
Abstract
Background
The high cost of fermentation, purification, cold storage and transportation, short shelf life, and sterile delivery methods of biopharmaceuticals, is a matter for producers and consumers as well. Since the FDA has now approved plant cells for large-scale, cost-effective biopharmaceutical production, the isolation and lyophilization of transplastomic chloroplasts can cover concerns about limitations. DARPins are engineered small single-domain proteins that have been selected to bind to HER2 with high affinity and specificity. HER2 is an oncogene involved in abnormal cell growth in some cancers and the target molecule for cancer immunotherapy.
Results
In this study, we reported the prolonged stability and functionality of DARPin G3 in lyophilized transplastomic tobacco leaves and chloroplasts. Western blot analysis of lyophilized leaves and chloroplasts stored at room temperature for up to nine months showed that the DARPin G3 protein was stable and preserved proper folding. Lyophilization of leaves and isolated chloroplasts increased DARPin G3 protein concentrations by 16 and 32-fold, respectively. The HER2-binding assay demonstrated that the chloroplast-made DARPin G3 can maintain its stability and binding activity without any affinity drop in lyophilized leaf materials throughout this study for more than nine months at room temperature.
Conclusion
Lyophilization of chloroplasts expressing DARPin G3 would further reduce costs and simplify downstream processing, purification, and storage. Compressed packages of lyophilized chloroplasts were much more effective than lyophilized transplastomic leaves considering occupied space and downstream extraction and purification of DARPin G3 after nine months. These methods facilitate any relevant formulation practices for these compounds to meet any demand-oriented needs.
Similar content being viewed by others

Background
The biological production of bioactive designed proteins in bulk quantities is one of the methods to meet the growing needs in the protein-related market and industries. Reducing production costs in mass production systems is a major challenge for all manufacturing companies. Among the recombinant protein production platforms, transplastomic plant-made pharmaceuticals are highly demanded and can be one of the most cost-effective routes for meeting the needs of rapidly growing recombinant drug companies. Although the currently used production systems based on mammalian, insect, yeast, or bacterial cell cultures have been developed and improved in accordance with current good manufacturing practice [1], plant-based protocols are facing some ups and downs in the commercialization process. Here we examined the potential of lyophilization in the long-term storage of centrifugally compressed isolated chloroplasts as well as intact leaves by develo** transplastomic tobacco plants expressing the antibody mimetic DARPin G3. It is assumed that the longevity and low-cost preservation methods remove any negative speculation on the efficiency of plant-based protocols in the formulation of any medicinal recipes. Typically, focusing on a small number of platforms makes meeting the unique requirements of certain target proteins difficult; this is especially true for recombinant proteins that are needed in small quantities for individual patients as well as large quantities for mass production or where rapid scale-up is required [2].
Compared to bacterial, yeast, and mammalian cell culture systems, the production of pharmaceutical proteins in plants offers several advantages, including low cost, a high production rate, easy scale-up with relatively low demand and a simplified procedure, and importantly, a low risk of product contamination by human or animal pathogens and endotoxins, resulting in increased patient safety [3,4,5]. Additionally, plants are able to perform appropriate eukaryotic protein post-translational modifications such as glycosylation, disulfide bond formation, folding, and multimeric assembly, which are frequently crucial for the biological activity and stability of many mammalian proteins [5,6,7].
Although stable nuclear transformation is useful for variety research, it is inefficient for protein accumulation, leading to low expression levels that have hampered the production of biopharmaceuticals at industrial levels in plants as compared to those produced by other methods [8, 9]. Instead, plastidial biopharming as a highly efficient bioreactor offers several unique advantages over nuclear transformation, which make the system ideal for plant-made biopharmaceuticals. This includes multiple genes transferring into the chloroplast genome in a single transformation event; high-level expression of transgenes due to high copy numbers of the plastome in each plant cell (several thousands); a lack of gene silencing and positional effects due to site-specific integration, which minimizes the number of events required for screening; and the absence of epigenetic effects and transgene containment due to maternal inheritance of plastids [10].
