
Abstract
Focal cortical dysplasia (FCD) is a malformation of the cerebral cortex with poorly-defined epileptogenic zones (EZs), and poor surgical outcome in FCD is associated with inaccurate localization of the EZ. Hence, identifying novel epileptogenic markers to aid in the localization of EZ in patients with FCD is very much needed. High-throughput gene expression studies of FCD samples have the potential to uncover molecular changes underlying the epileptogenic process and identify novel markers for delineating the EZ. For this purpose, we, for the first time performed RNA sequencing of surgically resected paired tissue samples obtained from electrocorticographically graded high (MAX) and low spiking (MIN) regions of FCD type II patients and autopsy controls. We identified significant changes in the MAX samples of the FCD type II patients when compared to non-epileptic controls, but not in the case of MIN samples. We found significant enrichment for myelination, oligodendrocyte development and differentiation, neuronal and axon ensheathment, phospholipid metabolism, cell adhesion and cytoskeleton, semaphorins, and ion channels in the MAX region. Through the integration of both MAX vs non-epileptic control and MAX vs MIN RNA sequencing (RNA Seq) data, PLP1, PLLP, UGT8, KLK6, SOX10, MOG, MAG, MOBP, ANLN, ERMN, SPP1, CLDN11, TNC, GPR37, SLC12A2, ABCA2, ABCA8, ASPA, P2RX7, CERS2, MAP4K4, TF, CTGF, Semaphorins, Opalin, FGFs, CALB2, and TNC were identified as potential key regulators of multiple pathways related to FCD type II pathology. We have identified novel epileptogenic marker elements that may contribute to epileptogenicity in patients with FCD and could be possible markers for the localization of EZ.
Similar content being viewed by others


Introduction
Focal cortical dysplasia is the most commonly encountered developmental malformation that causes drug resistant focal epilepsy, particularly in children [1]. Its anatomopathological position and cellular appearance are highly variable and influence not only the cortical architecture and unique neuronal subpopulations, but also the junction of gray-white matter and sub-cortical white matter regions [2, 3]. The most frequent subtype is FCD type II, mainly localized in the frontal and parietal lobes and can range from either small and almost invisible bottom‐of‐sulcus dysplasia to larger dysplastic regions covering more than a single gyrus. Focal cortical dysplasia type II is marked by gross histopathological changes, i.e., dysmorphic neurons (FCD type IIA) and additional balloon cells (FCD type IIB) [4, 5]. Because dysplastic tissue contains atypical neuronal networks that are highly susceptible to abnormal excitation, FCD is thought to be intrinsically epileptogenic. Despite the introduction of new anti-epileptic drugs (AEDs) in the last two decades, over 30% of epilepsy patients have recurring seizures and many have undesirable side effects. Surgery is an effective alternative treatment as it offers seizure freedom or a significant reduction in seizures for those patients with drug-resistant epilepsy (DRE). Epilepsy surgical outcome is influenced by a number of factors, including epilepsy type, underlying pathology, and the most significant accurate localization of the epileptogenic zone (EZ) and precise details of its association with the eloquent cortex for complete and safe removal using a variety of clinical, neuroimaging, and neurophysiological tests [6,7,8].
FCD is a diffuse lesion with poorly defined epileptogenic zones. Thus, incomplete resection has been consistently known to be a poor prognostic factor. Clinical history, comprehensive semiology analysis, long-term video-EEG recording, inter-ictal and ictal EEG analysis, neuroimaging, and neuropsychological examination are all part of the pre-surgical evaluation process and each modality gives distinct and complementary information. Because no single currently available approach can consistently diagnose EZ, and each modality has its own set of limitations, comprehensive examinations are required to analyse the EZ's various characteristics [8, 9]. The functional involvement of the dysplastic cortex in the epileptogenic network cannot be identified through MRI alone. FCDs can be microscopic (or MRI negative), which means they may go undetected even with high resolution MRI. The lesions are subtle in these cases; morphological features may vary only marginally from normal tissue [10, 11]. fMRI or magnetoencephalography (MEG) detects classical and aberrant distributed functional networks but may be falsely suppressed in the postictal period. The absence of a visible lesion is one of the greatest challenges in epilepsy surgery; dysplastic tissue looks similar to normal brain tissue and can be missed, unless intracranial electrode application and intraoperative electrocorticography (EcoG) recordings are performed [6, 12].
Despite the use of all available invasive and non-invasive approaches, the epileptogenic zone cannot be fully identified, and patients do not benefit in more than 30% of these cases, owing to the inability to accurately locate the EZ [13]. A more precise framework for identifying EZ can be provided by molecular and cellular biomarkers combined with imaging and electrical investigations [13, 14]. Aberrant gene expression and epigenetic alterations such as DNA methylation have been reported in different epilepsy pathologies, including FCD [15,16,17,18,19,20]. These studies have helped us to better understand the molecular mechanism of epileptogenesis, but the search for biomarkers to localize the EZ accurately has not ended yet.
This has kindled interest in unveiling the mechanisms of epileptogenicity in these lesions. Human tissue samples, on the other hand, restrict experimental design because age and gender-matched control samples from non-epileptic patients are rarely available for comparison. However, having a better understanding of how seizures are generated in the dysplastic human neocortex ultimately requires an examination of the available human tissue samples. Surgically resected human dysplastic tissue can be a good model to study the mechanism of epileptogenicity in these patients. Intracranial EcoG recording is usually performed in FCD cases to identify the extent of the epileptogenic zone and its complete excision. Tissues with different EcoG grades have been removed during surgery from the same patients. These tissues from the same patient can be an ideal model to extrapolate the mechanism of epiletogenicity in FCD type II, which in turn helps to delineate the epileptozenic zone in FCD patients. Hence, the present study was designed to study the transcriptomic profile of surgically resected paired tissue samples obtained from electrocorticographically graded high- (MAX) and low-spiking (MIN) regions of FCD type II patients and autopsy controls. The current study’s findings are discussed to gain better understanding of the epileptogenicity in FCD and the localization of EZ.
Materials and methods
Patient and control samples
The patients who were diagnosed to have DRE due to FCD type II and underwent electrocorticography (ECoG)-guided surgery were included in the study. Pre-surgical assessment was done for each patient, and the pathology was determined by analysing convergent data on MRI, video EEG (vEEG), fluoro-2-deoxyglucose positron emission tomography (FDG-PET) and magnetoencephalography (MEG), further confirmed by histopathological examinations by neuropathologists. Patients with dual pathology were excluded.
Based on ECoG recordings, the regions were graded from scores of 1 to 5 [21, 22], with grade 2 and above reported as a high spiking zone (MAX) and grade 1 as a low spiking zone (MIN) (Additional file 1: Fig. S1). Surgical resection of ECoG graded cortical samples was performed as per the previously reported protocol [22, 23]. The MAX region was defined as cortical regions of MEG abnormality, the greatest positron emission tomography hypometabolism, the most severe magnetic resonance imaging architectural abnormalities, and the most abnormal ECoG findings. The MIN region was defined as less severely involved based on neuroimaging and ECoG, but it was part of the planned resection. Resected tissue samples from the MAX and MIN regions from the same patients were collected for transcriptomic analysis. Details of neuroimaging techniques used and their scores are listed in Table 1. Based on EcoG grade, MRI, PET and MEG data, E018, E019, E070, E075, E077, E273, E460, and E578 were categorized under the MIN region, and E006, E028, E045, E084, SampleE1, E115, E135, E536 and E593 were categorized under the MAX region.
As there are no “ideal” or acceptable non-epilepsy controls for such studies involving epilepsy surgery, we have used histologically normal cortex tissues obtained from the frontal lobes of the post-mortem cases without any history of seizures or other neurological disorders as non-epileptic controls. All the autopsies were performed within 8 h of death. All the patients included in the study were seizure-free post-operatively (Class I Engel outcome). Part of the resected tissue were stored in 4% paraformaldehyde for histopathological examination and remaining parts were immediately frozen and stored at − 80 °C until further use.
The study was conducted in accordance with the Declaration of Helsinki and was approved by the Institute Ethics Committee, AIIMS, New Delhi. Informed and written consent was obtained from all the patients, their parents, or legal guardians if the patients were underage.
