
Abstract
Inflammasomes are macromolecular platforms formed in response to damage-associated molecular patterns (DAMPs) and pathogen-associated molecular patterns, whose formation would cause maturation of interleukin-1 (IL-1) family members and gasdermin D (GSDMD), leading to IL-1 secretion and pyroptosis respectively. Several kinds of inflammasomes detecting different types of dangers have been found. The activation of inflammasomes is regulated at both transcription and posttranscription levels, which is crucial in protecting the host from infections and sterile insults. Present findings have illustrated that inflammasomes are involved in not only infection but also the pathology of tumors implying an important link between inflammation and tumor development. Generally, inflammasomes participate in tumorigenesis, cell death, metastasis, immune evasion, chemotherapy, target therapy, and radiotherapy. Inflammasome components are upregulated in some tumors, and inflammasomes can be activated in cancer cells and other stromal cells by DAMPs, chemotherapy agents, and radiation. In some cases, inflammasomes inhibit tumor progression by initiating GSDMD-mediated pyroptosis in cancer cells and stimulating IL-1 signal-mediated anti-tumor immunity. However, IL-1 signal recruits immunosuppressive cell subsets in other cases. We discuss the conflicting results and propose some possible explanations. Additionally, we also summarize interventions targeting inflammasome pathways in both preclinical and clinical stages. Interventions targeting inflammasomes are promising for immunotherapy and combination therapy.
Similar content being viewed by others


Background
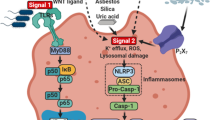
One of the crucial functions of the innate immune system is to recognize DAMPs and PAMPs by pattern recognition receptors (PRRs) during microbial infection and sterile damage [1]. Some PRRs, such as Toll-like receptors (TLRs), are located in the cytoplasm membrane and endosome membrane to supervise extracellular and endosomal dangers [2]. In the cytosol, nucleotide-binding leucine-rich repeat receptors (NLRs), absent in melanoma 2 (AIM2), and pyrin are able to recognize cytosolic DAMPs and PAMPs [3]. Distinct from TLRs that eventually elevate pro-inflammatory cytokines, type I interferons, and chemokines at the transcription level, NLRs (NACHT, leucine-rich repeat and pyrin domain-containing 1 (NLRP1), NOD-, LRR- and pyrin domain-containing 3 (NLRP3), and NLR family apoptosis inhibitory protein (NAIP)/NLR family CARD domain-containing 4 (NLRC4)), AIM2, and pyrin initiate posttranslational mechanisms by assembling inflammasomes, a group of multicomponent complexes [4, 5]. Briefly, inflammasome sensors recruit caspase-1 family members with or without the assistance of apoptosis-associated speck-like protein-containing CARD (ASC) to initiate auto-cleavage of caspase-1. The activated caspase-1 cleaves precursors of GSDMD and IL-1 family members to release these cytokines and induce pyroptosis. The canonical and non-canonical inflammasome pathways are summarized in Fig. 1.

Overview of inflammasome activation
Canonical inflammasomes are composed of sensors, ASC, and caspase-1 [5]. Once activated, these inflammasome sensors oligomerize and recruit ASC to form an “ASC speck” through pyrin–pyrin (PYD–PYD) interaction [6, 7]. Then caspase-1 is recruited to ASC through CARD–CARD interaction [6, 7]. However, exceptions have been reported in the activation of NLRP1 and NLRC4. CARD domain of the NLRP1 directly recruits caspase-1 through CARD–CARD interaction without ASC [8, 9]. However, human NLRP1 also recruits ASC through the PYD domain [10]. For NLRC4, caspase-1 can be recruited to NLRC4 with (through CARD–CARD interaction between NLRC4 and ASC [11]) or without (through CARD–CARD interaction between NLRC4 and caspase-1 [12]) ASC, although differences in size and duration of activated inflammasomes have been observed between these two kinds of NLRC4 inflammasomes [13]. The recruited caspase-1 (also known as caspase-11 in mice) dimerizes and autoclaves to generate p33/p10 species with full protease activity [13]. The cleaved caspase-1 is able to process pro-IL-1β at D26 and D116 and pro-IL-18 at D36 to produce active IL-1β and IL-18 [14]. GSDMD is also cleaved by the caspase-1 to release the amino-terminal domain of GSDMD, which inserts into the plasma membrane to form GSDMD pores leading to pyroptosis [15,16,17]. The GSDMD oligomerization relies on mitochondria reactive oxygen species (mtROS) provoked by the Ragulator-Rag complex and its downstream mTORC1 [18]. In some cases, cleaved GSDMD can insert into mitochondrial membranes [19, 20]. The leakage of mtROS switches pyroptosis into necroptosis [19]. Through GSDMD pores, are mature IL-1β and IL-18 released into the extracellular environment [21, 22]. A good question is how the GSDMD pores distinguish mature IL-1β and IL-18 from their precursors. A recently published cryo-electron microscopy analysis shows a predominantly negatively charged conduit of the GSDMD pore that favors the passage of mature IL-1β and IL-18 and sequestrates negatively charged IL-1 precursors containing acidic domain [159, 203, 246]. These agents inhibit NLRP3 in different mechanisms, some of which remain elusive.
The most commonly used NLRP3 inhibitor in preclinical experiments is MCC950 (CRID3), which inhibits NLRP3 with nM potency without interfering with other inflammasome sensors [246]. Mechanistically, MCC950 interacts with Walker B motif of NACHT domain that is close to the ATP binding pocket, thereby blocking the hydrolysis of ATP and suppressing NLRP3 activation [247]. This specific blockage is consistent no matter whether in wild-type or mutated NLRP3 [248]. Another structural research has illustrated that the sulfonylurea group of MCC950 interacts with the Walker A motif of NLRP3 and it is sandwiched between Arg351 and Arg578 resulting in stabilized NACHT and LLR domains relative to each other [63]. MCC950 is initially developed as a potential therapeutic agent for CAPS, as well as other autoinflammatory and autoimmune diseases [246]. Later research works illustrate the potential anti-tumor effect of MCC950 against pancreatic cancer and head and neck squamous cell carcinoma [159, 203]. Similar to MCC950, the target of CY-09 is Walker A motif of NACHT, which binds ATP [249]. Another inhibitor that targets NACHT domain is tranilast [250]. However, it also suppresses TGF-β, MAPK, and NF-κB signals [251]. Present results have demonstrated that tranilast inhibits malignant behaviors of NSCLC and gastric cancer [252, 253], but the authors do not clarify whether these effects are related to inhibited inflammasome signal. Other inhibitors targeting NACHT domain include 3,4-methylenedioxy-β-nitrostyrene (MNS) [254] and oridonin [255]. MNS also binds to LRR domain [254]. Interestingly, isoliquiritigenin and glycyrrhizin are able to inhibit NLRP3 through both signal 1 (TLR4) and signal 2 (NLRP3) [256], but their inhibitory potency is not as powerful as that of MCC950. There is also an indirect NLRP3 inhibitor, ibrutinib [257]. Ibrutinib inhibits the generation of phosphorylated Bruton tyrosine kinase (BTK) that directly interacts with NLRP3 and ASC leading to the formation of inflammasomes [257]. Another indirect NLRP3 modulator is resveratrol that suppresses the expression of NLRP3 in renal cancer cells [258]. In a word, present findings imply that NACHT domain is the key target for inhibitors.
Although NLRP3 inhibitors alone have shown anti-tumor effects, some attempts highlighted the potential combination of NLRP3 inhibitors with other therapeutic methods. OLT1177 disrupts IL-1β/IL-6/STAT3 axis in TME resulting in reduced tumor growth through attenuating immunosuppressive activities in MDSCs [202], and the anti-tumor effect is further enhanced in combination with anti-PD-1 [245]. In addition to enhancing therapeutic effects, NLRP3 inhibitors may protect against the side effects of chemotherapy and radiotherapy. Resveratrol reduces doxorubicin-induced cardiac injury and systemic inflammation through downregulating NLRP3 inflammasomes [259]. Likewise, bowel inflammation after irradiation is also suppressed by resveratrol through a similar mechanism [228]. More research works are needed to explore other possibilities of such a combination.
It is suppressing that although many kinds of NLRP3 inhibitors have been invented, only a few of these agents have entered clinical trials for tumor therapy. ACT001 combined with anti-PD-1 or ACT001 alone has been applied for a phase I/II trial against glioblastoma. This agent is primarily developed for Parkinson’s disease [260]. Others are some agents that have been reported to inhibit NLRP3, including glycyrrhizin and andrographolides. Whether their anti-tumor effects are underpinned by NLRP3 inhibition remains to be further evaluated.
AIM2 inhibitors
Compared with NLRP3, limited AIM2 inhibitors have been found. Two agents are able to inhibit AIM2, but they are not specific inhibitors. Glycyrrhizin suppresses both AIM2 and NLRP3 [256]. Methylene blue is a broad-spectrum inflammasome inhibitor against NLRP3, NLRC4, AIM2, and non-canonical inflammasomes [261]. Fortunately, andrographolide has shown a promising effect for the future clinical application that it reduces radiation-induced lung inflammation and fibrosis by preventing AIM2 from entering the nucleus and sensing DNA damage [224]. Although potential anti-tumor effects of glycyrrhizin, andrographolide, and methylene blue would be evaluated in colon cancer, breast cancer, and liver cancer during clinical trials, specific AIM2 inhibitors are in need.
NLRC4 inhibitors
Few specific NLRC4 inhibitors are available. Instead, two inflammasome inhibitors with limited selectivity have been reported to suppress NLRC4. Sulforaphane attenuates the activation of both NLRC4 and NLRP3 at μM potency, which limits inflammation during peritonitis [262]. Methylene blue, a broad-spectrum inflammasome inhibitor, blocks NLRC4, NLRP3, AIM2, and non-canonical inflammasomes, which improves the survival rate of mice challenged with LPS [261]. Considering that many NLRP3 inhibitors target NACHT and that NACHT domain also exists in the NLRC4 inflammasomes, future selective NLRC4 inhibitors might be the derivate of the NLRP3 inhibitors.
Caspase-1 inhibitors
Owing to the upsurge of the study in caspase-related signals, many inhibitors targeting caspase family members have been developed, some of which are able to inhibit caspase-1. As the inhibitors of the common downstream protein of inflammasomes, caspase-1 inhibitors restrain not only NLRP3-derived but also AIM2-derived inflammasome signals. For example, VX-765 inhibits NLRP3/caspase-1/GSDMD-induced pyroptosis in NSCLC [263], and it also attenuates AIM2-mediated cell migration in NSCLC [264]. An interesting question is whether caspase-1 inhibitors promote or suppress cancer cell growth. Direct inhibition of pyroptosis has been reported in NSCLC by VX-765 [263], liver cancer by Ac-YVAD-CMK [265], and prostate cancer by Z-YVAD-fmk [266]. On the contrary, the caspase-1 inhibitor, thalidomide, impedes tumor growth in melanoma by suppressing caspase-1 in MDSCs [267]. Thus, non-selective administration of caspase-1 inhibitors may promote tumor growth, while selective caspase-1 inhibition in MDSCs may attenuate tumor development. Of note, in the early years when inflammasome signal was not intensively studied, caspase-1 mediated cell death was regarded as apoptosis [268, 269]. These findings should be updated to clarify the kind of cell death. Additionally, the relationship between pyroptosis of cancer cells and tumor growth should be further studied. Because DAMPs from dead cancer cells may elicit inflammasomes in adjacent myeloid cells and probably cancer cells resulting in the recruitment of MDSCs that facilitate tumor growth. At present, thalidomide alone or plus other agents have entered clinical evaluation against multiple myeloma, prostate cancer, and other advanced cancer.
ASC inhibitors
Although MCC950 has been demonstrated to selectively block the NACHT domain of NLRP3, it is also able to downregulate protein expression of ASC, caspase-1, IL-1β, and IL-18 [270]. This is an in vivo study that tests protein expression in tissues. Thus, it is possible that MCC950 directly inhibits NLRP3-mediated pyroptosis and IL-1β and IL-18 secretion causing reduced infiltration of macrophages [270]. Decreased number of macrophages in tissues may explain the downregulated protein levels of the inflammasome components. However, another research has found that MCC950 inhibits both NLRP3 and AIM2-derived inflammasome formation [271]. The MCC950-mediated ASC suppression is possibly through Glutathione S-Transferase Omega 1 (GSTO1), a putative target of MCC950 [271]. In a word, there is a lack of selective and direct ASC inhibitors.