Transplastomic leaf lyophilization offers additional advantages that further reduce costs and facilitate processing, purification, storage, and immunization. Furthermore, this method increases the concentration of the recombinant products, making them more stable (retaining appropriate folding, disulfide bond formation, and functional efficacy) even after long-term room-temperature storage, eliminating the need for costly cold chains and simplifying transportation [11].
Antibody mimetics, which are small, designed amino acid sequences to mimic structural features of antibody complementarity-determining regions, have inhibitory properties similar to mAbs [12]. These antibody substitutes have a completely different protein topology, which provides advantages such as single-chain buildup (enabling the simple production and subsequent construction of fusion proteins); small size (for good tissue penetration); and, in many cases, high thermostability (enabling long-term storage at room temperature without loss of activity) [13]. Designed ankyrin repeat proteins (DARPins) are one of several antibody mimetics with binding functions that have been developed in recent years [14].
Human epidermal growth factor receptor-2 (HER2) has tyrosine kinase activity that has been linked to the growth of malignancies of various origins and is a validated target for monoclonal antibodies and kinase inhibitors [15, 16]. Utilizing a new approach with DARPins as alternative binders can cause stronger cytotoxic effects on the HER2-addicted breast cancer cell lines [17, 18]. HER2 is overexpressed in approximately 20–30% of breast cancer tumors (Yan et al., 2015). Trastuzumab and Pertuzumab, two currently available monoclonal antibodies and tyrosine kinase inhibitor drugs, rarely achieve complete disease control (Escrivá-de-Romaní et al., 2018). However, the production of antibodies is comparatively expensive and challenging. Additionally, the massive size of immunoglobulin G is rather large for therapeutic applications that require efficient tissue penetration (Binz et al., 2003). DARPin G3, with a low molecular weight (14–15 kDa) and picomolar affinity for HER2 (91 pmol/L), can be used as a molecular imaging agent to visualize HER2-positive tumors (Jost et al., 2013; Zahnd et al., 2007; Zahnd et al., 2010).
We recently and for the first time developed tobacco transplastomic lines producing high levels of DARPin G3 as an antibody mimetic for HER2 targeting in HER2-positive cancers [19]. Our previous study’s results demonstrated that the DARPin G3 produced in chloroplasts binds HER2 with high affinity, as previously reported for this DARPin G3 expressed in E. coli [20] or P. pastoris [17], as shown using an enzyme-linked immunosorbent assay (ELISA). The HER2-specific binding of chloroplast-made DARPin G3 on the cancer cell membrane was revealed by flow cytometry and immunofluorescent microscopy.
The present research was designed and conducted to evaluate the bioactivity and longevity of bio-encapsulated chloroplast-made DARPin G3 from lyophilized intact chloroplasts and leaves. Our findings show that even after nine months of room-temperature storage, the chloroplast-made DARPin G3 protein remained stable and could be detected at high levels in lyophilized chloroplasts and leaves. An enzyme-linked immunosorbent assay confirmed the binding affinity of chloroplast-made DARPin G3, purified from freeze-dried leaf materials, to HER2 receptors. Immunofluorescent microscopy revealed the functionality of the HER2-specific binding of chloroplast-made DARPin G3 on the adenocarcinoma cell surface. According to these results, DARPin G3 preservation in lyophilized leaf materials, particularly in lyophilized chloroplasts, would simplify processing, purification, and storage, lowering costs and enabling the practical production of scaffold proteins in chloroplast-based expression systems.
Results
Chloroplast isolation
Isolation of chloroplasts was performed for comparative analysis of the DARPin G3 content in fresh and lyophilized chloroplasts and intact leaves. The purity and integrity of the isolated chloroplasts were examined and confirmed using optical microscopy, showing the above 99% intact chloroplasts in the sample (Fig. 1A). Based on the volumetric calculation of a single typical chloroplast (3 μm in diameter) and photosynthetic parenchyma cell (40–80 μm width and length), considering 100–200 chloroplasts per cell, the chloroplast content of each cell was estimated at around 3.4–6.8%. The volumetric estimation is an acceptable approximation for the isolated chloroplasts content of 6.38%, which is achieved by mass estimation based on the fresh weight of leaves and isolated chloroplasts. The isolated chloroplasts were used for protein extraction in subsequent analyses or for lyophilization.