RNA sequencing (RNA seq)
RNA extraction and sequencing were performed as described previously with some modifications [16]. Briefly, frozen brain samples were homogenized in RiboZol reagent (Amresco) and RNA was extracted using RNeasy Mini Kit (Qiagen) as per manufacturer’s instructions. An additional DNase1 digestion step was performed to ensure that the samples were not contaminated with genomic DNA. RNA quality was assessed using Bioanalyzer 2100 (Agilent). RNA libraries were prepared using TruSeq RNA Access Library Prep Kit (Illumina) and paired-end sequencing was performed on IlluminaHiseq 2500 platforms. Sequences were quality-checked using FastQC and low-quality bases and reads were excluded from further analysis. Sequences were aligned using HISATaligner against all known genes and transcripts of GRCh37/hg19 assembly.
In this comparative study we analyzed the RNA Sequencing data by three of the most frequently used software tools: Cuffdiff, DESeq2 and EdgeR [24, 25]. Significantly altered genes which were common in all three software tools were further used for downstream gene enrichment and network analysis. RNA Seq analysis was performed for three categories: (1) Between autopsy samples (A1 andA2) and samples from MIN region (E018, E019, E075 and E273); (2) Between autopsy samples (A1 andA2) and samples from MAX region (E006, E028, E115, SampleE1 and E135); and (3) Between samples from MIN region (E018, E019, E075 and E273) and samples from MAX region (E006, E028, E115, SampleE1, and E135).
Principal component analysis (PCA), pathway enrichment analysis and gene network analysis
Intersections of gene expression which were found to be significantly altered in all three RNA Seq analysis software were used for calculating and plotting the principal components using ClustVis [26]. Unit variance scaling was applied to genes and singular value decomposition with imputation was used to calculate the principal components. Samples were clustered using correlation distance and average linkage in heatmap. Common DEGs (based on fold-change (≥ 2) and FDR-adjusted p values (padj) in all three software packages were used for downstream gene enrichment and network analyses. Gene enrichment analysis was performed as described previously [27]. Briefly, the DEGs were used as an ordered query in g: Profiler with term size ranging from 3 to 350 and significance cut-off (FDR q val) set to < 0.05. Custom gene sets containing GO: BP terms and KEGG pathways were used.
Network analysis was performed to graphically display associations between DEGs, to show both direct and indirect interactions using Natural language processing-based (NLP) network discovery algorithms in gene spring software version 13.0 as described previously [16].
Validation by real-time PCR (RT-PCR)
Real-time PCR (RT-PCR) was performed to validate the differentially expressed genes using specific primers (Additional file 7: Table S1) for selected genes (TNC, MOBP, SLC12A2, CTGF, GPR37, KCNK10 and CARTPT) on 9 FCD (F1 to F9) patients and 8 control samples (A1 to A8). RNA was reverse transcribed using High-capacity cDNA Reverse Transcription Kit (ThermoFisher; catalogue# 4368814). Hypoxanthine phosphoribosyltransferase (HPRT) gene was used as an internal reference. Real-time PCR amplifications were performed in CFX 96 real-time systems (Bio-Rad) with Bio-Rad CFX software manager with the following cycling parameters: an initial hot start of 95 °C for 3 min followed by 40 cycles of 95 °C for 5 s and 60 °C for 30 s. The melting curve of each replicate was examined to confirm a single peak appearance. All samples were run in triplicates. The 2−ΔΔCq method was used to measure the mRNA expression of target genes based on the average Ct values across samples [28].
Histopathology
Tissues were fixed in 4% paraformaldehyde and embedded in paraffin wax for preparing 5-µm thick tissue sections. Haematoxylin and eosin (H&E) staining was performed as described previously [16]. One series of sections was stained with crystal violet (Sigma) according to the previously established protocol [29]. The slides were independently reviewed by two neuropathologists to confirm the pathology and evaluate any damage in control tissue.
Results
Clinical characteristics of patients and controls
A total of nine FCD Type II patients (three male and six females) patients were included in this study. For RNA Seq analysis, graded samples of five FCD type II patients (F1 to F5) and two controls (A1 to A2) were included. Subsequently, we used surgically resected graded tissues from 9 FCD type II patients (F1–F9) and eight controls (A1–A8) (including samples of RNA Seq analysis) for real-time PCR analysis. The detailed clinical characteristics of individuals are listed in Table 1.
The mean age of FCD type II patients was 14.77 ± 6.23 years (ranges from 5 to 22 years). Autopsy patient’s age ranged from 16 to 65 years (mean age 29.37 ± 15.01 years). Detailed histopathological investigations were performed on all the samples obtained for experiments (as mentioned in Table 1) to confirm the pathology (Fig. 1). Haematoxylin and eosin (HE), and crystal violet (CV) staining were performed to evaluate the histopathological features. Characteristic features of FCD type II patients were observed in all the patients. Cortical section from FCD type II patients showed dysmorphic neurons Fig. 1B, E and dysmorphic neurons with balloon cells Fig. 1C, F. Cortical sections from autopsy showed normal cytoarchitecture Fig. 1A, D.

Photomicrograph showing characteristic histopathological features of focal cortical dysplasia II. Representative image of the cortical section from FCD type II patients showing dysmorphic neurons B HE, × 400) and E CV staining × 400; and dysmorphic neurons with balloon cells C HE, × 400) and F CV staining × 400. Cortical sections from autopsy showed normal cytoarchitechure. A HE, × 400 and D CV staining × 400
Differentially expressed genes (DEGs)
RNAseq read summary is provided in the Fig. 2A. Cuffdiff analysis revealed 38 differentially expressed genes (16 up-regulated and 22 down-regulated) in MIN vs autopsy; 325 DEGs (224 up-regulated and 101 down-regulated) in MAX vs autopsy; and 550 DEGs (378 up-regulated and 172 down-regulated) in MAX vs MIN (Fig. 2B–D; Additional file 3). List of genes with significantly altered expression analysed by Cuffdiff is provided in the Additional file 3 As per DESeq2 analysis, a total of 171 genes were found to be differentially expressed (56 up-regulated and 115 down-regulated) in MIN vs autopsy; 660 genes (316 up-regulated and 344 down-regulated) in MAX vs autopsy; and 783 genes (582 up-regulated and 201 down-regulated) in MAX vs MIN (Fig. 2B–D; Additional file 4). List of DEGs analysed by DESeq2 is given in Additional file 4. EdgeR analysis demonstrated 53 DEGs (5 up-regulated and 48 down-regulated) in MIN vs autopsy; 865 DEGs (176 up-regulated and 689 down-regulated) in MAX vs autopsy; and 292 DEGs (240 up-regulated and 52 down-regulated) in MAX vs MIN (Fig. 2B–D). List of genes with significantly altered gene expression analysed by EdgeR is provided in Additional file 5. Most of the DEGs identified by each of three tools overlapped, DESeq2 detected more DEGs than the other tools. To avoid false positive results, intersection of DEGs from two or more tools is generally used for analysis [24, 25]. To get more accurate and precise findings, intersection of DEGs from Cuffdiff, DESeq2 and EdgeR was used for further analysis, details of commonly found DEGs among three tools is presented in Fig. 3 and Additional file 6. Only 6 genes (2 up-regulated and 4 down-regulated) were found to be differentially expressed in MIN vs autopsy, 109 DEGs (85 up- regulated and 24 down-regulated) were observed in MAX vs autopsy, and 158 DEGs (152 up-regulated and 6 down-regulated) were found to be significantly altered in MAX vs MIN. No gene was found to be common in all three groups. 49 genes were found to be common in MAX vs autopsy and MAX vs MIN. 4 genes were observed to be common in MIN vs autopsy and MAX vs autopsy (Fig. 3).