GSDMD inhibitors
GSDMD pores are the direct cause of pyroptosis and the exit for intracellular mature IL-1β and IL-18. In addition to blocking pyroptosis and secretion of pro-inflammatory cytokines, two GSDMD inhibitors, LDC7559 [272] and Disulfiram [273], also restrain inflammation through curbing NETosis, a special kind of cell death of neutrophils. Although GSDMD inhibitors, disulfiram and Bay 11–7082, potently suppress pyroptosis [274], they show anti-tumor effects through inducing ferroptosis [275] (by disulfiram) or apoptosis [276, 277] (by Bay 11–7082). In a word, the anti-inflammation effects of GSDMD inhibitors have been repeatedly proven, but their applications in tumor development remain to be further evaluated.
IL-1 signal inhibitors
Four targets of the IL-1 signal have been developed, including IL-1 receptor, IL-1α, IL-1β, and IL-18 that can be intervened by antagonists, antibodies, and binding proteins. These potent anti-inflammatory inhibitors are pleiotropic agents applied in various inflammation-related diseases, for example, rheumatoid arthritis [278] (by canakinumab), autoimmune disorders [279, 280] (by rilonacept), and cardiac remodeling [281] (by gevokizumab). Blocking IL-1 signals might promote or inhibit tumor development. IL-18BP, a binding protein targeting IL-18, limits anti-tumor immunity [282]. However, anakinra, an IL-1 receptor antagonist, reduces IL-1β and downstream production of cancer-promoting IL-22 [283]. Similarly, anti-inflammatory therapy in patients with atherosclerosis by canakinumab reduces lung cancer incidence [284]. This effect has been proven to be underpinned by the reduced tumor-promoting inflammation [285]. For tumor therapies, anakinra gives rise to cytotoxic/NK cell transcriptional pathways and hampers innate inflammation in breast cancer patients receiving chemotherapy [286]. Additionally, anakinra is reported to limit the mucosal barrier injury and the accompanying clinical symptoms induced by melphalan [287]. Although more frequent fatal infections and sepsis are recorded in the canakinumab treatment group, all-cause mortality does not differ significantly between the placebo and the canakinumab group [284]. On the contrary, the anakinra treatment seems to be more safety [286], and no adverse events or dose-limiting toxicities have been observed [287]. In another phase 2 clinical trial, anakinra is applied in patients receiving 5-FU plus bevacizumab therapy, and no grade 4/5 toxicity related to therapy occurs during the study [288]. Interestingly, anakinra abrogates cytokine release syndrome during CAR-T therapy implying its compelling clinical application [289]. A series of clinical trials testing the prevention of CAR-T cell-mediated toxicity by anakinra have been launched, such as NCT04432506, NCT04150913, and NCT04148430. At present, most interventions targeting inflammasome pathways for cancer therapies listed in Table 3 are based on IL-1 signal inhibitors, possibly owing to the ready-made agents for other non-malignant diseases. For example, the therapeutic effects of canakinumab in lung cancer, colon cancer, breast cancer, pancreatic cancer, renal cancer, and leukemia would be evaluated in a number of clinical trials. It is a compelling topic to test whether the combination of these IL-1 signal inhibitors with other therapies can be beneficial for patients.
Inflammasome activators
Although many results support that activated inflammasomes show anti-tumor effects directly through inducing pyroptosis and indirectly through stimulating immune cells, limited inflammasome activators are developed at present. Polyphyllin VI induces pyroptosis by activating NLRP3 in NSCLC cells [263]. Similarly, 17β-estradiol provokes pyroptosis via NLRP3 in liver cancer cells [265, 290]. Another NLRP3 activator, BMS-986299, shows potential anticancer effects, but the details are largely unknown [3]. BMS-986299 have entered a phase I trial to explore its safety and effectiveness in patients with solid tumor or advanced tumor. An alternative strategy is to supply the downstream IL-1 cytokines directly. Since IL-18 is likely to be beneficial for anti-tumor immunity [182, 282], recombinant IL-18 has been applied in several clinical trials such as NCT00659178, NCT00107718, and NCT00500058. In the clinical trial NCT04684563, CAR-T cells targeting CD19 and expressing IL-18 are applied in patients with chronic lymphocytic leukemia or non-Hodgkin lymphoma. Present results indicate that more efforts should be paid to develop inflammasome activators. Considering that inflammasomes may initiate pyroptosis in tumor cells and that IL-1β and IL-18 have been shown to activate T cells and NK cells, inflammasome activators may improve the effects of immune checkpoint inhibitors.
Conclusions
In this review, we summarize the mechanisms that activate canonical and non-canonical inflammasome pathways. More importantly, we discuss the roles of canonical and non-canonical inflammasomes in tumorigenesis, tumor cell death, tumor metastasis, immune evasion, chemotherapy, and radiotherapy. Finally, we review the interventions targeting the inflammasome pathways in preclinical and clinical stages.
A good question is how the inflammasomes are activated in TME. Expression levels of inflammasome components have been compared between healthy and tumor tissues [117, 134, 138, 170, 183]. Mice deficient in certain components of the inflammasome pathway [138, 182, 183] or inflammasome inhibitors [196, 291, 292] have been applied to reveal the various influences of inflammasomes on tumor behaviors. Inflammasome activators (such as ATP, H2O2, monosodium urate, and Mycoplasma hyorhinis) have been used in vitro to confirm that inflammasomes can be activated in certain cell subsets [117, 172, 175, 184]. However, little is known about the direct activators of inflammasomes in TME during tumor progression. The activators may be bacteria [130], cell debris [117], ATP [206], PKR [199], other unknown factors in TME, or more complicated cross-talk between cells. Novel techniques such as single-cell sequencing may improve our understanding of the details during inflammasome activation.
The inflammasome signal seems to be a conserved pathway, which even exists in bacteria [293]. Although similar mechanisms have been identified in different species, discrepancies in NLR homologous genes and inflammasome pathways between humans and mice have been found [294]. For example, Francisella tularensis activates NLRP3 in humans instead of mice [295]. Thus, more detailed comparisons are needed to answer the question of to what extent can the findings from mouse models be extended to human patients.
It seems fuzzy that inflammasome signals have conflicting effects in different research works. A possible explanation is that inflammasomes can be activated at different extents, which may result in distinct inflammation responses [16, 42]. Future research works should compare the outcomes of different extents of inflammasome activation in various cell subsets in TME. Through this way, we can make accurate decisions about whether and how inflammasomes should be activated or inhibited.
GSDMD-mediated pyroptosis is involved in cancer cell death during chemotherapy and radiotherapy; however, secreted IL-1β may recruit immunosuppressive cell subsets and initiate inflammation-related side effects. Thus, the combination of IL-1R signal inhibitors and chemotherapy or radiotherapy may improve outcomes. On the other hand, NLRP3 in DCs [206] and AIM2 in macrophages [196] have been shown to facilitate anti-tumor immunity. The combination of NLRP3 or AIM2 activators and immune checkpoint inhibitors is a compelling strategy for immunotherapy.
Availability of data and materials
The materials supporting our conclusion of this review are included within the article.
Abbreviations
- DAMPs:
-
Damage-associated molecular patterns
- PAMPs:
-
Pathogen-associated molecular patterns
- IL-1:
-
Interleukin-1
- GSDMD:
-
Gasdermin D
- PRRs:
-
Pattern recognition receptors
- TLRs:
-
Toll-like receptors
- NLRs:
-
Nucleotide-binding leucine-rich repeat receptors
- AIM2:
-
Absent in melanoma 2
- NLRP1:
-
NACHT, leucine-rich repeat and pyrin domain-containing 1
- NLRP3:
-
NOD-, LRR-, and pyrin domain-containing 3
- NAIP:
-
NLR family apoptosis inhibitory protein
- NLRC4:
-
NLR family CARD domain-containing 4
- ASC:
-
Apoptosis-associated speck-like protein-containing CARD
- PYD–PYD:
-
Pyrin–pyrin
- ESCRT:
-
Endosomal sorting complex required for transport
- LPS:
-
Lipopolysaccharide
- dectin-1:
-
Dendritic cell-associated C-type lectin-1
- DCs:
-
Dendritic cells
- Th:
-
T helper
- NK:
-
Natural killer
- LRR:
-
Leucine-rich repeat
- LeTx:
-
Lethal toxin
- DDP:
-
Dipeptidyl peptidase
- NF-κB:
-
Nuclear factor-kappa B
- TNF-α:
-
Tumor necrosis factor-α
- JNK-1:
-
C-Jun N-terminal kinase-1
- MyD88:
-
Myeloid differentiation factor 88
- mtROS:
-
Mitochondria reactive oxygen species
- BRCC3:
-
BRCA1/BRCA2-containing complex 3
- KAT5:
-
Lysine acetyltransferase 5
- NEK7:
-
NIMA-related kinase 7
- CAPS:
-
Cryopyrin-associated periodic syndrome
- T3SS:
-
Type III secretion system
- T4SS:
-
Type IV secretion system
- HIN200:
-
Hematopoietic interferon-inducible nuclear antigens with 200 amino acid repeat
- PKN1:
-
Protein kinase N1
- PKN2:
-
Protein kinase N2
- TME:
-
Tumor microenvironment
- SMAD:
-
Small mothers against decapentaplegic
- c-myc:
-
V-myc myelocytomatosis viral oncogene homolog
- TP53:
-
Tumor protein p53
- bcl-2:
-
B-cell lymphoma-2
- Bax:
-
Bcl-2-associated X protein
- gp130:
-
Glycoprotein 130
- STAT3:
-
Signal transducer and activator of transcription 3
- MDSCs:
-
Myeloid-derived suppressor cells
- IFN-γ:
-
Interferon gamma
- 4-NQO:
-
4-Nitroquinoline 1-oxide
- NSCLC:
-
Non-small cell lung cancer
- EGFR:
-
Epidermal growth factor receptor
- ERK:
-
Extracellular signal-regulated kinase
- PI3K:
-
Phosphatidylinositol 3-kinase
- HIF-1\(\mathrm{\alpha }\) :
-
Hypoxia-inducible factor-1\(\mathrm{\alpha }\)
- CXCL2:
-
C-X-C motif chemokine ligand 2
- TAMs:
-
Tumor-associated macrophages
- VEGF:
-
Vascular endothelial growth factor
- S1PR1:
-
S1P receptor 1
- Vegfa:
-
Vascular endothelial growth factor A
- BRCA1:
-
Breast cancer susceptibility gene 1
- P2Y2R:
-
P2Y2 receptor
- MMP-9:
-
Matrix metallopeptidase-9
- GSK3β:
-
Glycogen synthase kinase 3β
- CCDN1:
-
Cyclin D1
- SNAIL1:
-
Snail family transcriptional repressor 1
- AP-1:
-
Activator protein-1
- SCLC:
-
Small cell lung cancer
- EMT:
-
Epithelial–mesenchymal transition
- AKR1C1:
-
Aldo–keto reductase 1C1
- CCL5:
-
C-C motif chemokine ligand 5
- CXCL9:
-
C-X-C motif chemokine ligand 9
- PD-L1:
-
Programmed cell death-ligand 1
- MHC-I:
-
Major histocompatibility complex class I
- Tregs:
-
Regulatory T cells
- JAK:
-
Janus kinase
- CAR:
-
Chimeric antigen receptor
- PKR:
-
Protein kinase R
- PD-1:
-
Programmed cell death protein-1
- HSP70:
-
Heat shock protein 70
- Wnt5a:
-
Wnt family member 5A
- CXCL5:
-
C-X-C motif chemokine ligand 5
- CXCR2:
-
C-X-C motif chemokine receptor 2
- P2X7:
-
P2 purinergic receptors
- 5-FU:
-
Fluorouracil
- BRAF:
-
B-Raf proto-oncogene
- PTEN:
-
Phosphatase and tensin homolog
- MAPK:
-
Mitogen-activated kinase-like protein
- CXCR2:
-
C-X-C motif chemokine receptor 2
- cGAS:
-
Cyclic GMP–AMP synthase
- SPARC:
-
Secreted protein acidic and rich in cysteine
- Siglec-1:
-
Sialic acid binding Ig-like lectin 1, sialoadhesin
- MNS:
-
3,4-Methylenedioxy-β-nitrostyrene
- BTK:
-
Bruton tyrosine kinase
- GSTO1:
-
Glutathione S-transferase omega 1
References
Newton K, Dixit V. Signaling in innate immunity and inflammation. Cold Spring Harb Perspect Biol. 2012;4(3):a006049.