Preparation of lyophilized powdered tobacco leaves and chloroplasts expressing the DARPin G3 protein. A freshly extracted chloroplasts along with a microscopic image of them; (B) lyophilized chloroplasts; (C) fresh leaves; and (D) lyophilized leaves
Lyophilization
Lyophilization of DARPin G3-expressing tobacco leaves and isolated chloroplasts was performed to facilitate long-term storage at room temperature for the subsequent purification and evaluation of DARPin G3 stability and activity (Fig. 1B-D). We observed approximately a 93% and 48% decrease in weight due to the removal of water content in lyophilized leaves and lyophilized chloroplastic pellets, respectively.
DARPin G3 quantitation
DARPin G3 accumulation, integrity, and stability in the fresh and lyophilized leaves and chloroplasts of transplastomic plants after nine months of storage at room temperature were investigated by Western blot analysis. A Western blot was performed with cellular and chloroplastic protein extracts from fresh and lyophilized materials of transplastomic plants. As the N-terminal of DARPin G3 is equipped with a histidine-glutamate (HE)3-tag, DARPin G3 was probed with a rabbit anti-His-tag antibody as the primary antibody and an HRP-conjugated goat anti-rabbit antibody as the secondary antibody on SDS-PAGE under reducing conditions. The DARPin G3 protein with an accurate molecular mass of ~ 15 kDa was detected in lyophilized chloroplasts and leaves of transplastomic plants (Fig. 2A). Western blots showed an apparent difference in DARPin G3 intensity between equal protein loading (50 µg) from fresh and lyophilized leaves against fresh and lyophilized chloroplasts. In addition to the higher number of chloroplasts per cell and the higher polyploidy level of each chloroplast, higher DARPin G3 concentrations and amounts in chloroplastic samples are consistent with the fact that chloroplast proteins comprise 40% of total cellular protein [21].

Accumulation and quantification of DARPin G3 in fresh and lyophilized materials. A Immunoblot of DARPin G3 protein in fresh (F) and lyophilized (L) tobacco leaves (L) and chloroplasts (Ch). TSP extracted from a mature leaf of wild-type (WT) was also loaded as a control. A normalized amount of fresh and lyophilized leaves and chloroplasts was used for protein extraction, and an equal amount (50 µg) of protein extract was loaded. DARPin G3 and molecular weight marker (Broad Range Protein Molecular Weight Markers) positions are indicated on the margins. B ELISA analysis for the chloroplast-made DARPin G3 content in fresh and lyophilized materials. The total soluble protein of the wild-type (WT) tobacco plant was used as a negative control. Data are the means ± SD of three independent experiments. Error bars represent the standard deviation of the mean
The quantitation of the chloroplast-made DARPin G3 in fresh and lyophilized leaves and chloroplasts of tobacco transplastomic plants was performed by ELISA. The expressed amounts of chloroplast-made DARPin G3 protein accounted for approximately 20% of TSP in both fresh and lyophilized leaves of transplastomic plants. On this basis, the production yield of DARPin G3 was estimated to be 3.4 mg/g of fresh leaf weight and 55.2 mg/g of lyophilized leaf weight in the transplastomic plants. The DARPin G3 protein content in fresh and lyophilized chloroplasts was estimated to be approximately 33% of TSP, which is representative of 56.1 mg/g fresh chloroplast weight and 111.3 mg/g lyophilized chloroplasts weight (Fig. 2B). Therefore, the content of DARPin G3 increased 16- and 32-fold in lyophilized leaves and chloroplasts when compared to fresh leaves, respectively. The ELISA approach proved transgene expression without imposing a yield penalty. In addition, the significant difference in the accumulation of DARPin G3 protein in intact cells and chloroplasts further confirms the cellular/chloroplastic protein ratio again.
These analyses revealed that chloroplast-made DARPin G3 remained soluble and stable even after lyophilization and storage for at least nine months at an ambient temperature comparable to their respective total extracts from fresh leaves and chloroplasts.