A RNA Seq summary of samples from patients and controls B, C and D. The Venn diagram depicts both unique and common DEGs identified by three softwares: CuffDiff, DESeq2 and EdgeR in three groups: MIN vs autopsy (B), MAX vs autopsy (C), and MAX vs MIN (D)

Details of differentially expressed genes in all three groups (MIN vs autopsy, MAX vs autopsy, and MAX vs MIN), were found to be common in CuffDiff, DESeq2, and EdgeR analysis software
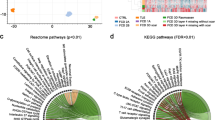
The PCA result indicated that MIN and MAX region of FCD type II patients could be separated by their transcriptome profile by unsupervised clustering. Dimensionality reduction using principal component analysis segregated FCD type II samples and autopsy samples into distinct clusters with PC1 (85.5% for autopsy and MAX, and 89.2% for MIN and MAX) accounting for most of the variance (Fig. 4).

Differential gene expression in FCD type II patients. The PCA analysis and heatmap was generated using ClustVis. For Peer Review Singular value decomposition with imputation was used to calculate the PCs with maximum variance explained by PC1 (89.2% and 85.5% respectively). Plotting of the first two PCs (PC1 and PC2) segregated FCD MAX and control samples; and FCD MIN and FCD MAX samples into distinct clusters. Unit variance scaling is applied to rows containing the log-normalized counts of differentially expressed genes (padj1) and the columns are clustered using correlation distance and average linkage in the heatmap
Pathway enrichment analysis and network analysis
Detailed pathway g: Profiler enrichment results are provided in the Table 2 (FDR q value < 0.05). Pathway enrichment did not provide any result for MIN vs autopsy. Pathway enrichment scores revealed enrichment of altered genes in the four pathways related to myelination, oligodendrocytes development and neuronal and axon ensheathment in MAX vs autopsy, represented by PLP1, MAG, UGT8, CD9, PLLP, SH3TC2. Myelination, ensheathment of neurons and axons, oligodendrocyte development and differentiation, gliogenesis, glial cell development and differentiation, phospholipid biosynthesis, cell adhesion and cytoskeleton proteins, neurogenesis, nervous system development, small molecule transport, and ion channels are among the 44 significantly enriched pathways for MAX vs MIN. Genes related to myelination, oligodendrocytes development and differentiation, neuronal and axon ensheathment included PLP1, SOX10, MAG, MOG, MOBP, KLK6, UGT8, CLDN11, ASPA, SH3TC2, NKX6-2, FA2H,CTGF, SEPT 4, CDKN1C, GLDN, SPP1, CNTN2, TMEM98, TMEM10 (OPALIN), ANLN, ERMN, ENPP2, CD9, ABCA2, ABCA8, SPP1, GPR37 and TF. All these were found to be up-regulated. Among these 9 genes (KLK6, ASPA, SOX10, CNTN2, CLDN11, ERMN, NKX6-2, FA2H, SEPT4, SPP1, MOG, NKX6-2 and MOBP) were found to be significantly up-regulated only in MAX vs MIN, whereas CTGF was found to be up-regulated only in MAX vs autopsy. PLLP, UGT8, ABCA2, PLD1, ELOVL1, CERS2, S1PR5, PLPPR1, SPTLC1, ENPP2, ENPP4, ENPP6, NPC1, FA2H, LRP2, P2RX7, S1PR5 and GAL3ST1 were reported to be involved in phospholipid biosynthesis. Of these, ABCA2, PLD1, ELOVL1, FA2H, CERS2, P2RX7, and GAL3ST1 were found to be up-regulated in MAX vs MIN only, whereas expression of SPTLC1 was observed to be up-regulated only in MAX vs autopsy. Genes related to ion and water channels i.e., SLC12A2, SLC45A3, SLC26A9, SLC44A1, SLC26A9, AQP1, KCNK10, KCNH8, P2RX7, SGK1, SGK2, and SLC6A2 were found to be up-regulated. Among these, SLC12A2, SLC45A3, SGK1 and SLC25A41 were found to be up-regulated only in MAX vs autopsy, however, AQP1, P2RX7, SLC6A2, SLC26A9, SLC44A1, SLC45A3, SLC5A11 and SLC26A9 were found to be up-regulated only in MAX vs MIN. Semaphorins (SEMA4D, SEMA3B and SEMA6A) were found be up-regulated only in MAX vs MIN. Cell signaling molecules of various functions, MAP4K4, TNC, FGF1, FGF17, TGFA, ATF3, MCAM, TGFBRII were found to be up-regulated, GRP and CALB2 were found to be down-regulated. Of these, FGF1, MAP4K4, ATF3, CALB2 and GRP were significantly altered only in MAX vs MIN, whereas expression of FGF17 and TGFBR2 were significantly up-regulated only in MAX vs autopsy. Extracellular matrix (ECM) related genes TJP2, CLDN9, CLDN11, SPP1, and GJB1 were also found to be up-regulated. TJP2, SPP1, CLDN11 were found to be up-regulated only in MAX vs MIN whereas expression of CLDN9 was up-regulated in only MAX vs autopsy.
Various genes i.e., DAAM2, CERS2, SLC45A3, PLP1, ASPA, SH3TC2, MAG, FA2H, CNTN2, TMEM98, ABCA2, CD9, BOK, SOX10, NKX6-2 were also involved in gliogenesis, glial cell development and differentiation as evident by pathway enrichment analysis. Pathway enrichment analysis also demonstrated significant enrichment for neurogenesis and nervous system development represented by AQP1, MOG, PLP1, KLK6, UGT8, CLDN11, SPP1, GLDN, GJB1, ASPA, MOBP, SH3TC2, MAG, SEMA4D, SEMA6A, SEMA3B, TNC, SOX10, MAP4K4, UGT8.
Network analysis also revealed the interaction of various genes including several of above-mentioned genes. Network analysis demonstrated associations of CTGF, MAG, TNC, SLC12A2, SLC6A2, SGK1, SPTLC1, NPC1 and TF in MAX vs autopsy and MAP4K4, CNTN2, P2RX7, KCNH8, TNC, GRP, FGF1, TGFA, MAG, PLP1, PLD1, ABCA2, ABCA8, AQP1, ATF3, ELOVL1, MOG and NPC in MAX vs MIN, further strengthen their role in pathophysiology of FCD type II (Fig. 5).

Gene network analysis showing associations between significantly modulated DEGs: A Direct interactions for MAX vs autopsy control B Indirect interactions for MAX vs autopsy control. C Direct interactions for MAX vs MIN D Indirect interactions for MAX vs MIN. Genes found modulated in our study are shown in filled circles. Different coloured edges with arrows shows the direction of interactions. Details of the graphical display of these associations are provided in Additional file 2: Fig. S2
Validation of data by real-time PCR
The mRNA levels of TNC, SLC12A2, CTGF, KCNK10, MOBP, and GPR37 were significantly up-regulated in MAX compared to autopsy controls (fold-change ≥ 2; p value < 0.05), whereas CARTPT was down-regulated (fold-change ≥ 2; p value < 0.05) (Fig. 6). The mRNA levels of TNC, KCNK10, MOBP, SLC12A2 and GPR37, were significantly up-regulated in MAX compared to MIN (fold-change ≥ 2; p value < 0.05), whereas CARTPT was significantly down-regulated. CTGF expression was relatively higher in MAX as compare to MIN, but it was not statistically significant (Fig. 6). Only TNC expression was significantly higher in MIN as compare to autopsy (fold-change ≥ 2; p value < 0.05) (Fig. 6).