Balka KR, De Nardo D. Understanding early TLR signaling through the myddosome. J Leukoc Biol. 2019;105(2):339–51.
Chauhan D, Vande Walle L, Lamkanfi M. Therapeutic modulation of inflammasome pathways. Immunol Rev. 2020;297(1):123–38.
Lamkanfi M, Dixit VM. Mechanisms and functions of inflammasomes. Cell. 2014;157(5):1013–22.
Broz P, Dixit VM. Inflammasomes: mechanism of assembly, regulation and signalling. Nat Rev Immunol. 2016;16(7):407–20.
Cai X, Chen J, Xu H, et al. Prion-like polymerization underlies signal transduction in antiviral immune defense and inflammasome activation. Cell. 2014;156(6):1207–22.
Lu A, Magupalli VG, Ruan J, et al. Unified polymerization mechanism for the assembly of ASC-dependent inflammasomes. Cell. 2014;156(6):1193–206.
Frew BC, Joag VR, Mogridge J. Proteolytic processing of Nlrp1b is required for inflammasome activity. PLoS Pathog. 2012;8(4): e1002659.
Faustin B, Lartigue L, Bruey JM, et al. Reconstituted NALP1 inflammasome reveals two-step mechanism of caspase-1 activation. Mol Cell. 2007;25(5):713–24.
Martinon F, Burns K, Tschopp J. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-β. Mol Cell. 2002;10(2):417–26.
Proell M, Gerlic M, Mace PD, et al. The CARD plays a critical role in ASC foci formation and inflammasome signalling. Biochem J. 2013;449(3):613–21.
Poyet JL, Srinivasula SM, Tnani M, et al. Identification of Ipaf, a human caspase-1-activating protein related to Apaf-1. J Biol Chem. 2001;276(30):28309–13.
Boucher D, Monteleone M, Coll RC, et al. Caspase-1 self-cleavage is an intrinsic mechanism to terminate inflammasome activity. J Exp Med. 2018;215(3):827–40.
Afonina IS, Muller C, Martin SJ, et al. Proteolytic processing of interleukin-1 family cytokines: variations on a common theme. Immunity. 2015;42(6):991–1004.
Sborgi L, Ruhl S, Mulvihill E, et al. GSDMD membrane pore formation constitutes the mechanism of pyroptotic cell death. EMBO J. 2016;35(16):1766–78.
Ding J, Wang K, Liu W, et al. Pore-forming activity and structural autoinhibition of the gasdermin family. Nature. 2016;535(7610):111–6.
Liu X, Zhang Z, Ruan J, et al. Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature. 2016;535(7610):153–8.
Evavold CL, Hafner-Bratkovic I, Devant P, et al. Control of gasdermin D oligomerization and pyroptosis by the ragulator-rag-mTORC1 pathway. Cell. 2021;184(17):4495–511.
Weindel CG, Martinez EL, Zhao X, et al. Mitochondrial ROS promotes susceptibility to infection via gasdermin D-mediated necroptosis. Cell. 2022;185(17):3214–31.
Rogers C, Erkes DA, Nardone A, et al. Gasdermin pores permeabilize mitochondria to augment caspase-3 activation during apoptosis and inflammasome activation. Nat Commun. 2019;10(1):1689.
Chan AH, Schroder K. Inflammasome signaling and regulation of interleukin-1 family cytokines. J Exp Med. 2020;217(1):e20190314.
He WT, Wan H, Hu L, et al. Gasdermin D is an executor of pyroptosis and required for interleukin-1beta secretion. Cell Res. 2015;25(12):1285–98.
**a S, Zhang Z, Magupalli VG, et al. Gasdermin D pore structure reveals preferential release of mature interleukin-1. Nature. 2021;593(7860):607–11.
**ang H, Zhu F, Xu Z, et al. Role of inflammasomes in kidney diseases via both canonical and non-canonical pathways. Front Cell Dev Biol. 2020;8:106.
Shi J, Zhao Y, Wang Y, et al. Inflammatory caspases are innate immune receptors for intracellular LPS. Nature. 2014;514(7521):187–92.
Vigano E, Diamond CE, Spreafico R, et al. Human caspase-4 and caspase-5 regulate the one-step non-canonical inflammasome activation in monocytes. Nat Commun. 2015;6:8761.
Ross C, Chan AH, Von Pein J, et al. Dimerization and auto-processing induce caspase-11 protease activation within the non-canonical inflammasome. Life Sci Alliance. 2018;1(6): e201800237.
Yang J, Zhao Y, Shao F. Non-canonical activation of inflammatory caspases by cytosolic LPS in innate immunity. Curr Opin Immunol. 2015;32:78–83.
Kayagaki N, Stowe IB, Lee BL, et al. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature. 2015;526(7575):666–71.
Shi J, Zhao Y, Wang K, et al. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature. 2015;526(7575):660–5.
Ruhl S, Broz P. Caspase-11 activates a canonical NLRP3 inflammasome by promoting K(+) efflux. Eur J Immunol. 2015;45(10):2927–36.
Kobayashi T, Ogawa M, Sanada T, et al. The Shigella OspC3 effector inhibits caspase-4, antagonizes inflammatory cell death, and promotes epithelial infection. Cell Host Microbe. 2013;13(5):570–83.
Knodler LA, Crowley SM, Sham HP, et al. Noncanonical inflammasome activation of caspase-4/caspase-11 mediates epithelial defenses against enteric bacterial pathogens. Cell Host Microbe. 2014;16(2):249–56.
Wiggins KA, Parry AJ, Cassidy LD, et al. IL-1alpha cleavage by inflammatory caspases of the noncanonical inflammasome controls the senescence-associated secretory phenotype. Aging Cell. 2019;18(3): e12946.
Mandal R, Barron JC, Kostova I, et al. Caspase-8: the double-edged sword. Biochim Biophys Acta Rev Cancer. 2020;1873(2): 188357.
Bossaller L, Chiang PI, Schmidt-Lauber C, et al. Cutting edge: FAS (CD95) mediates noncanonical IL-1beta and IL-18 maturation via caspase-8 in an RIP3-independent manner. J Immunol. 2012;189(12):5508–12.
Gringhuis SI, Kaptein TM, Wevers BA, et al. Dectin-1 is an extracellular pathogen sensor for the induction and processing of IL-1beta via a noncanonical caspase-8 inflammasome. Nat Immunol. 2012;13(3):246–54.
Ketelut-Carneiro N, Ghosh S, Levitz SM, et al. A dectin-1-caspase-8 pathway licenses canonical caspase-1 inflammasome activation and interleukin-1beta release in response to a pathogenic fungus. J Infect Dis. 2018;217(2):329–39.
Vajjhala PR, Lu A, Brown DL, et al. The Inflammasome adaptor ASC induces procaspase-8 death effector domain filaments. J Biol Chem. 2015;290(49):29217–30.
Antonopoulos C, Russo HM, El Sanadi C, et al. Caspase-8 as an effector and regulator of NLRP3 inflammasome signaling. J Biol Chem. 2015;290(33):20167–84.
Man SM, Tourlomousis P, Hopkins L, et al. Salmonella infection induces recruitment of caspase-8 to the inflammasome to modulate IL-1beta production. J Immunol. 2013;191(10):5239–46.
Rühl S, Shkarina K, Demarco B, et al. ESCRT-dependent membrane repair negatively regulates pyroptosis downstream of GSDMD activation. 2018.
Aglietti RA, Estevez A, Gupta A, et al. GsdmD p30 elicited by caspase-11 during pyroptosis forms pores in membranes. Proc Natl Acad Sci U S A. 2016;113(28):7858–63.
Magupalli VG, Negro R, Tian Y, et al. HDAC6 mediates an aggresome-like mechanism for NLRP3 and pyrin inflammasome activation. Science. 2020;369(6510):eaas8995.
Martinon F, Burns K, Tschopp J. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol Cell. 2002;10(2):417–26.
Man SM, Kanneganti TD. Regulation of inflammasome activation. Immunol Rev. 2015;265(1):6–21.
Masters SL, Gerlic M, Metcalf D, et al. NLRP1 inflammasome activation induces pyroptosis of hematopoietic progenitor cells. Immunity. 2012;37(6):1009–23.
Boyden ED, Dietrich WF. Nalp1b controls mouse macrophage susceptibility to anthrax lethal toxin. Nat Genet. 2006;38(2):240–4.
Okondo MC, Johnson DC, Sridharan R, et al. DPP8 and DPP9 inhibition induces pro-caspase-1-dependent monocyte and macrophage pyroptosis. Nat Chem Biol. 2017;13(1):46–53.
Bauernfried S, Scherr MJ, Pichlmair A, et al. Human NLRP1 is a sensor for double-stranded RNA. Science. 2021;371(6528):eabd0811.
Robinson KS, Toh GA, Rozario P, et al. ZAKα-driven ribotoxic stress response activates the human NLRP1 inflammasome. Science. 2022;377(6603):328–35.
Hollingsworth LR, Sharif H, Griswold AR, et al. DPP9 sequesters the C terminus of NLRP1 to repress inflammasome activation. Nature. 2021;592(7856):778–83.
Chui AJ, Okondo MC, Rao SD, et al. N-terminal degradation activates the NLRP1B inflammasome. Science. 2019;364(6435):82–5.
Sandstrom A, Mitchell PS, Goers L, et al. Functional degradation: a mechanism of NLRP1 inflammasome activation by diverse pathogen enzymes. Science. 2019;364(6435):eaau1330.
Huang M, Zhang X, Toh GA, et al. Structural and biochemical mechanisms of NLRP1 inhibition by DPP9. Nature. 2021;592(7856):773–7.
Van Opdenbosch N, Gurung P, Vande Walle L, et al. Activation of the NLRP1b inflammasome independently of ASC-mediated caspase-1 autoproteolysis and speck formation. Nat Commun. 2014;5:3209.
Witola WH, Mui E, Hargrave A, et al. NALP1 influences susceptibility to human congenital toxoplasmosis, proinflammatory cytokine response, and fate of toxoplasma gondii-infected monocytic cells. Infect Immun. 2011;79(2):756–66.
Drutman SB, Haerynck F, Zhong FL, et al. Homozygous NLRP1 gain-of-function mutation in siblings with a syndromic form of recurrent respiratory papillomatosis. Proc Natl Acad Sci U S A. 2019;116(38):19055–63.
Zhong FL, Mamai O, Sborgi L, et al. Germline NLRP1 mutations cause skin inflammatory and cancer susceptibility syndromes via inflammasome activation. Cell. 2016;167(1):187–202.
Magitta NF, Boe Wolff AS, Johansson S, et al. A coding polymorphism in NALP1 confers risk for autoimmune Addison’s disease and type 1 diabetes. Genes Immun. 2009;10(2):120–4.
Duncan JA, Bergstralh DT, Wang Y, et al. Cryopyrin/NALP3 binds ATP/dATP, is an ATPase, and requires ATP binding to mediate inflammatory signaling. Proc Natl Acad Sci U S A. 2007;104(19):8041–6.
Andreeva L, David L, Rawson S, et al. NLRP3 cages revealed by full-length mouse NLRP3 structure control pathway activation. Cell. 2021;184(26):6299–312.
Hochheiser IV, Pilsl M, Hagelueken G, et al. Structure of the NLRP3 decamer bound to the cytokine release inhibitor CRID3. Nature. 2022;604(7904):184–9.