Purification of chloroplast-made DARPin G3
When DARPin G3 is used as an injectable protein, the purification of chloroplast-made DARPin G3 protein is necessary. For this purpose, a histidine-tag was incorporated into the chloroplastic coding sequence of the DARPin G3-expressing cassette. The chloroplast-made DARPin G3 was partially purified from the total soluble protein of fresh and lyophilized materials with the Ni-NTA Purification System. The Coomassie blue-stained SDS-PAGE of purified protein showed a 15 kDa band corresponding to the DARPin G3 protein. SDS-PAGE analysis of purified protein when an equal volume of purified protein was loaded showed that the fresh and lyophilized chloroplasts contained the highest amount of DARPin G3 (Fig. 3A). The 55 kDa band corresponds to the large subunit of Rubisco encoded by the chloroplast rbcL gene, which is the most abundant protein in nature. It requires further purification steps to be removed from the partially purified protein. The purified DARPin G3 was used for the binding assay to the HER2 extracellular domain and to HER2 on the cancer cell surface.

A SDS-PAGE analysis of chloroplast-made DARPin G3 purified from fresh leaves (F.L), lyophilized leaves (L.L), fresh chloroplasts (F.Ch) and lyophilized chloroplasts (L.Ch) of transplastomic tobacco plants. TSP extracted from a mature leaf of wild-type (WT) was also loaded as a control. On the margins, the positions of molecular weight markers (Broad Range Protein Molecular Weight Markers), DARPin G3, and RbcL are indicated. B HER2-ECD binding activity and (C) affinity of chloroplast-made DARPin G3 purified from fresh and lyophilized leaves and chloroplasts of transplastomic tobacco plants. The total soluble protein of the wild-type (WT) tobacco plant and the bovine serum albumin (BSA, 1%, w/v) were used as negative controls. Data are the means ± SD of three independent experiments. Error bars represent the standard deviation of the mean
Binding affinity of chloroplast-made DARPin G3 to the HER2 receptor
To evaluate whether the DARPin G3 protein purified from lyophilized leaves and chloroplasts of transplastomic tobacco plants preserved its biological function of binding to the HER2 receptor, we performed a HER2-binding ELISA assay. Purified DARPin G3 from lyophilized materials showed strong binding affinity to HER2. As shown in Fig. 3B, binding activity in ELISA did not show detectable differences among chloroplast-made DARPin G3 purified from lyophilized materials compared to those purified from fresh leaf and chloroplasts of transplastomic plants (Fig. 3B). The binding affinity test by ELISA using the purified chloroplast-made DARPin G3 from lyophilized leaves and chloroplasts exhibits a sub-nanomolar range of affinity for HER2 (Fig. 3C), which is comparable with DARPin G3 previously produced in E. coli [22] and P. pastoris [17]. The efficacy of binding to the HER2 receptor was retained after lyophilization and long-term storage for nine months at ambient temperature. Wild-type plants and BSA didn’t show binding to the HER2 receptor.
HER2 specificity of chloroplast-made DARPin G3
After a binding affinity test, the ability of chloroplast-made DARPin G3 from lyophilized leaves and chloroplasts to bind to the HER2 receptor on the cell surface was assessed in vitro using flow cytometry and immunofluorescent microscopy. Two human breast adenocarcinoma cell lines with different levels of HER2 expression were used for these purposes. The SKBR-3 line has been shown to strongly overexpress HER2, and MDA-MB-231, which has not been shown to overexpress HER2.
The flow cytometry results of the treated breast cancer cell lines demonstrated that chloroplast-made DARPin G3 from lyophilized leaves and chloroplasts bound to HER2-positive human breast cancer cells (SKBR-3), but did not bind to HER2-negative human breast adenocarcinoma cells (MDA-MB-231) (Fig. 4). Their performance was comparable with DARPin G3 purified from fresh leaves and chloroplasts and is in agreement with the formerly described DARPin G3 expressed in E. coli [20] or P. pastoris [17].

Characterization of HER2 binding of chloroplast-made DARPin G3 from fresh and lyophilized leaves and chloroplasts of tobacco transplastomic plants by flow cytometry in human HER2-positive (SKBR3) and human HER2-negative breast cancer cells (MDA-MB-231). Cells incubated with PBS or anti-His-tag antibody followed by FITC-conjugated antibody were used as unstained and omission controls, respectively. Data from representative experiments are shown
Cellular binding of the chloroplast-made DARPin G3, purified from lyophilized leaves and chloroplasts, to HER2 on the cell surface was visualized by immunofluorescent microscopy using FITC tags. In contrast to MDA-MB-231 cells, we found a significant signal of FITC staining throughout the cell membrane in SKBR-3 cells (Fig. 5). The control experiment, which did not include the DARPin G3 or FITC-conjugated antibody, revealed no FITC staining at all. Some intracellular signals suggest that HER2 internalization has resulted in the internalization of at least a fraction of DARPin G3 proteins [23,24,25].