Real-time PCR validation of selected DEGs. The expression level of up-regulated mRNAs (TNC, MOBP, CTGF, SLC12A2, GPR37, and KCNK10) and down-regulated mRNA (CARTPT) as identified in the RNAseq data were further validated by real-time PCR. Relative changes in gene expression were calculated using the ΔΔCq method with HPRT as a reference gene. Error bar is mean ± SD based on nine patients and eight control samples, and each sample is analyzed in triplicates. Mean increase in transcripts levels are statistically significant (One-way ANOVA followed by the Tukeys’ post hoc test; *p < 0.05; **p < 0.01; a = MIN vs autopsy, b = MAX vs autopsy; c = MAX vs MIN)
Discussion
In the present study, we utilized ECoG-graded clinically well-characterized brain tissue resected during surgical treatment of drug-resistant FCD type II patients to evaluate all elements of the transcriptome towards develo** a better understanding of the molecular mechanisms underlying the pathogenesis of FCD type II with the ultimate goal of identifying novel markers to help to localize EZ. Only a few high throughput profiling studies have been conducted on focal cortical dysplasia patients [15, 17,18,19,20]. Integrated genome-wide DNA methylation and RNAseq analysis identifies aberrant signalling pathways related to receptor tyrosine kinases (RTK), EGFR, PDGFRA in FCD type II patients [15]. Contrary to our results, transcriptomic profiling of dysplastic human temporal neocortex demonstrated the down-regulation of myelination-associated transcripts [17]. Other transcriptomic studies compared acutely high-spiking cortical areas against non-spiking cortical areas to look at seizure-induced gene expression [19, 20]. Similar to our results, Arion et al. (2006) demonstrated up-regulation of myelination associated genes. They also demonstrated the down-regulation of multiple GABA system-related genes (GABRA5, GABRB3, ABAT) and alterations in transcripts related to various signalling cascades in the spiking samples from temporal lobe patients [19], but we did not observe any alterations in GABA related gene expression. Similarly, Dachet et al. (2015) compared acutely high-spiking versus non-spiking cortical areas in neocortical epilepsy patients. They demonstrated increased expression of genes related to endothelial, red and white blood cells, neurons, and microglia, but a decreased expression of oligodendrocyte-specific transcripts in high spiking cortical regions [20]. Here, we are discussing the role of the DEGs and their possible association with epileptogenesis in FCD type II by grou** them into specific pathways.
Myelination, axon and neuronal ensheathment, oligodendrocyte development and differentiation
Oligodendrocyte-specific and myelination-associated genes were one of the dominating functional groups found to be up-regulated in MAX of dysplastic tissues in FCD type II patients compared to MIN and autopsy control. These include PLP1, SOX10, MAG, MOG, MOBP, KLK6, UGT8, CLDN11, ASPA, SH3TC2, NKX6-2, CTGF, SEPT 4, CDKN1C, GLDN, SPP1, CNTN2, TMEM98, TMEM10 (OPALIN), ANLN, ERMN, ENPP2, CD9, ABCA2, ABCA8, SPP1 and TF.
PLP1, MAG, MOG, MOBP, TMEM10 (OPALIN), ASPA, ABCA2, TF, GLDN, and SOX10 all play important roles in the oligodendrocytes (OLs) differentiation and myelination [30,31,32,33,34,35,36,37]. KLK6, a serine protease, may rapidly hydrolyze major myelin and blood brain barrier proteins and promote oligodendrogliopathy, neuronal injury and astrogliosis [38, 39]. ANLN from oligodendrocytes disrupts myelin septin assembly, causing the appearance of abnormal myelin outfoldings. ERMN plays a significant role in cytoskeletal rearrangements during the late wrap** and/or compaction phases of myelin assembly [40, 41]. Contrary to this study, many of these genes, GLDN, MOBP, UGT8, ASPA, TMEM10 (OPALIN), MOG, ERMN, and CLDN11 were found to be down-regulated in dysplastic human temporal neocortex [17]. Similar to this study, increased expression of MOG, PLP1, ABCA2, FA2H, TF, ASPA was demonstrated in high-spiking regions of cortical areas of temporal lobe epilepsy patients [19].
ECM related genes, i.e., TJP2, CLDN9, CLDN11, SPP1, and GJB1 (CX32), were found to be up-regulated in the MAX region of FCD patients. Lee et al. (2007) demonstrated the up-regulation of UGT8, MOG, TJP2, and ENPP2 in temporal lobe epilepsy patients [42]. These molecules are important for maintaining the proper physiological ambience for the timely development of oligodendrocyte precursor cells (OPCs) into myelinating OLs [43,44,45]. SH3TC2/KIAA1985, ABCA2 and ABCA8 are supposed to be involved in cargo transport for myelin formation [46, 47]. GPR37 (G protein-coupled receptor 37) negatively regulates oligodendrocyte differentiation and myelination [48]. Likewise, increased expression of GPR37 was reported in the high-spiking region of temporal lobe epilepsy patients [19]. A mutation in CNTN2 may be associated with adult myoclonic epilepsy [49].
CTGF expression was found to be up-regulated in MAX of surgically resected sample of patients compared to autopsy controls. CTGF/CCN2 negatively regulates myelination through the mTOR pathway [50]. Mutations in mTOR pathway genes were reported in FCD [51, 52]. Our previous study also demonstrated differential epigenetic regulation of the mTOR pathway in FCD [15]. Overtly active mTOR signaling may lead to insufficient myelination associated with FCD type II. CTGF has also been linked to astrogenesis, astrocyte activation, and neuro-inflammation [53].
In the present study, we have demonstrated the increased expression of genes related to myelination, remyelination or demyelination, suggesting that both phenomena are prevalent in patients. Demyelination is compensated for by remyelinating factors, and a delicate balance between them must be disrupted, resulting in myelin pathology, which may contribute to the epileptogenicity of this cortical malformation. OLs' inability to synthesize functional myelin could also be a factor. Up-regulated expression of several OL differentiation related genes could be due to an increased number of OPCs and differentiating OLs. It could be due to a compensatory mechanism to suppress epileptiform activity. Reductions in the number of oligodendroglial cells and myelin content have been reported in FCD, but the results remain controversial. An increased number of oligodendendroglia was also reported in patients with temporal lobe epilepsy and malformations of cortical development [54,55,56,57]. Scholl et al. (2017) suggested that impaired oligodendroglial turnover is associated with myelin pathology in focal cortical dysplasia and tuberous sclerosis complex. Proliferative oligodendroglia was identified in FCD IIA, IIB, and TSC, suggestive of a reactive phenomenon due to insufficient maturation or delayed maturation that prevents adequate myelination [55].
Recent studies show that neuronal activity can influence the generation of new oligodendrocytes (oligodendrogenesis) and myelination. Changes in myelination in cortical white matter are mostly reported, but alterations in myelination of grey matter have also been demonstrated [58]. During epileptogenesis, various kinds of synchronous sub-threshold excitatory stimuli allow their temporal summation in the post synaptic neurons [59]. This summation could be a direct result of axons with poorly distributed conduction velocities, resulting in synchronous action potential firing. The conduction velocity of an axon is mainly related to its diameter and the myelin sheath. Therefore, a direct relation might exist between epileptic seizure susceptibility and abnormal myelin content. Conversely, previous studies have indicated that neurological disorders associated with abnormal myelin content are accompanied by a higher susceptibility to epileptic seizures [60,61,62,63]. Several studies have indicated that epilepsy is also associated with myelin abnormalities [64,65,66,67,68,69]. Oligodendrocytes also control potassium accumulation in white matter and seizure susceptibility [70]. A subset of CNS oligodendrocytes expresses glutamine synthetase and directly modulates glutamatergic excitatory neurotransmission [71]. The findings presented here highlight avenues for potential therapeutic interventions targeting aberrant oligodendrogenesis and myelination.
Phospholipid biosynthesis
RNA Seq data highlights the perturbation of key metabolism processes in lipid metabolism, especially phospholipid biosynthesis in the MAX region of the FCD type II patients. Altered lipid levels and/or distribution have been reported in a variety of neurodegenerative diseases. [43, 72, 73]. PLLP, UGT8, ABCA2, PLD1, ELOVL1, CERS2, S1PR5, PLPPR1, SPTLC1, ENPP2, NPC1, FA2H, LRP2, S1PR5 and GAL3ST1 gene expression were found to be up-regulated in this study. PLLP, CERS2, UGT8, ASPA and GAL3ST1 contribute to various processes related to myelin synthesis [74]. PLD1, ELOVL1, NPC1, SPTLC1, FA2H, LRP2, and S1PR5 contribute to the synthesis of fatty acids, sphingolipids, and intracellular trafficking of lipid molecules [75,76,77]. Our data demonstrated dysregulation in lipid metabolism, i.e. phospholipid biosynthesis and trafficking, which in turn ameliorates the signalling pathways related to lipid molecules and can affect diverse cellular functions. Apart from these, phospholipids are known to be important regulators of many channels, mitochondrial function, excitotoxicity, impaired neuronal transport, cytoskeletal defects, inflammation, and reduced neurotransmitter release [72]. Future studies on these altered genes could provide us with promising targets with the potential to delineate the epileptogenic zone in FCD type II.