Bauernfeind FG, Horvath G, Stutz A, et al. Cutting edge: NF-kappaB activating pattern recognition and cytokine receptors license NLRP3 inflammasome activation by regulating NLRP3 expression. J Immunol. 2009;183(2):787–91.
Franchi L, Eigenbrod T, Nunez G. Cutting edge: TNF-alpha mediates sensitization to ATP and silica via the NLRP3 inflammasome in the absence of microbial stimulation. J Immunol. 2009;183(2):792–6.
Marucha PT, Zeff RA, Kreutzer DL. Cytokine regulation of IL-1 beta gene expression in the human polymorphonuclear leukocyte. J Immunol. 1990;145(9):2932–7.
Song N, Liu ZS, Xue W, et al. NLRP3 phosphorylation is an essential priming event for inflammasome activation. Mol Cell. 2017;68(1):185–97.
Juliana C, Fernandes-Alnemri T, Kang S, et al. Non-transcriptional priming and deubiquitination regulate NLRP3 inflammasome activation. J Biol Chem. 2012;287(43):36617–22.
Py BF, Kim MS, Vakifahmetoglu-Norberg H, et al. Deubiquitination of NLRP3 by BRCC3 critically regulates inflammasome activity. Mol Cell. 2013;49(2):331–8.
Lamkanfi M, Dixit VM. Inflammasomes and their roles in health and disease. Annu Rev Cell Dev Biol. 2012;28:137–61.
Ng J, Hirota SA, Gross O, et al. Clostridium difficile toxin-induced inflammation and intestinal injury are mediated by the inflammasome. Gastroenterology. 2010;139(2):542–52.
Lee MS, Kwon H, Lee EY, et al. Shiga toxins activate the NLRP3 inflammasome pathway to promote both production of the proinflammatory cytokine interleukin-1beta and apoptotic cell death. Infect Immun. 2016;84(1):172–86.
Munoz-Planillo R, Kuffa P, Martinez-Colon G, et al. K(+) efflux is the common trigger of NLRP3 inflammasome activation by bacterial toxins and particulate matter. Immunity. 2013;38(6):1142–53.
Platnich JM, Chung H, Lau A, et al. Shiga toxin/lipopolysaccharide activates caspase-4 and gasdermin D to trigger mitochondrial reactive oxygen species upstream of the NLRP3 inflammasome. Cell Rep. 2018;25(6):1525–36.
Hornung V, Bauernfeind F, Halle A, et al. Silica crystals and aluminum salts activate the NALP3 inflammasome through phagosomal destabilization. Nat Immunol. 2008;9(8):847–56.
Murakami T, Ockinger J, Yu J, et al. Critical role for calcium mobilization in activation of the NLRP3 inflammasome. Proc Natl Acad Sci U S A. 2012;109(28):11282–7.
Shimada K, Crother TR, Karlin J, et al. Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis. Immunity. 2012;36(3):401–14.
Subramanian N, Natarajan K, Clatworthy MR, et al. The adaptor MAVS promotes NLRP3 mitochondrial localization and inflammasome activation. Cell. 2013;153(2):348–61.
Zhao K, Zhang Y, Xu X, et al. Acetylation is required for NLRP3 self-aggregation and full activation of the inflammasome. 2019.
**ao L, Magupalli VG, Wu H. Cryo-EM structures of the active NLRP3 inflammasome disc. Nature. 2023;613(7944):595–600.
Van Gorp H, Van Opdenbosch N, Lamkanfi M. Inflammasome-dependent cytokines at the crossroads of health and autoinflammatory disease. Cold Spring Harb Perspect Biol. 2019;11(1):a028563.
Hoffman HM, Mueller JL, Broide DH, et al. Mutation of a new gene encoding a putative pyrin-like protein causes familial cold autoinflammatory syndrome and Muckle-Wells syndrome. Nat Genet. 2001;29(3):301–5.
Brydges SD, Broderick L, McGeough MD, et al. Divergence of IL-1, IL-18, and cell death in NLRP3 inflammasomopathies. J Clin Invest. 2013;123(11):4695–705.
Lee GS, Subramanian N, Kim AI, et al. The calcium-sensing receptor regulates the NLRP3 inflammasome through Ca2+ and cAMP. Nature. 2012;492(7427):123–7.
Bauer R, Rauch I. The NAIP/NLRC4 inflammasome in infection and pathology. Mol Aspects Med. 2020;76: 100863.
Broz P, von Moltke J, Jones JW, et al. Differential requirement for caspase-1 autoproteolysis in pathogen-induced cell death and cytokine processing. Cell Host Microbe. 2010;8(6):471–83.
Mariathasan S, Newton K, Monack DM, et al. Differential activation of the inflammasome by caspase-1 adaptors ASC and Ipaf. Nature. 2004;430(6996):213–8.
Mascarenhas DPA, Cerqueira DM, Pereira MSF, et al. Inhibition of caspase-1 or gasdermin-D enable caspase-8 activation in the Naip5/NLRC4/ASC inflammasome. PLoS Pathog. 2017;13(8): e1006502.
Amer A, Franchi L, Kanneganti TD, et al. Regulation of Legionella phagosome maturation and infection through flagellin and host Ipaf. J Biol Chem. 2006;281(46):35217–23.
Miao EA, Mao DP, Yudkovsky N, et al. Innate immune detection of the type III secretion apparatus through the NLRC4 inflammasome. Proc Natl Acad Sci U S A. 2010;107(7):3076–80.
Kofoed EM, Vance RE. Innate immune recognition of bacterial ligands by NAIPs determines inflammasome specificity. Nature. 2011;477(7366):592–5.
Zhao Y, Yang J, Shi J, et al. The NLRC4 inflammasome receptors for bacterial flagellin and type III secretion apparatus. Nature. 2011;477(7366):596–600.
Yang J, Zhao Y, Shi J, et al. Human NAIP and mouse NAIP1 recognize bacterial type III secretion needle protein for inflammasome activation. Proc Natl Acad Sci U S A. 2013;110(35):14408–13.
Kortmann J, Brubaker SW, Monack DM. Cutting edge: inflammasome activation in primary human macrophages is dependent on flagellin. J Immunol. 2015;195(3):815–9.
Romberg N, Al Moussawi K, Nelson-Williams C, et al. Mutation of NLRC4 causes a syndrome of enterocolitis and autoinflammation. Nat Genet. 2014;46(10):1135–9.
Canna SW, de Jesus AA, Gouni S, et al. An activating NLRC4 inflammasome mutation causes autoinflammation with recurrent macrophage activation syndrome. Nat Genet. 2014;46(10):1140–6.
Kumari P, Russo AJ, Shivcharan S, et al. AIM2 in health and disease: inflammasome and beyond. Immunol Rev. 2020;297(1):83–95.
Bae JH, Jo SI, Kim SJ, et al. Circulating cell-free mtDNA contributes to AIM2 inflammasome-mediated chronic inflammation in patients with type 2 diabetes. Cells. 2019;8(4):328.
Lee S, Karki R, Wang Y, et al. AIM2 forms a complex with pyrin and ZBP1 to drive PANoptosis and host defence. Nature. 2021;597(7876):415–9.
Rathinam VA, Jiang Z, Waggoner SN, et al. The AIM2 inflammasome is essential for host defense against cytosolic bacteria and DNA viruses. Nat Immunol. 2010;11(5):395–402.
Smith S, Jefferies C. Role of DNA/RNA sensors and contribution to autoimmunity. Cytokine Growth Factor Rev. 2014;25(6):745–57.
Hu B, ** C, Li HB, et al. The DNA-sensing AIM2 inflammasome controls radiation-induced cell death and tissue injury. Science. 2016;354(6313):765–8.
Woerner SM, Kloor M, Schwitalle Y, et al. The putative tumor suppressor AIM2 is frequently affected by different genetic alterations in microsatellite unstable colon cancers. Genes Chromosom Cancer. 2007;46(12):1080–9.
Man SM, Zhu Q, Zhu L, et al. Critical role for the DNA sensor AIM2 in stem cell proliferation and cancer. Cell. 2015;162(1):45–58.
Dihlmann S, Tao S, Echterdiek F, et al. Lack of absent in melanoma 2 (AIM2) expression in tumor cells is closely associated with poor survival in colorectal cancer patients. Int J Cancer. 2014;135(10):2387–96.
Malik HS, Bliska JB. The pyrin inflammasome and the Yersinia effector interaction. Immunol Rev. 2020;297(1):96–107.
Park YH, Wood G, Kastner DL, et al. Pyrin inflammasome activation and RhoA signaling in the autoinflammatory diseases FMF and HIDS. Nat Immunol. 2016;17(8):914–21.
Gao W, Yang J, Liu W, et al. Site-specific phosphorylation and microtubule dynamics control Pyrin inflammasome activation. Proc Natl Acad Sci. 2016;113(33):E4857–66.
Garlanda C, Dinarello CA, Mantovani A. The interleukin-1 family: back to the future. Immunity. 2013;39(6):1003–18.
Nold MF, Nold-Petry CA, Zepp JA, et al. IL-37 is a fundamental inhibitor of innate immunity. Nat Immunol. 2010;11(11):1014–22.
Rider P, Carmi Y, Guttman O, et al. IL-1alpha and IL-1beta recruit different myeloid cells and promote different stages of sterile inflammation. J Immunol. 2011;187(9):4835–43.
Werman A, Werman-Venkert R, White R, et al. The precursor form of IL-1alpha is an intracrine proinflammatory activator of transcription. Proc Natl Acad Sci U S A. 2004;101(8):2434–9.
Dinarello CA. Overview of the IL-1 family in innate inflammation and acquired immunity. Immunol Rev. 2018;281(1):8–27.
Schroder K, Hertzog PJ, Ravasi T, et al. Interferon-gamma: an overview of signals, mechanisms and functions. J Leukoc Biol. 2004;75(2):163–89.
Elinav E, Strowig T, Kau AL, et al. NLRP6 inflammasome regulates colonic microbial ecology and risk for colitis. Cell. 2011;145(5):745–57.
Cao X, Xu J. Insights into inflammasome and its research advances in cancer. Tumori. 2019;105(6):456–64.
Ershaid N, Sharon Y, Doron H, et al. NLRP3 inflammasome in fibroblasts links tissue damage with inflammation in breast cancer progression and metastasis. Nat Commun. 2019;10(1):4375.
Weichand B, Popp R, Dziumbla S, et al. S1PR1 on tumor-associated macrophages promotes lymphangiogenesis and metastasis via NLRP3/IL-1beta. J Exp Med. 2017;214(9):2695–713.
Das S, Shapiro B, Vucic EA, et al. Tumor cell-derived IL1beta promotes desmoplasia and immune suppression in pancreatic cancer. Cancer Res. 2020;80(5):1088–101.
Bruchard M, Mignot G, Derangere V, et al. Chemotherapy-triggered cathepsin B release in myeloid-derived suppressor cells activates the Nlrp3 inflammasome and promotes tumor growth. Nat Med. 2013;19(1):57–64.
Tu CE, Hu Y, Zhou P, et al. Lactate and TGF-beta antagonistically regulate inflammasome activation in the tumor microenvironment. J Cell Physiol. 2021;236(6):4528–37.
Ju M, Bi J, Wei Q, et al. Pan-cancer analysis of NLRP3 inflammasome with potential implications in prognosis and immunotherapy in human cancer. Brief Bioinform. 2020.
Zheng T, Wang X, Yue P, et al. Prognostic inflammasome-related signature construction in kidney renal clear cell carcinoma based on a pan-cancer landscape. Evid Based Complement Alternat Med. 2020;2020:3259795.
Elinav E, Nowarski R, Thaiss C, et al. Inflammation-induced cancer: crosstalk between tumours, immune cells and microorganisms. Nat Rev Cancer. 2013;13:759–71.
Verma D, Bivik C, Farahani E, et al. Inflammasome polymorphisms confer susceptibility to sporadic malignant melanoma. Pigment Cell Melanoma Res. 2012;25(4):506–13.