Binding of purified chloroplast-made DARPin G3 from fresh and lyophilized leaves and chloroplasts to HER2 on the SKBR-3 as HER2-positive and MDA-MB-231 as HER2-negative cell lines by immunofluorescent microscopy. Bound DARPin G3 was detected by incubating with an anti-His-tag antibody followed by a FITC-conjugated antibody, which showed HER2-specific targeting on the cell surface. Laser power and gain were kept constant, and brightness and contrast settings were adjusted equally. The scale bar represents 20 μm
Retention of full activity and function of chloroplast-made DARPin G3, demonstrated by flow cytometry and immunofluorescent microscopy analysis, confirmed that once tobacco leaves and chloroplasts are lyophilized, DARPin G3 can be stabilized in plant cells at room temperature for over nine months.
Discussion
Chloroplast transformation provides an opportunity for accurate incorporation of the target gene(s) into a predetermined region of the plastid genome through the mechanism of double homologous recombination. The chloroplast’s ability to correct protein folding and the formation of disulfide bridges, as well as its prokaryotic nature, are important features for the production of recombinant proteins through transplastomic strategies. The stability of expressed proteins is a major factor influencing the expression system’s overall viability. For proteins that do not require glycosylation, the chloroplast expression system is ideal for increasing protein yield, accumulation, and stability [53]. Homoplasmic shoots were regenerated three times under stringent spectinomycin/streptomycin selection pressure. The preliminary confirmation of transgenesis and homoplasmic status of transplastomic plants was confirmed by polymerase chain reaction and Southern blot analysis, respectively. After three rounds of selection, inheritance assays of transplastomic seeds displayed a homogeneous population of antibiotic-resistant seedlings, confirming that they are homoplasmic [54].

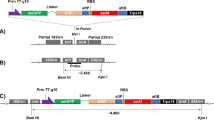
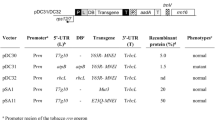
Physical map of fine structure for the chloroplast-specific DARPin G3 expression cassette, Nt-Prrn: ribosomal RNA operon promoter from tobacco; T7g10 5´ UTR: 5´ untranslated region of bacteriophage T7 gene 10; DARPin G3: coding sequence of DARPin G3, TrrnB: rrnB 3´ untranslated region from E. coli; PpsbA: promoter and 5´ UTR of the psbA gene; aadA: aminoglycoside 3´- adenylytransferase gene; TpsbA: terminator of the psbA gene
Isolation of chloroplasts
To prepare chloroplast proteins for downstream analysis, chloroplasts were separated from mature, green, and fully grown leaves from the mid-section of DARPin G3 transplastomic and wild-type tobacco plants, which were kept in the dark for 48 h to destarch the plastids. The leaves of transplastomic and wild-type plants, after removing the midrib, were finely ground and homogenized with 3 volumes (v/w) of ice-cold chloroplast isolation buffer (50 mM Tris-HCl, 0.35 M mannitol, 5 mM disodium EDTA, 0.1% BSA (w/v), and 1.0 mM 2-mercaptoethanol) using a motor-driven blender. The homogenate was centrifuged at 4 °C for 20 min at 1000 × g to pellet cell debris and nuclei after being passed through three layers of Miracloth. To isolate chloroplasts, the supernatant was decanted into fresh tubes and centrifuged at 4 °C for 20 min at 2500 × g. After discarding the supernatant, the green pellet was washed twice in the isolation buffer before final centrifugation for 10 min at 1500 × g. The supernatant was discarded, and the chloroplast pellet was utilized, either to extract total soluble proteins or by lyophilization.