Ion channels
Ion channel dysfunction, either caused by mutations or acquired, has been associated with epilepsy. Many AEDs tend to manipulate the ion permeability of these channels to modify neuronal excitability [78]. In the present study, we have demonstrated the up-regulation of AQP1, KCNK10, KCNH8, P2RX7, SGK1, SGK2, SLC12A2, SLC6A2, SLC44A1, SLC45A3, SCLC5A11, SCL26A9, CLCA4 and SEPT4 in MAX of FCD type II patients.
AQP1 functions as a water channel protein, whereas KCNK10 and KCNH8 are potassium channels for neurotransmitter release, neuronal excitability, and electrolyte transport [79, 80]. SGK1 and SGK2 are reported to be involved in the regulation of a wide variety of ion channels, i.e., potassium, sodium, and chloride channels, membrane transporters, cell growth, survival and proliferation [81]. Activation of the P2X7 receptor has been associated with neuronal excitability, microglia activation and neuro-inflammatory responses. Increased expression of the P2X7 receptor has been demonstrated in animal models of epilepsy. P2X7 receptor ligands may be considered as a therapeutic target for DRE [82]. High SLC12A2 expression results in elevated Cl- concentration inside the cell, leading to net Cl− outflow and subsequent depolarization when GABA activates GABAARs [83, 84]. Increased expression of SLC12A2 has been reported in surgically resected tissue specimens from FCD patients [85]. SLC26A9, a highly selective chloride ion channel, CLCA4, calcium sensitive chloride channel, SLC45A3, SLC5A11 may be involved in ion transport and neurotransmitter release in FCD [78]. SLC44A1, a choline transporter, may contribute to membrane synthesis and myelin production. Alterations in ion channel gene expression might affect the ionic homeostasis of ions involved in epileptic activity within dysplastic tissues. So, it could serve as a potential biomarker to identify the EZ in FCD patients, but confirmatory studies on a larger cohort are needed.
Cell signaling molecules of various functions
Aside from these, several genes related to diverse cellular functions were found to be altered in this study, including semaphorins, fibroblast growth factors (FGFs), MAP4K4, ATF3, TNC, CALB2 and GRP. Here, we demonstrated the up-regulation of SEMA3B, SEMA4D and SEMA6A in MAX compared to MIN. SEMA3B-NRP1 mediated immune response and apoptosis have been reported, and their involvement in neuro-inflammation and cell death in epileptic conditions cannot be ruled out [86, 87]. SEMA4D/CD100 may regulate oligodendrocyte differentiation by promoting apoptosis [88]. SEMA4D also promotes inhibitory synapse formation and alleviates seizures in an animal model of epilepsy [89]. SEMA6A is considered to be a positive regulator of oligodendrocyte differentiation and myelination [90].
Other than semaphorins, the expression of MAP4K4, TNC, FGF1, FGF17, TGFA, ATF3, MCAM was found to be up-regulated and GRP and CALB2 were down-regulated in the study. MAP4K4 plays a specific role in activating the MAPK8/JNK pathway, which has also been found to be up-regulated in high-spiking cortical areas of TLE patients [19, 91]. Increased expression of TNC is highly associated with glial reactivity and reduced myelination, and also participates in Notch signalling [92]. As the FGF system is involved in the development of specific brain circuits in the hippocampus and cortex associated with epileptogenesis, increased expression of FGF1 and FGF17 was very much expected. FGF17 can activate numerous transcription factors involved in intra-cortical wiring. FGF1 has also been linked to a role in an animal model of epilepsy. Contrary to this, FGF1 has been shown to have anti-convulsant properties in kainate-induced epilepsy [93]. ATF-3 expression has been correlated with seizure frequency in epilepsy patients [94]. Loss of CALB2 (Calretinin) expression in hippocampal interneurons was shown in the dentate gyrus of patients with epilepsy [95]. Contrary to this, an increase in the number of calretinin-positive cells was observed by Blumcke et al. (1999) in patients with temporal lobe epilepsy [96]. Further studies on a greater number of samples are required for absolute findings.
There is evidence that GRP mediated signalling might play a role in regulating cognitive functions such as emotional responses, social interaction, memory, and feeding behaviour. Alterations in GRP or GRPR expression or function have been reported in patients with neurodegenerative, neurodevelopmental, and psychiatric disorders [97].
The small sample size of this study which does not include age and gender matched cases and controls is one of its limitations. Only two autopsy samples have been included in the present study. The age range of FCD patients is from 5 to 22 years, whereas autopsy patients range from 16 to 65 years. It’s very difficult to obtain age and gender-matched autopsy samples as per the inclusion criteria. Surgically resected tissue samples obtained for this study were from patients suffering from seizures for many years. Therefore, it is difficult to delineate and relate the transcriptional changes to underlying epileptogenic changes and to seizure activity. Further in vitro and in vivo studies are needed to determine whether the identified transcriptional changes are epileptogenic or a symptom of seizure activity.
The patients with FCD were on a combination of anti-epileptic drugs which may affect the expression of certain genes. AEDs selectively reduce the excitability of neurons and provide appropriate seizure control in epileptic patients by acting on a variety of biological targets. AEDs have a variety of modes of action, which can be classified based on their regulatory roles in voltage-gated ion channels and synaptic excitability control. However, recent research has revealed that AEDs can act as epigenetic modifiers to regulate gene expression [98]. Changes in gene expression caused by Valproate were seen in the peripheral blood of patients with newly diagnosed epilepsy [99]. The antiepileptic drug levetiracetam selectively modifies kindling-induced alterations in gene expression in the temporal lobe of rats [100]. These studies suggest that AEDs may have modulatory effects on the expression of certain genes. Hence, the contribution of AEDs to changes in gene expression cannot be ruled out. The findings of this study suggest that myelin and/or oligodendrocyte cells are involved in the epileptogenic process. Further exploration of the altered pathways may provide potential markers to aid in specifying the EZ in FCD patients. To date, there have been several preclinical and human studies presenting clear evidence that myelin content could be associated with epilepsy, epileptic seizures and epileptogenesis. Attempts to restore the process of myelination through pharmacological intervention could represent another promising therapeutic strategy for FCD as there is no evidence that administering these drugs to human patients can prevent seizures [58, 62]. Even with potential limitations, our study shows a tight association between ECoG grading of samples and the expression pattern of PLP1, PLLP, UGT8, KLK6, SOX10, MOG, MAG, MOBP, ANLN, ERMN, SPP1, TF, FA2H, CLDN11, TNC, GPR37, GRP, ABCA2, ABCA8, ASPA, P2RX7 (P2X7), CERS2, MAP4K4, OPALIN, Semaphorins, FGF1, CALB2, and TNC in patients with FCD. These genes could be further studied as a potential biomarker for the identification of epileptogenic margins in these patients. The primary reason for poor surgical outcomes in patients with FCD is the inaccurate localization of the epileptogenic margins. These results further support that EcoG-guided resection is likely to have a better outcome in terms of achieving seizure freedom post-operatively [22, 101].
Availability of data and materials
RNAseq data and sequences are submitted at NCBI BioProject (http://www.ncbi.nlm.nih.gov/bioproject/PRJNA369732).
Abbreviations
- AEDs:
-
Anti-epileptic drugs
- ALS:
-
Amyotrophic lateral sclerosis
- DEGs:
-
Differentially expressed genes
- DRE:
-
Drug-resistant epilepsy
- EZ:
-
Epileptogenic zone
- ECOG:
-
Electrocorticography
- EEG:
-
Electroencephalogram
- FCD:
-
Focal cortical dysplasia
- MCD:
-
Malformation of cortical development
- MSA:
-
Multiple system atrophy
- OLs:
-
Oligodendrocytes
- OPCs:
-
Oligodendrocytes precursor cells
- RNAseq:
-
RNA sequencing
- MRI:
-
Magnetic resonance imaging
- PCA:
-
Principal component analysis
- PET:
-
Positron emission tomography
- MEG:
-
Magnetoencephalography
- GO:
-
Gene ontology
References
Palmini A, Holthausen H. Focal malformations of cortical development: a most relevant etiology of epilepsy in children. Handb Clin Neurol. 2013;111:549–65.