Castano-Rodriguez N, Kaakoush NO, Goh KL, et al. The NOD-like receptor signalling pathway in Helicobacter pylori infection and related gastric cancer: a case-control study and gene expression analyses. PLoS ONE. 2014;9(6): e98899.
Miskiewicz A, Szparecki G, Durlik M, et al. The Q705K and F359L single-nucleotide polymorphisms of NOD-like receptor signaling pathway: association with chronic pancreatitis, pancreatic cancer, and periodontitis. Arch Immunol Ther Exp. 2015;63(6):485–94.
Zhao X, Zhang C, Hua M, et al. NLRP3 inflammasome activation plays a carcinogenic role through effector cytokine IL-18 in lymphoma. Oncotarget. 2017;8(65):108571–83.
Deswaerte V, Nguyen P, West A, et al. Inflammasome adaptor ASC suppresses apoptosis of gastric cancer cells by an IL18-mediated inflammation-independent mechanism. Cancer Res. 2018;78(5):1293–307.
Ikuta T, Kobayashi Y, Kitazawa M, et al. ASC-associated inflammation promotes cecal tumorigenesis in aryl hydrocarbon receptor-deficient mice. Carcinogenesis. 2013;34(7):1620–7.
Tu S, Bhagat G, Cui G, et al. Overexpression of interleukin-1beta induces gastric inflammation and cancer and mobilizes myeloid-derived suppressor cells in mice. Cancer Cell. 2008;14(5):408–19.
Chow MT, Sceneay J, Paget C, et al. NLRP3 suppresses NK cell-mediated responses to carcinogen-induced tumors and metastases. Cancer Res. 2012;72(22):5721–32.
Feng X, Luo Q, Zhang H, et al. The role of NLRP3 inflammasome in 5-fluorouracil resistance of oral squamous cell carcinoma. J Exp Clin Cancer Res. 2017;36(1):81.
Wei Q, Mu K, Li T, et al. Deregulation of the NLRP3 inflammasome in hepatic parenchymal cells during liver cancer progression. Lab Invest. 2014;94(1):52–62.
Ojcius DM, Gasparoto TH, de Oliveira CE, et al. Inflammasome activation is critical to the protective immune response during chemically induced squamous cell carcinoma. PLoS ONE. 2014;9(9): e107170.
Allen IC, TeKippe EM, Woodford RM, et al. The NLRP3 inflammasome functions as a negative regulator of tumorigenesis during colitis-associated cancer. J Exp Med. 2010;207(5):1045–56.
Zaki MH, Vogel P, Body-Malapel M, et al. IL-18 production downstream of the Nlrp3 inflammasome confers protection against colorectal tumor formation. J Immunol. 2010;185(8):4912–20.
Sharma D, Malik A, Guy CS, et al. Pyrin Inflammasome regulates tight junction integrity to restrict colitis and tumorigenesis. Gastroenterology. 2018;154(4):948–64.
Flood B, Manils J, Nulty C, et al. Caspase-11 regulates the tumour suppressor function of STAT1 in a murine model of colitis-associated carcinogenesis. Oncogene. 2019;38(14):2658–74.
Dagenais M, Dupaul-Chicoine J, Douglas T, et al. The interleukin (IL)-1R1 pathway is a critical negative regulator of PyMT-mediated mammary tumorigenesis and pulmonary metastasis. Oncoimmunology. 2017;6(3): e1287247.
Hu B, Elinav E, Huber S, et al. Inflammation-induced tumorigenesis in the colon is regulated by caspase-1 and NLRC4. Proc Natl Acad Sci U S A. 2010;107(50):21635–40.
Liu W, Luo Y, Dunn JH, et al. Dual role of apoptosis-associated speck-like protein containing a CARD (ASC) in tumorigenesis of human melanoma. J Invest Dermatol. 2013;133(2):518–27.
Drexler SK, Bonsignore L, Masin M, et al. Tissue-specific opposing functions of the inflammasome adaptor ASC in the regulation of epithelial skin carcinogenesis. Proc Natl Acad Sci U S A. 2012;109(45):18384–9.
Gao J, Qiu X, ** G, et al. Downregulation of GSDMD attenuates tumor proliferation via the intrinsic mitochondrial apoptotic pathway and inhibition of EGFR/Akt signaling and predicts a good prognosis in nonsmall cell lung cancer. Oncol Rep. 2018;40(4):1971–84.
Wang WJ, Chen D, Jiang MZ, et al. Downregulation of gasdermin D promotes gastric cancer proliferation by regulating cell cycle-related proteins. J Dig Dis. 2018;19(2):74–83.
Rao Z, Zhu Y, Yang P, et al. Pyroptosis in inflammatory diseases and cancer. Theranostics. 2022;12(9):4310–29.
Yan H, Luo B, Wu X, et al. Cisplatin induces pyroptosis via activation of MEG3/NLRP3/caspase-1/GSDMD pathway in triple-negative breast cancer. Int J Biol Sci. 2021;17(10):2606–21.
Wang L, Li K, Lin X, et al. Metformin induces human esophageal carcinoma cell pyroptosis by targeting the miR-497/PELP1 axis. Cancer Lett. 2019;450:22–31.
Yue E, Tuguzbaeva G, Chen X, et al. Anthocyanin is involved in the activation of pyroptosis in oral squamous cell carcinoma. Phytomedicine. 2019;56:286–94.
Sannino F, Sansone C, Galasso C, et al. Pseudoalteromonas haloplanktis TAC125 produces 4-hydroxybenzoic acid that induces pyroptosis in human A459 lung adenocarcinoma cells. Sci Rep. 2018;8(1):1190.
Wang F, Liu W, Ning J, et al. Simvastatin suppresses proliferation and migration in non-small cell lung cancer via pyroptosis. Int J Biol Sci. 2018;14(4):406–17.
Johnson DC, Taabazuing CY, Okondo MC, et al. DPP8/DPP9 inhibitor-induced pyroptosis for treatment of acute myeloid leukemia. Nat Med. 2018;24(8):1151–6.
Pizato N, Luzete BC, Kiffer L, et al. Omega-3 docosahexaenoic acid induces pyroptosis cell death in triple-negative breast cancer cells. Sci Rep. 2018;8(1):1952.
Nadeem S, Yang C, Du Y, et al. A virus-spike tumor-activatable pyroptotic agent. Small. 2021;17(8): e2006599.
Ploetz E, Zimpel A, Cauda V, et al. Metal-organic framework nanoparticles induce pyroptosis in cells controlled by the extracellular pH. Adv Mater. 2020;32(19): e1907267.
Qiao L, Wu X, Zhang J, et al. alpha-NETA induces pyroptosis of epithelial ovarian cancer cells through the GSDMD/caspase-4 pathway. FASEB J. 2019;33(11):12760–7.
Yokoyama S, Cai Y, Murata M, et al. A novel pathway of LPS uptake through syndecan-1 leading to pyroptotic cell death. Elife. 2018;7:e37854.
Serna N, Alamo P, Ramesh P, et al. Nanostructured toxins for the selective destruction of drug-resistant human CXCR4(+) colorectal cancer stem cells. J Control Release. 2020;320:96–104.
Yaw ACK, Chan EWL, Yap JKY, et al. The effects of NLRP3 inflammasome inhibition by MCC950 on LPS-induced pancreatic adenocarcinoma inflammation. J Cancer Res Clin Oncol. 2020;146(9):2219–29.
Viallard C, Larrivee B. Tumor angiogenesis and vascular normalization: alternative therapeutic targets. Angiogenesis. 2017;20(4):409–26.
Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–74.
Karki R, Kanneganti TD. Diverging inflammasome signals in tumorigenesis and potential targeting. Nat Rev Cancer. 2019;19(4):197–214.
Van Gorp H, Lamkanfi M. The emerging roles of inflammasome-dependent cytokines in cancer development. EMBO Rep. 2019;20(6):e47575.
Saijo Y, Tanaka M, Miki M, et al. Proinflammatory cytokine IL-1 beta promotes tumor growth of Lewis lung carcinoma by induction of angiogenic factors: in vivo analysis of tumor-stromal interaction. J Immunol. 2002;169(1):469–75.
Jung YJ, Isaacs JS, Lee S, et al. IL-1beta-mediated up-regulation of HIF-1alpha via an NFkappaB/COX-2 pathway identifies HIF-1 as a critical link between inflammation and oncogenesis. FASEB J. 2003;17(14):2115–7.
Voronov E, Shouval DS, Krelin Y, et al. IL-1 is required for tumor invasiveness and angiogenesis. Proc Natl Acad Sci U S A. 2003;100(5):2645–50.
Carmi Y, Voronov E, Dotan S, et al. The role of macrophage-derived IL-1 in induction and maintenance of angiogenesis. J Immunol. 2009;183(7):4705–14.
Kolb R, Phan L, Borcherding N, et al. Obesity-associated NLRC4 inflammasome activation drives breast cancer progression. Nat Commun. 2016;7:13007.
Carmi Y, Dotan S, Rider P, et al. The role of IL-1beta in the early tumor cell-induced angiogenic response. J Immunol. 2013;190(7):3500–9.
Kong H, Wang Y, Zeng X, et al. Differential expression of inflammasomes in lung cancer cell lines and tissues. Tumour Biol. 2015;36(10):7501–13.
Poli G, Brancorsini S, Cochetti G, et al. Expression of inflammasome-related genes in bladder cancer and their association with cytokeratin 20 messenger RNA. Urol Oncol. 2015;33(12):505.
Lee HE, Lee JY, Yang G, et al. Inhibition of NLRP3 inflammasome in tumor microenvironment leads to suppression of metastatic potential of cancer cells. Sci Rep. 2019;9(1):12277.
Bent R, Moll L, Grabbe S, et al. Interleukin-1 beta-a friend or foe in malignancies? Int J Mol Sci. 2018;19(8):2155.
Horio D, Minami T, Kitai H, et al. Tumor-associated macrophage-derived inflammatory cytokine enhances malignant potential of malignant pleural mesothelioma. Cancer Sci. 2020;111(8):2895–906.
** H, Ko YS, Kim HJ. P2Y2R-mediated inflammasome activation is involved in tumor progression in breast cancer cells and in radiotherapy-resistant breast cancer. Int J Oncol. 2018;53(5):1953–66.
Genin M, Clement F, Fattaccioli A, et al. M1 and M2 macrophages derived from THP-1 cells differentially modulate the response of cancer cells to etoposide. BMC Cancer. 2015;15:577.
Li Y, Cao F, Li M, et al. Hydroxychloroquine induced lung cancer suppression by enhancing chemo-sensitization and promoting the transition of M2-TAMs to M1-like macrophages. J Exp Clin Cancer Res. 2018;37(1):259.
Yang D, Cao X, Wang F, et al. LFG-500, a novel synthetic flavonoid, suppresses epithelial-mesenchymal transition in human lung adenocarcinoma cells by inhibiting NLRP3 in inflammatory microenvironment. Cancer Lett. 2017;400:137–48.
Tulotta C, Lefley DV, Freeman K, et al. Endogenous production of IL1B by breast cancer cells drives metastasis and colonization of the bone microenvironment. Clin Cancer Res. 2019;25(9):2769–82.
Matsumoto R, Tsuda M, Yoshida K, et al. Aldo-keto reductase 1C1 induced by interleukin-1beta mediates the invasive potential and drug resistance of metastatic bladder cancer cells. Sci Rep. 2016;6:34625.
Zhang Y, Yang H, Sun M, et al. Alpinumisoflavone suppresses hepatocellular carcinoma cell growth and metastasis via NLRP3 inflammasome-mediated pyroptosis. Pharmacol Rep. 2020;72(5):1370–82.
Dupaul-Chicoine J, Arabzadeh A, Dagenais M, et al. The Nlrp3 inflammasome suppresses colorectal cancer metastatic growth in the liver by promoting natural killer cell tumoricidal activity. Immunity. 2015;43(4):751–63.
Deng Q, Geng Y, Zhao L, et al. NLRP3 inflammasomes in macrophages drive colorectal cancer metastasis to the liver. Cancer Lett. 2019;442:21–30.
Xu Y, Li H, Chen W, et al. Mycoplasma hyorhinis activates the NLRP3 inflammasome and promotes migration and invasion of gastric cancer cells. PLoS ONE. 2013;8(11): e77955.