Lyophilization
Fully expanded leaves from transplastomic and wild-type tobacco plants expressing DARPin G3, after excision of the midrib, were sliced into small pieces measuring roughly 1 cm2, frozen in liquid nitrogen, and then lyophilized in a Christ Alpha 1–2 LDplus Freeze Dry System in a vacuum (0.036 mBar) at -55 °C for 72 h. Lyophilization of isolated chloroplasts was performed in vacuum (0.036 mBar) at -50 °C for 72 h. The lyophilized materials were stored at room temperature for nine months and were used for the protein extraction after being ground in a grinder for 2 min at maximum speed three times (each time, pulse on for 15 s and off for 30 s). In the case of lyophilized leaves, the ground materials were subjected to sieving (mesh size: 100).
Protein immunoblot analysis
Total soluble protein was extracted from fresh and lyophilized leaves and chloroplasts of transplastomic and wild-type tobacco plants. 100 mg of fresh, fully expanded leaves grounded in the presence of liquid nitrogen or 100 mg of isolated chloroplasts were combined with 500 µl of cold extraction buffer (PBS 1X, pH 7.4, 150 mM NaCl, and 1X EDTA-free protease inhibitor), vigorously vortexed, and incubated for 30 min at 4 °C. Due to the weight reduction during freeze-drying (93% for leaves and 48% for chloroplast pellets), Lyophilized leaf tissue and chloroplasts were normalized with fresh leaf tissue and fresh isolated chloroplast weight, so that 7 mg of lyophilized leaves and 52 mg of lyophilized chloroplasts were used by adding 500 µl of extraction buffer and vortexed for 1 h at 4 °C for rehydration. The crude extracts were centrifuged at 13,000 rpm at 4 °C for 10 min to pellet cell debris. Supernatants were collected, and total protein concentrations were quantified by the Bradford protein assay (Bradford, 1976) using bovine serum albumin (BSA; Sigma Aldrich) as a standard.
As the normalized amount of fresh and lyophilized leaves and chloroplasts was used for protein extraction, we loaded gels on the basis of an equal amount of protein extract for Western blotting. About 50 µg of total soluble protein from each sample was boiled in sample buffer and separated by a 12% sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) in reducing conditions. Separated proteins were electro–transferred onto a nitrocellulose membrane (Bio-Rad) using Mini TransBlot (Bio-Rad), following the manufacturer’s instructions. Chloroplast-made DARPin G3 was detected using rabbit anti-His-tag antibody as a primary antibody and goat anti-rabbit conjugated with horseradish peroxidase (HRP) antibody as a secondary antibody. The bands were detected with the addition of a DAB-peroxidase substrate solution.
ELISA quantification of chloroplast-made DARPin G3 proteins
An enzyme-linked immunosorbent assay (ELISA) was used to measure the amounts of chloroplast-made DARPin G3 expression in fresh and lyophilized leaves and fresh and lyophilized chloroplasts of transplastomic and wild-type tobacco plants. About 50 µg of total soluble proteins in extraction buffer (PBS 1X, pH 7.4, 150 mM NaCl, and 1X EDTA-free protease inhibitor) from fresh and lyophilized materials were bound to a 96-well polyvinyl chloride microtiter plate overnight at 4 °C. Background was blocked for two hours at 37 °C with 300 µl of blocking buffer (1% BSA (w/v) in 1X PBS buffer containing 0.1% Tween 20 (w/v)). Wells were incubated for 2 h at 37 °C with rabbit anti-His-tag antibody diluted to 1:1000 with blocking buffer as the primary antibody, and then three times washed with PBS-T (PBS buffer containing 0.1% Tween 20 (w/v)). The blots were then incubated with a goat anti-rabbit antibody conjugated with HRP in blocking buffer at a dilution of 1:10000 as a secondary antibody for 2 h at 37 °C. The plate was washed again as stated above and developed using the 3, 3, 5, 5-tetramethylbenzidine (TMB) peroxidase substrate solution (200 mM citrate buffer, pH 3.95, 1% TMB, 0.01% H2O2). The reaction was terminated with 100 µl of 2 M H2SO4, and the optical density of each well was measured using an ELISA reader at 450 nm. For a standard curve, purified 15 kDa His-tagged standard protein was added in a serial dilution ranging from 3 to 25 ng/ml to the microplate and processed as described above. The standard curve was used to quantify the amount of DARPin G3 protein in the total soluble proteins that remained in freeze-dried materials compared to fresh tissue. The yield of DARPin G3 was calculated and expressed as a percentage of total soluble protein (TSP) and as the amount of DARPin G3 protein (mg/g).