Muhlebner A, Coras R, Kobow K. Neuropathologic measurements in focal cortical dysplasias: validation of the ILAE 2011 classification system and diagnostic implications for MRI. Acta Neuropathol. 2012;123:259–72.
Blumcke I, Thom M, Aronica E. The clinicopathologic spectrum of focal cortical dysplasias: a consensus classification proposed by an ad hoc task force of the ILAE diagnostic methods commission. Epilepsia. 2011;52:158–74.
Chassoux F, Landre E, Mellerio C. Type II focal cortical dysplasia: electro-clinical phenotype and surgical outcome related to imaging. Epilepsia. 2012;53:349–58.
Chassoux F, Rodrigo S, Semah F. FDG-PET improves surgical outcome in negative MRI Taylor-type focal cortical dysplasias. Neurology. 2010;75:2168–75.
Tassi L, Colombo N, Garbelli R. Focal cortical dysplasia: neuropathological subtypes, EEG, neuroimaging and surgical outcome. Brain. 2002;125:1719–32.
Kabat J, Karol P. Focal cortical dysplasia—review. Pol J Radiol. 2012;77:35–43.
Lee SK, Kim DW. Focal cortical dysplasia and epilepsy surgery. J Epilepsy Res. 2013;3:43–7.
** P, Wu D, Li X, Ren L, Wang Y. Towards precision medicine in epilepsy surgery. Ann Transl Med. 2016;4:24.
Krsek P, Maton B, Jayakar P, et al. Incomplete resection of focal cortical dysplasia is the main predictor of poor postsurgical outcome. Neurology. 2009;72:217–23.
Chapman K, Wyllie E, Najm I, et al. Seizure outcome after epilepsy surgery in patients with normal preoperative MRI. J Neurol Neurosurg Psychiatry. 2005;76:710–3.
Guerrini R, Duchowny M, Jayakar P, Krsek P, Kahane P, Tassi L, et al. Diagnostic methods and treatment options for focal cortical dysplasia. Epilepsia. 2015;56:1669–86.
Dixit AB, Banerjee J, Tripathi M, Chandra PS. Presurgical epileptogenic network analysis: a way to enhance epilepsy surgery outcome. Neurol India. 2015;63:743–50.
Krsek P, Pieper T, Karlmeier A, et al. Different presurgical characteristics and seizure outcomes in children with focal cortical dysplasia type I or II. Epilepsia. 2009;50:125–37.
Dixit AB, Sharma D, Tripathi M, Srivastava A, Paul D, Prakash D, et al. Genome-wide DNA methylation and RNAseq analyses identify aberrant signalling pathways in focal cortical dysplasia (FCD) type II. Sci Rep. 2018;8:17976.
Dixit AB, Banerjee J, Srivastava A, Tripathi M, Sarkar C, Kakkar A, Jain M, Chandra PS. RNA-seq analysis of hippocampal tissues reveals novel candidate genes for drug refractory epilepsy in patients with MTLE-HS. Genomics. 2016;107:178–88.
Donkels C, Pfeifer D, Janz P, Huber S, Nakagawa J, Prinz M, et al. Whole transcriptome screening reveals myelination deficits in dysplastic human temporal neocortex. Cereb Cortex. 2017;27:1558–72.
Katja K, Mark Z, Harikrishnan K, Ishant K, Angelika M, Feucht M, et al. Genomic DNA methylation distinguishes subtypes of human focal cortical dysplasia. Epilepsia. 2019;60:1091–103.
Arion D, Sabatini M, Unger T, Pastor J, Alonso-Nanclares L, Ballesteros-Yanez I, Garcia Sola R, Munoz A, Mirnics K, DeFelipe J. Correlation of transcriptome profile with electrical activity in temporal lobe epilepsy. Neurobiol Dis. 2006;22:374–87.
Dachet F, Bagla S, Keren-Aviram G, Morton A, Balan K, Saadat L, Valyi-Nagy T, Kupsky W, Song F, Dratz E, et al. Predicting novel histopathological microlesions in human epileptic brain through transcriptional clustering. Brain. 2015;138:356–70.
Mathern GW, Cepeda C, Hurst RS, Flores-Hernandez J, Mendoza D, Levine MS. Neurons recorded from pediatric epilepsy surgery patients with cortical dysplasia. Epilepsia. 2000;41:S162–7.
Tripathi M, Garg A, Gaikwad S, Bal CS, Chitra S, Prasad K, et al. Intra-operative electrocorticography in lesional epilepsy. Epilepsy Res. 2010;89:133–41.
Dwivedi R, Ramanujam B, Chandra PS, Sapra S, Gulati S, Kalaivani M, et al. Surgery for drug-resistant epilepsy in children. N Engl J Med. 2017;377:1639–47.
Zhang ZH, Jhaveri DJ, Marshall VM, Bauer DC, Edson J, Narayanan RK, et al. A comparative study of techniques for differential expression analysis on RNA-Seq data. PLoS ONE. 2014;9: e103207.
Seyednasrollah F, Laiho A, Elo LL. Comparison of software packages for detecting differential expression in RNA-seq studies. Brief Bioinform. 2015;16:59–70.
Metsalu T, Vilo J. ClustVis: a web tool for visualizing clustering of multivariate data using principal 548 component analysis and heatmap. Nucleic Acids Res. 2015;43:W566–70.
Reimand J, Isserlin R, Voisin V, Kucera M, Tannus-Lopes C, Rostamianfar A, et al. Pathway enrichment 558 analysis and visualization of omics data using g: profiler, GSEA, cytoscape and EnrichmentMap. Nat Protoc. 2019;14:482–517.
Schmittgen TD, Livak KJ. Analyzing real-time PCR data by the comparative C(T) method. Nat Protoc. 2008;3:1101–8.
de Nascimento AL, Dos Santos NF, Campos Pelágio F, Aparecida Teixeira S, de Moraes Ferrari EA, Langone F. Neuronal degeneration and gliosis time-course in the mouse hippocampal formation after pilocarpine-induced status epilepticus. Brain Res. 2012;1470:98–110.
Donkels C, Peters M, FariñaNúñez MT, Nakagawa JM, Kirsch M, Vlachos A, et al. Oligodendrocyte lineage and myelination are compromised in the gray matter of focal cortical dysplasia type IIa. Epilepsia. 2020;61:2012171–84.
Dugas JC, Tai YC, Speed TP, Ngai J, Barres BA. Functional genomic analysis of oligodendrocyte differentiation. J Neurosci. 2006;26:10967–83.
Yool DA, Klugmann M, McLaughlin M, Vouyiouklis DA, Dimou L, Barrie JA, et al. Myelin proteolipid proteins promote the interaction of oligodendrocytes and axons. J Neurosci Res. 2001;63:151–64.
Tatar CL, Appikatla S, Bessert DA, Paintlia AS, Singh I, Skoff RP. Increased Plp1 gene expression leads to massive microglial cell activation and inflammation throughout the brain. ASN Neuro. 2010;2: e00043.
Stolt CC, Rehberg S, Ader M, Lommes P, Riethmacher D, Schachner M, Bartsch U, Wegner M. Terminal differentiation of myelin-forming oligodendrocytes depends on the transcription factor Sox10. Genes Dev. 2002;16:165–70.
Heinen A, Kremer D, Göttle P, Kruse F, Hasse B, Lehmann H, Hartung HP, Küry P. The cyclin-dependent kinase inhibitor p57kip2 is a negative regulator of Schwann cell differentiation and in vitro myelination. Proc Natl Acad Sci USA. 2008;105:8748–53.
Quarles RH. Myelin-associated glycoprotein (MAG): past, present and beyond. J Neurochem. 2007;87:3267–76.
Montague P, McCallion AS, Davies RW, Griffiths IR. Myelin-associated oligodendrocytic basic protein: a family of abundant CNS myelin proteins in search of a function. Dev Neurosci. 2006;28:479–87.
Scarisbrick IA, Blaber SI, Lucchinetti CF, Genain CP, Blaber M, Rodriguez M. Activity of a newly identified serine protease in CNS demyelination. Brain. 2002;125:1283–96.