Huang Q, Lan F, Wang X, et al. IL-1β-induced activation of p38 promotes metastasis in gastric adenocarcinoma via upregulation of AP-1/c-fos, MMP2 and MMP9. Mol Cancer. 2014;13:18.
Zhang D, Jiang Q, Ge X, et al. RHOV promotes lung adenocarcinoma cell growth and metastasis through JNK/c-Jun pathway. Int J Biol Sci. 2021;17(10):2622–32.
Wang JF, Zhao K, Chen YY, et al. NKCC1 promotes proliferation, invasion and migration in human gastric cancer cells via activation of the MAPK-JNK/EMT signaling pathway. J Cancer. 2021;12(1):253–63.
Chen Q, Lei JH, Bao J, et al. BRCA1 deficiency impairs mitophagy and promotes inflammasome activation and mammary tumor metastasis. Adv Sci. 2020;7(6):1903616.
Perez-Yepez EA, Ayala-Sumuano JT, Lezama R, et al. A novel beta-catenin signaling pathway activated by IL-1beta leads to the onset of epithelial-mesenchymal transition in breast cancer cells. Cancer Lett. 2014;354(1):164–71.
Jeon M, Han J, Nam SJ, et al. Elevated IL-1beta expression induces invasiveness of triple negative breast cancer cells and is suppressed by zerumbone. Chem Biol Interact. 2016;258:126–33.
Wang Y, Kong H, Zeng X, et al. Activation of NLRP3 inflammasome enhances the proliferation and migration of A549 lung cancer cells. Oncol Rep. 2016;35(4):2053–64.
Ohashi K, Wang Z, Yang YM, et al. NOD-like receptor C4 inflammasome regulates the growth of colon cancer liver metastasis in NAFLD. Hepatology. 2019;70(5):1582–99.
Farshchian M, Nissinen L, Siljamäki E, et al. Tumor cell-specific AIM2 regulates growth and invasion of cutaneous squamous cell carcinoma. Oncotarget. 2017;8(28):45825–36.
Wang Y, Li Z, Teng M, et al. Dihydroartemisinin inhibits activation of the AIM2 inflammasome pathway and NF-kappaB/HIF-1alpha/VEGF pathway by inducing autophagy in A431 human cutaneous squamous cell carcinoma cells. Int J Med Sci. 2021;18(12):2705–15.
Chai D, Shan H, Wang G, et al. AIM2 is a potential therapeutic target in human renal carcinoma and suppresses its invasion and metastasis via enhancing autophagy induction. Exp Cell Res. 2018;370(2):561–70.
Chai D, Zhang Z, Shi SY, et al. Absent in melanoma 2-mediating M1 macrophages facilitate tumor rejection in renal carcinoma. Transl Oncol. 2021;14(4): 101018.
Cervantes-Villagrana RD, Albores-Garcia D, Cervantes-Villagrana AR, et al. Tumor-induced neurogenesis and immune evasion as targets of innovative anti-cancer therapies. Signal Transduct Target Ther. 2020;5(1):99.
Reeves E, James E. Antigen processing and immune regulation in the response to tumours. Immunology. 2017;150(1):16–24.
Theivanthiran B, Evans KS, DeVito NC, et al. A tumor-intrinsic PD-L1/NLRP3 inflammasome signaling pathway drives resistance to anti-PD-1 immunotherapy. J Clin Invest. 2020;130(5):2570–86.
Lasithiotaki I, Tsitoura E, Samara KD, et al. NLRP3/Caspase-1 inflammasome activation is decreased in alveolar macrophages in patients with lung cancer. PLoS ONE. 2018;13(10): e0205242.
Sallman DA, List A. The central role of inflammatory signaling in the pathogenesis of myelodysplastic syndromes. Blood. 2019;133(10):1039–48.
Tengesdal IW, Dinarello A, Powers NE, et al. Tumor NLRP3-derived IL-1beta Drives the IL-6/STAT3 axis resulting in sustained MDSC-mediated immunosuppression. Front Immunol. 2021;12: 661323.
Chen L, Huang CF, Li YC, et al. Blockage of the NLRP3 inflammasome by MCC950 improves anti-tumor immune responses in head and neck squamous cell carcinoma. Cell Mol Life Sci. 2018;75(11):2045–58.
van Deventer HW, Burgents JE, Wu QP, et al. The inflammasome component NLRP3 impairs antitumor vaccine by enhancing the accumulation of tumor-associated myeloid-derived suppressor cells. Cancer Res. 2010;70(24):10161–9.
Nakamura K, Kassem S, Cleynen A, et al. Dysregulated IL-18 is a key driver of immunosuppression and a possible therapeutic target in the multiple myeloma microenvironment. Cancer Cell. 2018;33(4):634–48.
Ghiringhelli F, Apetoh L, Tesniere A, et al. Activation of the NLRP3 inflammasome in dendritic cells induces IL-1beta-dependent adaptive immunity against tumors. Nat Med. 2009;15(10):1170–8.
Hu B, Ren J, Luo Y, et al. Augmentation of antitumor immunity by human and mouse CAR T cells secreting IL-18. Cell Rep. 2017;20(13):3025–33.
Lu F, Zhao Y, Pang Y, et al. NLRP3 inflammasome upregulates PD-L1 expression and contributes to immune suppression in lymphoma. Cancer Lett. 2021;497:178–89.
Timperi E, Focaccetti C, Gallerano D, et al. IL-18 receptor marks functional CD8+ T cells in non-small cell lung cancer. OncoImmunology. 2017;6(7):e1328337.
Li XY, Moesta AK, **ao C, et al. Targeting CD39 in cancer reveals an extracellular ATP- and inflammasome-driven tumor immunity. Cancer Discov. 2019;9(12):1754–73.
Dmitrieva-Posocco O, Dzutsev A, Posocco DF, et al. Cell-type-specific responses to interleukin-1 control microbial invasion and tumor-elicited inflammation in colorectal cancer. Immunity. 2019;50(1):166–80.
Sauter KA, Wood LJ, Wong J, et al. Doxorubicin and daunorubicin induce processing and release of interleukin-1beta through activation of the NLRP3 inflammasome. Cancer Biol Ther. 2011;11(12):1008–16.
Wong J, Tran LT, Magun EA, et al. Production of IL-1beta by bone marrow-derived macrophages in response to chemotherapeutic drugs: synergistic effects of doxorubicin and vincristine. Cancer Biol Ther. 2014;15(10):1395–403.
Voloshin T, Alishekevitz D, Kaneti L, et al. Blocking IL1beta pathway following paclitaxel chemotherapy slightly inhibits primary tumor growth but promotes spontaneous metastasis. Mol Cancer Ther. 2015;14(6):1385–94.
Westbom C, Thompson JK, Leggett A, et al. Inflammasome modulation by chemotherapeutics in malignant mesothelioma. PLoS ONE. 2015;10(12): e0145404.
Tang Z, Ji L, Han M, et al. Pyroptosis is involved in the inhibitory effect of FL118 on growth and metastasis in colorectal cancer. Life Sci. 2020;257: 118065.
Huang Y, Wang H, Hao Y, et al. Myeloid PTEN promotes chemotherapy-induced NLRP3-inflammasome activation and antitumour immunity. Nat Cell Biol. 2020;22(6):716–27.
Zheng Q, Yao D, Cai Y, et al. NLRP3 augmented resistance to gemcitabine in triple-negative breast cancer cells via EMT/IL-1beta/Wnt/beta-catenin signaling pathway. Biosci Rep. 2020;40(7).
Zeng QZ, Yang F, Li CG, et al. Paclitaxel enhances the innate immunity by promoting NLRP3 inflammasome activation in macrophages. Front Immunol. 2019;10:72.
De Ruysscher D, Niedermann G, Burnet NG, et al. Radiotherapy toxicity. Nat Rev Dis Primers. 2019;5(1):13.
Wei J, Wang H, Wang H, et al. The role of NLRP3 inflammasome activation in radiation damage. Biomed Pharmacother. 2019;118: 109217.
Zhang Q, Hu Q, Chu Y, et al. The influence of radiotherapy on AIM2 inflammasome in radiation pneumonitis. Inflammation. 2016;39(5):1827–34.
Christersdottir Björklund T, Pirault J, Gisterå A, et al. Prevention of radiotherapy-induced arterial inflammation by interleukin-1 blockade. Eur Heart J. 2019;40(30):2495–503.
Gao J, Peng S, Shan X, et al. Inhibition of AIM2 inflammasome-mediated pyroptosis by Andrographolide contributes to amelioration of radiation-induced lung inflammation and fibrosis. Cell Death Dis. 2019;10(12):957.
Fernandez-Gil B, Moneim AE, Ortiz F, et al. Melatonin protects rats from radiotherapy-induced small intestine toxicity. PLoS ONE. 2017;12(4): e0174474.
**ao J, Wang C, Yao JC, et al. Radiation causes tissue damage by dysregulating inflammasome-gasdermin D signaling in both host and transplanted cells. PLoS Biol. 2020;18(8): e3000807.
Liu YG, Chen JK, Zhang ZT, et al. NLRP3 inflammasome activation mediates radiation-induced pyroptosis in bone marrow-derived macrophages. Cell Death Dis. 2017;8(2): e2579.
Sun H, Cai H, Fu Y, et al. The protection effect of resveratrol against radiation-induced inflammatory bowel disease via NLRP-3 inflammasome repression in mice. Dose Response. 2020;18(2):1559325820931292.
Wu M, Shi J, He S, et al. cGAS promotes sepsis in radiotherapy of cancer by up-regulating caspase-11 signaling. Biochem Biophys Res Commun. 2021;551:86–92.
Sohn SH, Lee JM, Park S, et al. The inflammasome accelerates radiation-induced lung inflammation and fibrosis in mice. Environ Toxicol Pharmacol. 2015;39(2):917–26.
Golden EB, Apetoh L. Radiotherapy and immunogenic cell death. Semin Radiat Oncol. 2015;25(1):11–7.
Formenti SC, Demaria S. Combining radiotherapy and cancer immunotherapy: a paradigm shift. J Natl Cancer Inst. 2013;105(4):256–65.
Ma Y, Kepp O, Ghiringhelli F, et al. Chemotherapy and radiotherapy: cryptic anticancer vaccines. Semin Immunol. 2010;22(3):113–24.
** H, Kim HJ. NLRC4, ASC and caspase-1 are inflammasome components that are mediated by P2Y2R activation in breast cancer cells. Int J Mol Sci. 2020;21(9):3337.
Young HL, Rowling EJ, Bugatti M, et al. An adaptive signaling network in melanoma inflammatory niches confers tolerance to MAPK signaling inhibition. J Exp Med. 2017;214(6):1691–710.
Hajek E, Krebs F, Bent R, et al. BRAF inhibitors stimulate inflammasome activation and interleukin 1 beta production in dendritic cells. Oncotarget. 2018;9(47):28294–308.
Abbate A, Toldo S, Marchetti C, et al. Interleukin-1 and the inflammasome as therapeutic targets in cardiovascular disease. Circ Res. 2020;126(9):1260–80.
Dinarello CA. The IL-1 family of cytokines and receptors in rheumatic diseases. Nat Rev Rheumatol. 2019;15(10):612–32.
Jesus AA, Goldbach-Mansky R. IL-1 blockade in autoinflammatory syndromes. Annu Rev Med. 2014;65:223–44.
Ridker PM, Everett BM, Thuren T, et al. Antiinflammatory therapy with canakinumab for atherosclerotic disease. N Engl J Med. 2017;377(12):1119–31.
Ottaviani S, Molto A, Ea HK, et al. Efficacy of anakinra in gouty arthritis: a retrospective study of 40 cases. Arthritis Res Ther. 2013;15(5):R123.
Grebe A, Hoss F, Latz E. NLRP3 inflammasome and the IL-1 pathway in atherosclerosis. Circ Res. 2018;122(12):1722–40.
Martinez GJ, Celermajer DS, Patel S. The NLRP3 inflammasome and the emerging role of colchicine to inhibit atherosclerosis-associated inflammation. Atherosclerosis. 2018;269:262–71.