Affinity purification of chloroplast-made DARPin G3
The chloroplast-made DARPin G3 was purified directly from total soluble protein extracted from fresh and lyophilized leaves and chloroplasts of transplastomic tobacco plants using the QIAexpress Ni-NTA Protein Purification System (QIAGEN). Total soluble proteins were mixed with 4x binding buffer (2 M NaCl and 2X PBS, no imidazole) at a 4:1 ratio in batch mode and incubated for 2 h at 4 °C. For the washing and elution steps, the protein–resin complex was packed into a column. The column was then washed with the wash buffer (0.5 M NaCl and 0.5X PBS, no imidazole) and eluted in 0.5 mL fractions for three times with the elution buffer (0.5 M NaCl and 0.5X PBS containing 200 mM imidazole). The eluted fractions were dialyzed against PBS three times, aliquoted, and stored at − 20 °C. SDS-PAGE and Coomassie blue staining were performed to detect chloroplast-made DARPin G3.
HER2 receptor binding assay
A binding test was carried out according to our previously described method [19] to measure the affinity of chloroplast-made DARPin G3 from lyophilized materials of transplastomic tobacco plants for the extracellular domain (ECD) of HER2 in 96-well pre-coated plates with the HER2-ECD. 100 µL of 1 g/mL HER2-ECD (Sino Biological, 10,004-HCCH) were used for the coating, which was then kept at 4 °C overnight. After being washed twice with phosphate-buffered saline solution containing 0.1% tween-20 (PBS-T), the plate was blocked of non-specific binding sites on a shaker for 1 h at room temperature with PBS-T containing 1% BSA. The ELISA procedure used a serial dilution ranging from 10 to 100 nM of purified chloroplast-made DARPin G3 in PBS-T/BSA, which was applied to triplicate wells in 100 µL volumes and incubated at room temperature for 1 h with shaking. After that, each well was washed three times with 200 µL of PBS-T. A rabbit anti-His-tag antibody (1:1000 in PBS-T/BSA), which recognizes the N-terminal histidine-glutamate (HE)3-tag of the chloroplast-made DARPin G3, was used to probe the binding and was incubated for an hour at room temperature on a shaker. After three washes with PBS-T, the goat anti-rabbit antibody conjugated with HRP was incubated for one hour at room temperature on a shaker in a final volume of 100 µL of PBS-T/BSA at a dilution of 1:10000. Following this, each well was washed with 200 µL of PBS-T for triplets. 100 µL of TMB solution was used to create the ELISA, which was then incubated at room temperature until a satisfactory colorimetric shift was seen. The reaction was stopped by adding 100 µL of 2 N H2SO4, and absorbance readings were measured at 450 nm using an ELISA plate reader. A total soluble protein of wild-type plants and bovine serum albumin (BSA) served as negative controls.
Cell culture conditions
We examined the HER2 specificity of chloroplast-made DARPin G3 from fresh and lyophilized leaves and chloroplasts using two distinct breast cancer cell lines that express HER2 to varying extents. SKBR-3 cells, a highly expressed HER2 breast cancer cell line, and MDA-MB-231 cells, which hardly express HER2, were provided by the Immunology Research Center (Tabriz, Iran). SKBR-3 and MDA-MB-231 cells were cultured in cell culture flasks containing RPMI 1640 medium supplemented with 1% penicillin (10,000 units/ml), 1% streptomycin (10 mg/ml), and 10% fetal bovine serum (FBS) at 37 °C in a humidified incubator with 5% CO2.