Bando Y, Hagiwara Y, Suzuki Y, Yoshida K, Aburakawa Y, Kimura T, Murakami C, et al. Kallikrein 6 secreted by oligodendrocytes regulates the progression of experimental autoimmune encephalomyelitis. Glia. 2018;66:359–78.
Erwig MS, Patzig J, Steyer AM, Dibaj P, Heilmann M, Heilmann I, Jung RB, Kusch K, et al. Anillin facilitates septin assembly to prevent pathological outfoldings of central nervous system myelin. Elife. 2019;8: e43888.
Brockschniede D, Sabanay H, Riethmacher D, Peles E. Ermin, a myelinating oligodendrocyte-specific protein that regulates cell morphology. J Neurosci. 2006;26:757–62.
Lee TS, Mane S, Eid T, Zhao H, Lin A, Guan Z, et al. Gene expression in temporal lobe epilepsy is consistent with increased release of glutamate by astrocytes. Mol Med. 2007;1–2:1–13.
Marangon D, Boccazzi M, Lecca D, Fumagalli M. Regulation of oligodendrocyte functions: targeting lipid metabolism and extracellular matrix for myelin repair. J Clin Med. 2020;9:470.
Denninger AR, Breglio A, Maheras KJ, LeDuc G, Cristiglio V, Demé B, Gow A, Kirschner DA. Claudin-11 tight junctions in myelin are a barrier to diffusion and lack strong adhesive properties. Biophys J. 2015;109:1387–97.
Selvaraju R, Bernasconi L, Losberger C, Graber P, Kadi L, Avellana-Adalid V, et al. Osteopontin is up-regulated during in vivo demyelination and remyelination and enhances myelin formation in vitro. Mol Cell Neurosci. 2004;25:707–21.
Kim WS, Weickert CS, Garner B. Role of ATP-binding cassette transporters in brain lipid transport and neurological disease. J Neurochem. 2008;104:1145–66.
Wong JH, Halliday GM, Kim WS. Exploring myelin dysfunction in multiple system atrophy. Exp Neurobiol. 2014;23:337–44.
Yang HJ, Vainshtein A, Maik-Rachline G, Peles E. G protein-coupled receptor 37 is a negative regulator of oligodendrocyte differentiation and myelination. Nat Commun. 2016;7:10884.
Stogmann E, Reinthaler E, Eltawil S, El Etribi MA, Hemeda M, El Nahhas N, et al. Autosomal recessive cortical myoclonic tremor and epilepsy: association with a mutation in the potassium channel associated gene CNTN2. Brain. 2013;136:1155–60.
Ercan E, Han JM, Di Nardo A, Winden K, Han MJ, Hoyo L, Saffari A, et al. Neuronal CTGF/CCN2 negatively regulates myelination in a mouse model of tuberous sclerosis complex. J Exp Med. 2017;214:681–97.
D’Gama AM, Geng Y, Couto JA, Martin B, Boyle EA, LaCoursiere CM, et al. Mammalian target of rapamycin pathway mutations cause hemimegalencephaly and focal cortical dysplasia. Ann Neurol. 2015;77:720–5.
Jesus-Ribeiro J, Pires LM, Melo JD, Ribeiro IP, Rebelo O, Sales F, Freire A, Melo JB. Genomic and epigenetic advances in focal cortical dysplasia type I and type II: a sco** review. Front Neurosci. 2021;14: 580357.
Lu M, Yan XF, Si Y, Chen XZ. CTGF triggers rat astrocyte activation and astrocyte-mediated inflammatory response in culture conditions. Inflammation. 2019;42:1693–704.
Ettle B, Schlachetzki JCM, Winkler J. Oligodendroglia and myelin in neurodegenerative diseases: more than just bystanders? Mol Neurobiol. 2016;53:3046–62.
Scholl T, Mühlebner A, Ricken G, Gruber V, Fabing A, Samueli S, et al. Impaired oligodendroglial turnover is associated with myelin pathology in focal cortical dysplasia and tuberous sclerosis complex. Brain Pathol. 2017;27:770–80.
Shepherd C, Liu J, Goc J, Martinian L, Jacques TS, Sisodiya SM, et al. A quantitative study of white matter hypomyelination and oligodendroglial maturation in focal cortical dysplasia type II. Epilepsia. 2013;54:898–908.
Donkels C, Peters M, FariñaNúñez MT, Nakagawa JM, Kirsch M, Vlachos A, et al. Oligodendrocyte lineage and myelination are compromised in the gray matter of focal cortical dysplasia type IIa. Epilepsia. 2020;61:171–84.
Drenthen GS, Backes WH, Aldenkamp AP, Vermeulen RJ, Klinkenberg S, Jansen JFA. On the merits of non-invasive myelin imaging in epilepsy, a literature review. J Neurosci Methods. 2020;338: 108687.
Bromfield EB, Cavazos JE, Sirven JI, editors. An introduction to epilepsy. West Hartford (CT): American Epilepsy Society; 2006.
Gruntz K, Bloechliger M, Becker C, Jick SS, Fuhr P, Meier CR, Stephan R. Parkinson disease and the risk of epileptic seizures. Ann Neurol. 2018;83:363–74.
Hyde TM, Weinberger DR. Seizures and schizophrenia. Schizophr Bull. 1997;23:611–22.
Lapato AS, Szu JI, Hasselmann JPC, Khalaj AJ, Binder DK, Tiwari-Woodruff SK. Chronic demyelination-induced seizures. Neuroscience. 2017;346:409–22.
Pandis D, Scarmeas N. Seizures in Alzheimer disease: clinical and epidemiological data. Epilepsy Curr. 2012;12:184–7.
Drenthen GS, Fonseca Wald ELA, Backes WH, Debeij-Van Hall MHJA, Hendriksen JGM, Aldenkamp AP, Vermeulen RJ, Klinkenberg S, Jansen JFA. Lower myelin-water content of the frontal lobe in childhood absence epilepsy. Epilepsia. 2019;60:1689–96.
Moldovan K, Boxerman JL, Muircheartaigh JO, Dean D, Eyerly-webb S, Cosgrove GR, Pucci FG, Deoni SCL, Spader HS. Myelin water fraction changes in febrile seizures Clin. Neurol Neurosurg. 2018;175:61–7.
Spader HS, Ellermeier A, O’Muircheartaigh J, Dean DC, Dirks H, Boxerma JL, Cosgrove GR, Deoni SCL. Advances in myelin imaging with potential clinical application to pediatric imaging. Neurosurg Focus. 2013;34:E9.
Gibson EM, Purger D, Mount CW, Goldstein AK, Lin GL, Wood LS, et al. Neuronal activity promotes oligodendrogenesis and adaptive myelination in the mammalian brain. Science. 2014;344:1252304.
Noori R, Park D, Griffiths JD, Bells S, Frankland PW, Mabbott D, Lefebvre J. Activity-dependent myelination: a glial mechanism of oscillatory self-organization in large-scale brain networks. Proc Natl Acad Sci USA. 2020;117:13227–37.
Juliet Knowles K, Caroline S, Eleanor F, Lydia TT, Danielle F, Haojun XU, et al. Maladaptive myelination promotes epileptogenesis in absence epilepsy. bioRxiv. 2020. https://doi.org/10.1101/2020.08.20.260083.
Larson VA, Mironova Y, Vanderpool KG, Waisman A, Rash JE, Agarwal A, Bergles DE. Oligodendrocytes control potassium accumulation in white matter and seizure susceptibility. Elife. 2018. https://doi.org/10.7554/eLife.34829.
**n W, Mironova YA, Shen H, Marino RAM, Waisman A, Lamers WH, Bergles DE, Bonci A. Oligodendrocytes support neuronal glutamatergic transmission via expression of glutamine synthetase. Cell Rep. 2019;27:2262-2271 e2265.
Tracey TJ, Steyn FJ, Wolvetang EJ, Ngo ST. Neuronal lipid metabolism: multiple pathways driving functional outcomes in health and disease. Front Mol Neurosci. 2018;23:10.
Dimas P, Montani L, Pereira JA, Moreno D, Trötzmüller M, Gerber J, et al. CNS myelination and remyelination depend on fatty acid synthesis by oligodendrocytes. Elife. 2019;8: e44702.