Lusebrink E, Goody PR, Lahrmann C, et al. AIM2 stimulation impairs reendothelialization and promotes the development of atherosclerosis in mice. Front Cardiovasc Med. 2020;7: 582482.
Tengesdal IW, Menon DR, Osborne DG, et al. Targeting tumor-derived NLRP3 reduces melanoma progression by limiting MDSCs expansion. Proc Natl Acad Sci U S A. 2021;118(10):e2000915118.
Coll RC, Robertson AA, Chae JJ, et al. A small-molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseases. Nat Med. 2015;21(3):248–55.
Coll RC, Hill JR, Day CJ, et al. MCC950 directly targets the NLRP3 ATP-hydrolysis motif for inflammasome inhibition. Nat Chem Biol. 2019;15(6):556–9.
Tapia-Abellan A, Angosto-Bazarra D, Martinez-Banaclocha H, et al. MCC950 closes the active conformation of NLRP3 to an inactive state. Nat Chem Biol. 2019;15(6):560–4.
Jiang H, He H, Chen Y, et al. Identification of a selective and direct NLRP3 inhibitor to treat inflammatory disorders. J Exp Med. 2017;214(11):3219–38.
Huang Y, Jiang H, Chen Y, et al. Tranilast directly targets NLRP3 to treat inflammasome-driven diseases. EMBO Mol Med. 2018;10(4):e8689.
Darakhshan S, Pour AB. Tranilast: a review of its therapeutic applications. Pharmacol Res. 2015;91:15–28.
Takahashi K, Menju T, Nishikawa S, et al. Tranilast inhibits TGF-beta1-induced epithelial-mesenchymal transition and invasion/metastasis via the suppression of smad4 in human lung cancer cell lines. Anticancer Res. 2020;40(6):3287–96.
Saito H, Fushida S, Harada S, et al. Importance of human peritoneal mesothelial cells in the progression, fibrosis, and control of gastric cancer: inhibition of growth and fibrosis by tranilast. Gastric Cancer. 2018;21(1):55–67.
He Y, Varadarajan S, Muñoz-Planillo R, et al. 3,4-methylenedioxy-β-nitrostyrene inhibits NLRP3 inflammasome activation by blocking assembly of the inflammasome. J Biol Chem. 2014;289(2):1142–50.
He H, Jiang H, Chen Y, et al. Oridonin is a covalent NLRP3 inhibitor with strong anti-inflammasome activity. Nat Commun. 2018;9(1):2550.
Honda H, Nagai Y, Matsunaga T, et al. Isoliquiritigenin is a potent inhibitor of NLRP3 inflammasome activation and diet-induced adipose tissue inflammation. J Leukoc Biol. 2014;96(6):1087–100.
Liu X, Pichulik T, Wolz OO, et al. Human NACHT, LRR, and PYD domain-containing protein 3 (NLRP3) inflammasome activity is regulated by and potentially targetable through Bruton tyrosine kinase. J Allergy Clin Immunol. 2017;140(4):1054–67.
Tian X, Zhang S, Zhang Q, et al. Resveratrol inhibits tumor progression by down-regulation of NLRP3 in renal cell carcinoma. J Nutr Biochem. 2020;85: 108489.
Maayah ZH, Alam AS, Takahara S, et al. Resveratrol reduces cardiac NLRP3-inflammasome activation and systemic inflammation to lessen doxorubicin-induced cardiotoxicity in juvenile mice. FEBS Lett. 2021;595(12):1681–95.
Liu Q, Guo X, Huang Z, et al. Anti-neuroinflammatory effects of dimethylaminomylide (DMAMCL, i.e., ACT001) are associated with attenuating the NLRP3 in fl ammasome in MPTP-induced Parkinson disease in mice. Behav Brain Res. 2020;383:112539.
Ahn H, Kang SG, Yoon SI, et al. Methylene blue inhibits NLRP3, NLRC4, AIM2, and non-canonical inflammasome activation. Sci Rep. 2017;7(1):12409.
Lee J, Ahn H, Hong EJ, et al. Sulforaphane attenuates activation of NLRP3 and NLRC4 inflammasomes but not AIM2 inflammasome. Cell Immunol. 2016;306–307:53–60.
Teng JF, Mei QB, Zhou XG, et al. Polyphyllin VI induces caspase-1-mediated pyroptosis via the induction of ROS/NF-kappaB/NLRP3/GSDMD signal axis in non-small cell lung cancer. Cancers. 2020;12(1):193.
Zhang M, ** C, Yang Y, et al. AIM2 promotes non-small-cell lung cancer cell growth through inflammasome-dependent pathway. J Cell Physiol. 2019;234(11):20161–73.
Wei Q, Zhu R, Zhu J, et al. E2-induced activation of the NLRP3 inflammasome triggers pyroptosis and inhibits autophagy in HCC cells. Oncol Res. 2019;27(7):827–34.
Winter RN, Rhee JG, Kyprianou N. Caspase-1 enhances the apoptotic response of prostate cancer cells to ionizing radiation. Anticancer Res. 2004;24(3a):1377–86.
Zeng Q, Fu J, Korrer M, et al. Caspase-1 from human myeloid-derived suppressor cells can promote T cell-independent tumor proliferation. Cancer Immunol Res. 2018;6(5):566–77.
Salvucci O, Carsana M, Bersani I, et al. Antiapoptotic role of endogenous nitric oxide in human melanoma cells. Cancer Res. 2001;61(1):318–26.
Kobori M, Iwashita K, Shinmoto H, et al. Phloretin-induced apoptosis in B16 melanoma 4A5 cells and HL60 human leukemia cells. Biosci Biotechnol Biochem. 1999;63(4):719–25.
Chen YQ, Wang SN, Shi YJ, et al. CRID3, a blocker of apoptosis associated speck like protein containing a card, ameliorates murine spinal cord injury by improving local immune microenvironment. J Neuroinflamm. 2020;17(1):255.
Coll RC, Robertson A, Butler M, et al. The cytokine release inhibitory drug CRID3 targets ASC oligomerisation in the NLRP3 and AIM2 inflammasomes. PLoS ONE. 2011;6(12): e29539.
Sollberger G, Choidas A, Burn GL, et al. Gasdermin D plays a vital role in the generation of neutrophil extracellular traps. Sci Immunol. 2018;3(26):eaar6689.
Silva CM, Wanderley CW, Veras FP, et al. Gasdermin D inhibition prevents multiple organ dysfunction during sepsis by blocking NET formation. Blood. 2021;138(25):2702–13.
Hu JJ, Liu X, Zhao J, et al. Identification of pyroptosis inhibitors that target a reactive cysteine in gasdermin D. 2018.
Li Y, Chen F, Chen J, et al. Disulfiram/copper induces antitumor activity against both nasopharyngeal cancer cells and cancer-associated fibroblasts through ROS/MAPK and ferroptosis Pathways. Cancers. 2020;12(1):138.
Wang Y, Zhang XL, Sun CM. BAY-11-7082 induces apoptosis of multiple myeloma U266 cells through inhibiting NF-κB pathway. Eur Rev Med Pharmacol Sci. 2018;22(9):2564–71.
Dai Y, Pei XY, Rahmani M, et al. Interruption of the NF-kappaB pathway by bay 11–7082 promotes UCN-01-mediated mitochondrial dysfunction and apoptosis in human multiple myeloma cells. Blood. 2004;103(7):2761–70.
Alten R, Gram H, Joosten LA, et al. The human anti-IL-1 beta monoclonal antibody ACZ885 is effective in joint inflammation models in mice and in a proof-of-concept study in patients with rheumatoid arthritis. Arthritis Res Ther. 2008;10(3):R67.
Economides AN, Carpenter LR, Rudge JS, et al. Cytokine traps: multi-component, high-affinity blockers of cytokine action. Nat Med. 2003;9(1):47–52.
Dinarello CA. Setting the cytokine trap for autoimmunity. Nat Med. 2003;9(1):20–2.
Harouki N, Nicol L, Remy-Jouet I, et al. The IL-1beta antibody gevokizumab limits cardiac remodeling and coronary dysfunction in rats with heart failure. JACC Basic Transl Sci. 2017;2(4):418–30.
Zhou T, Damsky W, Weizman OE, et al. IL-18BP is a secreted immune checkpoint and barrier to IL-18 immunotherapy. Nature. 2020;583(7817):609–14.
Voigt C, May P, Gottschlich A, et al. Cancer cells induce interleukin-22 production from memory CD4(+) T cells via interleukin-1 to promote tumor growth. Proc Natl Acad Sci USA. 2017;114(49):12994–9.
Ridker PM, MacFadyen JG, Thuren T, et al. Effect of interleukin-1beta inhibition with canakinumab on incident lung cancer in patients with atherosclerosis: exploratory results from a randomised, double-blind, placebo-controlled trial. Lancet. 2017;390(10105):1833–42.
Wong CC, Baum J, Silvestro A, et al. Inhibition of IL1beta by canakinumab may be effective against diverse molecular subtypes of lung cancer: an exploratory analysis of the CANTOS trial. Cancer Res. 2020;80(24):5597–605.
O’Shaughnessy J, Young RR, Levin MK, et al. Safety and immunologic activity of anakinra in HER2-negative metastatic breast cancer (MBC). J Clin Oncol. 2016;34(15_suppl):e14565–665.
Wardill HR, de Mooij CEM, Da Silva Ferreira AR, et al. Supporting the gastrointestinal microenvironment during high-dose chemotherapy and stem cell transplantation by inhibiting IL-1 signaling with anakinra. Sci Rep. 2022;12(1):6803.
Isambert N, Hervieu A, Rebe C, et al. Fluorouracil and bevacizumab plus anakinra for patients with metastatic colorectal cancer refractory to standard therapies (IRAFU): a single-arm phase 2 study. Oncoimmunology. 2018;7(9): e1474319.
Giavridis T, van der Stegen SJC, Eyquem J, et al. CAR T cell-induced cytokine release syndrome is mediated by macrophages and abated by IL-1 blockade. Nat Med. 2018;24(6):731–8.
Wei Q, Guo P, Mu K, et al. Estrogen suppresses hepatocellular carcinoma cells through ERbeta-mediated upregulation of the NLRP3 inflammasome. Lab Invest. 2015;95(7):804–16.
Raut PK, Kim SH, Choi DY, et al. Growth of breast cancer cells by leptin is mediated via activation of the inflammasome: critical roles of estrogen receptor signaling and reactive oxygen species production. Biochem Pharmacol. 2019;161:73–88.
Chu Q, Jiang Y, Zhang W, et al. Pyroptosis is involved in the pathogenesis of human hepatocellular carcinoma. Oncotarget. 2016;7(51):84658–65.
Johnson AG, Wein T, Mayer ML, et al. Bacterial gasdermins reveal an ancient mechanism of cell death. Science. 2022;375(6577):221–5.
Ariffin JK, Sweet MJ. Differences in the repertoire, regulation and function of Toll-like receptors and inflammasome-forming nod-like receptors between human and mouse. Curr Opin Microbiol. 2013;16(3):303–10.
Atianand MK, Duffy EB, Shah A, et al. Francisella tularensis reveals a disparity between human and mouse NLRP3 inflammasome activation. J Biol Chem. 2011;286(45):39033–42.
Chow MT, Tschopp J, Möller A, et al. NLRP3 promotes inflammation-induced skin cancer but is dispensable for asbestos-induced mesothelioma. Immunol Cell Biol. 2012;90(10):983–6.
Mori N, Yamada Y, Ikeda S, et al. Bay 11–7082 inhibits transcription factor NF-kappaB and induces apoptosis of HTLV-I-infected T-cell lines and primary adult T-cell leukemia cells. Blood. 2002;100(5):1828–34.
Chen L, Ruan Y, Wang X, et al. BAY 11–7082, a nuclear factor-kappaB inhibitor, induces apoptosis and S phase arrest in gastric cancer cells. J Gastroenterol. 2014;49(5):864–74.
Zhao J, Zhang H, Huang Y, et al. Bay11-7082 attenuates murine lupus nephritis via inhibiting NLRP3 inflammasome and NF-kappaB activation. Int Immunopharmacol. 2013;17(1):116–22.
