.
Wu SC, Zhang Y. Cyclin-dependent kinase 1 (CDK1)-mediated phosphorylation of enhancer of zeste 2 (Ezh2) regulates its stability. J Biol Chem. 2011;286(32):28511–9.
Wei Y, Chen YH, Li LY, Lang J, Yeh SP, Shi B, et al. CDK1-dependent phosphorylation of EZH2 suppresses methylation of H3K27 and promotes osteogenic differentiation of human mesenchymal stem cells. Nat Cell Biol. 2011;13(1):87–94.
Li Y, Gong J, Zhang L. PHD finger protein 19 expression in multiple myeloma: association with clinical features, induction therapy outcome, disease progression, and survival. J Clin Lab Anal. 2021;35(9):e23910.
Holmes AB, Corinaldesi C, Shen Q, Kumar R, Compagno N, Wang Z, et al. Single-cell analysis of germinal-center B cells informs on lymphoma cell of origin and outcome. J Exp Med. 2020;217(10).
King HW, Orban N, Riches JC, Clear AJ, Warnes G, Teichmann SA, et al. Single-cell analysis of human B cell maturation predicts how antibody class switching shapes selection dynamics. Sci Immunol. 2021;6(56).
Zhan F, Huang Y, Colla S, Stewart JP, Hanamura I, Gupta S, et al. The molecular classification of multiple myeloma. Blood. 2006;108(6):2020–8.
Acknowledgements
Gareth Morgan is supported by the Leukemia Lymphoma Society, the Multiple Myeloma Research Foundation and the Ammon Foundation.
Funding
The authors received no specific funding for this work.
Author information
Authors and Affiliations
Contributions
HG and GJM drafted the manuscript and finalized it with input and revision from EMB, PB, YW, JC, LW, MB, DA, BB, BAW, FED. HG, EMB, PB, MB performed the bioinformatics analysis. HG prepared the figures. All authors approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Supplementary Table 1.
Transcripts and protein isoforms of PHF19 reported in Ensembl, NCBI and protein human atlas databases.
Additional file 2: Supplementary Table 2.
Genes that represent PHF19 transcriptional signature in multiple myeloma.
Additional file 3: Supplementary Table 3.
Genes potentially regulated by PHF19 in hematopoietic stem cells and in MM cells.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Ghamlouch, H., Boyle, E.M., Blaney, P. et al. Insights into high-risk multiple myeloma from an analysis of the role of PHF19 in cancer. J Exp Clin Cancer Res 40, 380 (2021). https://doi.org/10.1186/s13046-021-02185-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13046-021-02185-1