Montani L. Lipids in regulating oligodendrocyte structure and function. Semin Cell Dev Biol. 2020. https://doi.org/10.1016/j.semcdb.2020.07.016.
Imgrund S, Hartmann D, Farwanah H, Eckhardt M, Sandhoff R, Degen J, et al. Adult ceramide synthase 2 (CERS2)-deficient mice exhibit myelin sheath defects, cerebellar degeneration, and hepatocarcinomas. J Biol Chem. 2009;284:33549–60.
Francis JS, Wojtas I, Markov V, Gray SJ, McCown TJ, Samulski RJ, et al. N-acetylaspartate supports the energetic demands of developmental myelination via oligodendroglialaspartoacylase. Neurobiol Dis. 2016;96:323–34.
Klein J. Functions and pathophysiological roles of phospholipase D in the brain. J Neurochem. 2005;94:1473–87.
Zhang S, Zhu Y, Cheng J, Tao J. Ion channels in epilepsy: blasting fuse for neuronal hyperexcitability. Intech Open. 2019. https://doi.org/10.5772/intechopen.83698.
Lehmann GL, Gradilone SA, Marinelli RA. Aquaporin water channels in central nervous system.Curr Neurovasc Res. 2004;1(4):293–303.
Villa C, Combi R. Potassium channels and human epileptic phenotypes: an updated overview. Front Cell Neurosci. 2016;10:81.
Florian L, Nathalie SS, Guiscard S, Undine L. Significance of SGK1 in the regulation of neuronal function. J Physiol. 2010;588:3349–54.
Engel T, Jimenez-Pacheco A, Miras-Portuga MT, Diaz-Hernandez M, Henshall DC. P2X7 receptor in epilepsy; role in pathophysiology and potential targeting for seizure control. Int J Physiol Pathophysiol Pharmacol. 2012;4:174–87.
Cepeda C, Andre VM, Wu N, Yamazaki I, Uzgil B, Vinters HV, Levine MS, Mathern GW. Immature neurons and GABA networks may contribute to epileptogenesis in pediatric cortical dysplasia. Epilepsia. 2007;48:79–85.
Banerjee J, Dey S, Dixit AB, Doddamani RS, Sharma MC, Garg A, Chandra PS, Tripathi M. GABAA receptor-mediated epileptogenicity in focal cortical dysplasia (FCD) depends on age at epilepsy onset. Front Cell Neurosci Cell Neurophysiol. 2020;14: 562811. https://doi.org/10.3389/fncel.2020.562811.
Liu R, Wang J, Liang S, Zhang G, Yang X. Role of NKCC1 and KCC2 in epilepsy: from expression to function. Front Neurol. 2019;10:1407.
Pasterkamp RJ, Giger RJ. Semaphorin function in neural plasticity and disease. Curr Opin Neurobiol. 2009;19:263–74.
Cohen RI, Rottkamp DM, Maric D, Barker JL, Hudson LD. A role for semaphorins and neuropilins in oligodendrocyte guidance. J Neurochem. 2003;85:1262–78.
Yamaguchi W, Tamai R, Furuyama T. Sema4D as an inhibitory regulator in oligodendrocyte development. Mol Cell Neurosci. 2012;49:290–9.
Acker DWM, Wong I, Kang M, Paradis S. Semaphorin 4D promotes inhibitory synapse formation and suppresses seizures in vivo. Epilepsia. 2018;59:1257–68.
Bernard F, Moreau-Fauvarque C, Heitz-Marchaland C, Zagar Y, Dumas L, Bernard F, et al. Role of transmembrane semaphorin Sema6A in oligodendrocyte differentiation and myelination. Glia. 2012;60:1590–604.
Larhammar M, Huntwork-Rodriguez S, Rudhard Y, Sengupta-Ghosh A, Lewcock JW. The Ste20 family kinases MAP4K4, MINK1, and TNIK converge to regulate stress-induced JNK signaling in neurons. J Neurosci. 2017;37:11074–84.
Jakovcevski I, Miljkovic D, Schachner M, Andjus PR. Tenascins and inflammation in disorders of the nervous system. Amino Acids. 2012;44:1115–27.
Cortney A, EmineEren-Koçak T, Inui EG, Watson SJ, Akil H. Dysregulated fibroblast growth factor (FGF) signaling in neurological and psychiatric disorders. Cell Dev Biol. 2016;53:136–43.
Pernhorst K, Herms S, Hoffmann P, Cichon S, Schulz H, Sander T, et al. TLR4, ATF-3 and IL8 inflammation mediator expression correlates with seizure frequency in human epileptic brain tissue. Seizure. 2013;22:675–8.
Tóth K, Erőss L, Vajda J, Halász P, Freund TF, Maglóczky Z. Loss and reorganization of calretinin-containing interneurons in the epileptic human hippocampus. Brain. 2010;133:2763–77.
Blumcke I, Beck H, Suter B, Hoffmann D, Fodisch HJ, Wolf HK, et al. An increase of hippocampal calretinin-immunoreactive neurons correlates with early febrile seizures in temporal lobe epilepsy. Acta Neuropathol. 1999;97:31–9.
Roesler R, Schwartsmann G. Gastrin-releasing peptide receptors in the central nervous system: role in brain function and as a drug target. Front Endocrinol (Lausanne). 2012;3:159.
Kong FC, Ma CL, Zhong MK. Epigenetic effects mediated by antiepileptic drugs and their potential application. Curr Neuropharmacol. 2020;18:153–66.
Gu J, Lynch BA, Anderson D, Klitgaard H, Lu S, Elashoff M, Ebert U, Potschka H, Löscher W. The antiepileptic drug levetiracetam selectively modifies kindling-induced alterations in gene expression in the temporal lobe of rats. Eur J Neurosci. 2004;19:334–45.
Rakitin A, Kõks S, Reimann E, Prans E, Haldre S. Changes in the peripheral blood gene expression profile induced by 3 months of valproate treatment in patients with newly diagnosed epilepsy. Front Neurol. 2015;6:188.
Ravat S, Iyer V, Panchal K, Muzumdar D, Kulkarni A. Surgical outcomes in patients with intraoperative Electrocorticography (EcoG) guided epilepsy surgery-experiences of a tertiary care centre in India. Int J Surg. 2016;36:420–8.
Acknowledgements
The authors are indebted to all the patients and their family for participating in this study.
Funding
This work was funded by MEG resource facility, Department of Biotechnology, Ministry of Science and Technology, Govt. of India [BT/MED/122/SP24580/2018] and Institute of Eminence Grant from University of Delhi (IOE/FRP/LS/2020/27), Delhi.
Author information
Authors and Affiliations
Contributions
A.S. and K.K. designed the research, performed research, analysed the data and wrote the paper. A.B.D. designed the research, and wrote the paper. V.D., D.S., and N.Y. performed research. M.T. designed the research. S.L. provided the autopsy samples for research. M.C.S. analysed the histopathological features. P.S.C. and R.D. provided clinical samples, designed the research. J.B. designed the research and wrote the paper. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
The present study was conducted in accordance with the Declaration of Helsinki and was approved by the Institute Ethics Committee, AIIMS, New Delhi. Informed and written consent was obtained from all the patients, their parents, or legal guardians if patients were underage.
Consent for publication
All donors or their parents/ legal guardians gave informed consent for the use of their tissue and medical files for research purposes and publication.
Competing interests
The authors declare no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Figure S1.
Representative image of EcoG recording of a 26-year-old male patient showing MAX and MIN region.
Additional file 2: Figure S2.
Symbols used in network analysis.
Additional file 3.
List of significantly altered genes analyzed by CuffDiff.
Additional file 4.
List of significantly altered genes analyzed by DESeq 2.
Additional file 5.
List of significantly altered genes analyzed by EdgeR.
Additional file 6.
List of common genes found significantly altered in all three softwares.
Additional file 7: Table S1.
List of primers used in the study.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Srivastava, A., Kumar, K., Banerjee, J. et al. Transcriptomic profiling of high- and low-spiking regions reveals novel epileptogenic mechanisms in focal cortical dysplasia type II patients. Mol Brain 14, 120 (2021). https://doi.org/10.1186/s13041-021-00832-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13041-021-00832-4