Irrera N, Vaccaro M, Bitto A, et al. BAY 11–7082 inhibits the NF-kappaB and NLRP3 inflammasome pathways and protects against IMQ-induced psoriasis. Clin Sci. 2017;131(6):487–98.
Chen S, Wang Y, Pan Y, et al. Novel role for tranilast in regulating NLRP3 ubiquitination, vascular inflammation, and atherosclerosis. J Am Heart Assoc. 2020;9(12): e015513.
Lonnemann N, Hosseini S, Marchetti C, et al. The NLRP3 inflammasome inhibitor OLT1177 rescues cognitive impairment in a mouse model of Alzheimer’s disease. Proc Natl Acad Sci U S A. 2020;117(50):32145–54.
Lunding LP, Skouras DB, Vock C, et al. The NLRP3 inflammasome inhibitor, OLT1177((R)), ameliorates experimental allergic asthma in mice. Allergy. 2022;77(3):1035-38.
Wang X, Sun K, Zhou Y, et al. NLRP3 inflammasome inhibitor CY-09 reduces hepatic steatosis in experimental NAFLD mice. Biochem Biophys Res Commun. 2021;534:734–9.
Zhang Y, Lin Z, Chen D, et al. CY-09 attenuates the progression of osteoarthritis via inhibiting NLRP3 inflammasome-mediated pyroptosis. Biochem Biophys Res Commun. 2021;553:119–25.
Liu H, Xu Y, Liang K, et al. Immune cells combined with NLRP3 inflammasome inhibitor exert better antitumor effect on pancreatic ductal adenocarcinoma. Front Oncol. 2020;10:1378.
Chen IH, Chang FR, Wu YC, et al. 3,4-Methylenedioxy-beta-nitrostyrene inhibits adhesion and migration of human triple-negative breast cancer cells by suppressing beta1 integrin function and surface protein disulfide isomerase. Biochimie. 2015;110:81–92.
**ao M, Li L, Li C, et al. 3,4-methylenedioxy-beta-nitrostyrene ameliorates experimental burn wound progression by inhibiting the NLRP3 inflammasome activation. Plast Reconstr Surg. 2016;137(3):566e-e575.
Xu L, Bi Y, Xu Y, et al. Oridonin inhibits the migration and epithelial-to-mesenchymal transition of small cell lung cancer cells by suppressing FAK-ERK1/2 signalling pathway. J Cell Mol Med. 2020;24(8):4480–93.
Yang J, Ren X, Zhang L, et al. Oridonin inhibits oral cancer growth and PI3K/Akt signaling pathway. Biomed Pharmacother. 2018;100:226–32.
Li M, Liu H, Shao H, et al. Glyburide attenuates B(a)p and LPS-induced inflammation-related lung tumorigenesis in mice. Environ Toxicol. 2021;36(8):1713–22.
Shim DW, Shin WY, Yu SH, et al. BOT-4-one attenuates NLRP3 inflammasome activation: NLRP3 alkylation leading to the regulation of its ATPase activity and ubiquitination. Sci Rep. 2017;7(1):15020.
Kim BH, Min YS, Choi JS, et al. Benzoxathiol derivative BOT-4-one suppresses L540 lymphoma cell survival and proliferation via inhibition of JAK3/STAT3 signaling. Exp Mol Med. 2011;43(5):313–21.
Lin ZH, Wang SY, Chen LL, et al. Methylene blue mitigates acute neuroinflammation after spinal cord injury through inhibiting nlrp3 inflammasome activation in microglia. Front Cell Neurosci. 2017;11:391.
Yi HJ, Lee JE, Lee DH, et al. The role of NLRP3 in traumatic brain injury and its regulation by pioglitazone. J Neurosurg. 2019;1–9.
Daniels MJ, Rivers-Auty J, Schilling T, et al. Fenamate NSAIDs inhibit the NLRP3 inflammasome and protect against Alzheimer’s disease in rodent models. Nat Commun. 2016;7:12504.
Guo C, Fulp JW, Jiang Y, et al. Development and characterization of a hydroxyl-sulfonamide analogue, 5-chloro-N-[2-(4-hydroxysulfamoyl-phenyl)-ethyl]-2-methoxy-benzamide, as a Novel NLRP3 inflammasome inhibitor for potential treatment of multiple sclerosis. ACS Chem Neurosci. 2017;8(10):2194–201.
Yin J, Zhao F, Chojnacki JE, et al. NLRP3 inflammasome inhibitor ameliorates amyloid pathology in a mouse model of alzheimer’s disease. Mol Neurobiol. 2018;55(3):1977–87.
Awad AS, Elariny HA, Sallam AS. The possible protective effect of colchicine against liver damage induced by renal ischemia-reperfusion injury: role of Nrf2 and NLRP3 inflammasome. Can J Physiol Pharmacol. 2020;98(12):849–54.
Shi Y, Lv Q, Zheng M, et al. NLRP3 inflammasome inhibitor INF39 attenuated NLRP3 assembly in macrophages. Int Immunopharmacol. 2021;92: 107358.
Liu F, Liu T, Sun M, et al. Maxing shigan decoction mitigates mycoplasma pneumonia-induced pyroptosis in A549 cells via the NLRP3 inflammasome. Infect Drug Resist. 2021;14:859–67.
Gong Y, Cao X, Gong L, et al. Sulforaphane alleviates retinal ganglion cell death and inflammation by suppressing NLRP3 inflammasome activation in a rat model of retinal ischemia/reperfusion injury. Int J Immunopathol Pharmacol. 2019;33:2058738419861777.
Shang S, Wang L, Zhang Y, et al. The beta-hydroxybutyrate suppresses the migration of glioma cells by inhibition of NLRP3 inflammasome. Cell Mol Neurobiol. 2018;38(8):1479–89.
Shippy DC, Wilhelm C, Viharkumar PA, et al. beta-Hydroxybutyrate inhibits inflammasome activation to attenuate Alzheimer’s disease pathology. J Neuroinflamm. 2020;17(1):280.
Goldberg EL, Asher JL, Molony RD, et al. beta-hydroxybutyrate deactivates neutrophil NLRP3 inflammasome to relieve gout flares. Cell Rep. 2017;18(9):2077–87.
Carbone S, Mauro AG, Prestamburgo A, et al. An orally available NLRP3 inflammasome inhibitor prevents western diet-induced cardiac dysfunction in mice. J Cardiovasc Pharmacol. 2018;72(6):303–7.
Marchetti C, Chojnacki J, Toldo S, et al. A novel pharmacologic inhibitor of the NLRP3 inflammasome limits myocardial injury after ischemia-reperfusion in the mouse. J Cardiovasc Pharmacol. 2014;63(4):316–22.
Baldwin AG, Rivers-Auty J, Daniels MJD, et al. Boron-based inhibitors of the NLRP3 inflammasome. Cell Chem Biol. 2017;24(11):1321–35.
Lim H, Min DS, Park H, et al. Flavonoids interfere with NLRP3 inflammasome activation. Toxicol Appl Pharmacol. 2018;355:93–102.
Liu W, Guo W, Wu J, et al. A novel benzo[d]imidazole derivate prevents the development of dextran sulfate sodium-induced murine experimental colitis via inhibition of NLRP3 inflammasome. Biochem Pharmacol. 2013;85(10):1504–12.
Wu D, Wu K, Zhu Q, et al. Formononetin administration ameliorates dextran sulfate sodium-induced acute colitis by inhibiting NLRP3 inflammasome signaling pathway. Mediat Inflamm. 2018;2018:1–12.
Li R, Lu K, Wang Y, et al. Triptolide attenuates pressure overload-induced myocardial remodeling in mice via the inhibition of NLRP3 inflammasome expression. Biochem Biophys Res Commun. 2017;485(1):69–75.
Guo W, Sun Y, Liu W, et al. Small molecule-driven mitophagy-mediated NLRP3 inflammasome inhibition is responsible for the prevention of colitis-associated cancer. Autophagy. 2014;10(6):972–85.
Gong Z, Zhao S, Zhou J, et al. Curcumin alleviates DSS-induced colitis via inhibiting NLRP3 inflammsome activation and IL-1beta production. Mol Immunol. 2018;104:11–9.
Abdullaha M, Mohammed S, Ali M, et al. Discovery of quinazolin-4(3 H)-ones as NLRP3 inflammasome inhibitors: computational design, metal-free synthesis, and in vitro biological evaluation. J Org Chem. 2019;84(9):5129–40.
Abderrazak A, Couchie D, Mahmood DF, et al. Anti-inflammatory and antiatherogenic effects of the NLRP3 inflammasome inhibitor arglabin in ApoE2.Ki mice fed a high-fat diet. Circulation. 2015;131(12):1061–70.
Flores J, Noel A, Foveau B, et al. Caspase-1 inhibition alleviates cognitive impairment and neuropathology in an Alzheimer’s disease mouse model. Nat Commun. 2018;9(1):3916.
Bauer C, Duewell P, Mayer C, et al. Colitis induced in mice with dextran sulfate sodium (DSS) is mediated by the NLRP3 inflammasome. Gut. 2010;59(9):1192–9.
Rudolphi K, Gerwin N, Verzijl N, et al. Pralnacasan, an inhibitor of interleukin-1β converting enzyme, reduces joint damage in two murine models of osteoarthritis. Osteoarthr Cartil. 2003;11(10):738–46.
Ross J, Brough D, Gibson RM, et al. A selective, non-peptide caspase-1 inhibitor, VRT-018858, markedly reduces brain damage induced by transient ischemia in the rat. Neuropharmacology. 2007;53(5):638–42.
Wang P, Pan B, Tian J, et al. Ac-FLTD-CMK inhibits pyroptosis and exerts neuroprotective effect in a mice model of traumatic brain injury. NeuroReport. 2021;32(3):188–97.
Sun Y, Guo Y. Expression of caspase-1 in breast cancer tissues and its effects on cell proliferation, apoptosis and invasion. Oncol Lett. 2018;15(5):6431–5.
Chai D, Qiu D, Zhang Z, et al. Absent in melanoma 2 enhances anti-tumour effects of CAIX promotor controlled conditionally replicative adenovirus in renal cancer. J Cell Mol Med. 2020;24(18):10744–55.
Chen-Deutsch X, Kutner A, Harrison JS, et al. The pan-caspase inhibitor Q-VD-OPh has anti-leukemia effects and can interact with vitamin D analogs to increase HPK1 signaling in AML cells. Leuk Res. 2012;36(7):884–8.
Lacy SE, Wu C, Ambrosi DJ, et al. Generation and characterization of ABT-981, a dual variable domain immunoglobulin (DVD-Ig(TM)) molecule that specifically and potently neutralizes both IL-1alpha and IL-1beta. MAbs. 2015;7(3):605–19.
Chevalier X, Conrozier T, Richette P. Desperately looking for the right target in osteoarthritis: the anti-IL-1 strategy. Arthritis Res Ther. 2011;13(4):124.
Han C, Yang Y, Guan Q, et al. New mechanism of nerve injury in Alzheimer’s disease: beta-amyloid-induced neuronal pyroptosis. J Cell Mol Med. 2020;24(14):8078–90.
Skrott Z, Mistrik M, Andersen KK, et al. Alcohol-abuse drug disulfiram targets cancer via p97 segregase adaptor NPL4. Nature. 2017;552(7684):194–9.
Acknowledgements
The figures in this review are created with BioRender.com
Funding
This work is supported by the National Science Fund for Excellent Young Scholars (No. 32122052) and the National Natural Science Foundation Regional Innovation and Development (No. U19A2003).
Author information
Authors and Affiliations
Contributions
XWi put forward the topic of the review. ZZ and XL performed the literature search and prepared the figures and tables. ZZ and YW prepared the main manuscript. YW helped with the revision of the review. All authors reviewed the manuscript and approved the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Zhang, Z., Li, X., Wang, Y. et al. Involvement of inflammasomes in tumor microenvironment and tumor therapies. J Hematol Oncol 16, 24 (2023). https://doi.org/10.1186/s13045-023-01407-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13045-023-01407-7