Flow cytometry
The binding specificity of the chloroplast-made DARPin G3 from fresh and lyophilized plant materials to HER2 on breast carcinoma cell lines was examined via flow cytometry by following the procedures as described by us [19]. In brief, two distinct cancer cell lines were individually prepared. After removing the medium, the cells were treated for 10 min with 5 ml of 0.2% EDTA. The cells were then transferred to tubes and centrifuged for 5 min at 4 °C at 1000 rpm. After removing the EDTA-containing supernatant, 5 ml of fresh medium was added. 1 ml of cells counted and diluted to 106 cells per ml were used for each test condition. After washing the cells with cold PBS containing 1% BSA, 100 nM chloroplast-derived DARPin G3 was added to the cells and incubated at 4 °C for 1 h. Following a cold PBS/BSA wash, cells were incubated for 1 h at 4 °C with 200 µl of rabbit anti-His-tag antibody diluted in PBS/BSA (1:1000). The cells were then washed with cold PBS/BSA, followed by incubation for 30 min at 4 °C in the presence of 200 µl of fluorescein isothiocyanate-coupled donkey (FITC) anti-rabbit antibody at a dilution of 1:1000. After washing with cold PBS, the cells were resuspended in 500 µl of cold PBS and analyzed on a MACSQuant 10 Flow Cytometer (Miltenyi Biotec, Germany) at a flow rate of 500 s−1. Fluorescence was detected at 525 nm after being excited with an argon laser at 488 nm. Cells were then gated according to size scatter, forward scatter, and pulse width, so only single cells were analyzed. Negative groups included cells that had not been treated or that had been treated with anti-His tag antibody followed by FITC donkey anti-rabbit IgG. A total of 10,000 cell events were recorded per sample. Fluorescence data was analyzed using the software FlowJo (Tree Star, Ashland, OR). Cells treated with PBS or antibodies in the absence of chloroplast-made DARPin G3 were used as controls.
Immunofluorescent microscopy
To begin, cells were seeded into sterile 96-well culture plates at a density of 1.0 × 104 in RPMI 1640 growth media and incubated at 37 °C and 5% CO2 for the duration of the night. After removing the medium, the cells were washed three times with ice-cold PBS containing 1% BSA. Cells were treated for 1 h at 4 °C with 200 nM chloroplast-made DARPin G3 in PBS/BSA. Following three washes with cold PBS/BSA, the cells were incubated for 1 h at 4 °C with rabbit anti-His-tag antibody (1:1000 diluted in PBS/BSA) as the primary antibody. Cells were washed and incubated for 30 min at 4 °C in the dark with FITC donkey anti-rabbit IgG (1:1000 diluted in PBS/BSA) as a secondary antibody. After three washes with cold PBS, cells were analyzed using the Citation 5 Cell Imaging Multimode Reader (BioTek, Winooski, VT). The laser at 469 nm excited the DARPin G3-FITC, and the fluorescence of FITC was registered at 525 nm.
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Abbreviations
- BSA:
-
Bovine serum albumin
- DARPin:
-
Designed Ankyrin Repeat Protein
- ELISA:
-
Enzyme-linked immunosorbent assay
- FITC:
-
Fluorescein isothiocyanate-coupled
- HER2:
-
Human epidermal growth factor receptor 2
- HRP:
-
Horseradish peroxidase
- PBS:
-
Phosphate Buffered Saline
- SDS-PAGE:
-
Sodium dodecyl sulphate–polyacrylamide gel electrophoresis
- TMB:
-
3,3',5,5'-Tetramethylbenzidine
References
Goodman M. Pharmaceutical industry financial performance. Nat Rev Drug Discovery. 2009;8(12):927.
Twyman M, Schillberg R., S., Fischer R. Optimizing the yield of recombinant pharmaceutical proteins in plants. Curr Pharm Design. 2013;19(31):5486–94.
Sharma AK, Sharma MK. Plants as bioreactors: recent developments and emerging opportunities. Biotechnol Adv. 2009;27(6):811–32.
Pogue GP, et al. Production of pharmaceutical-grade recombinant aprotinin and a monoclonal antibody product using plant‐based transient expression systems. Plant Biotechnol J. 2010;8(5):638–54.
Xu J, Ge X, Dolan MC. Towards high-yield production of pharmaceutical proteins with plant cell suspension cultures. Biotechnol Adv. 2011;29(3):278–99.
Faye L, Gomord V. Success stories in molecular farming—a brief overview. Wiley Online Library; 2010. pp. 525–8.
Gomord V, et al. Plant-specific glycosylation patterns in the context of therapeutic protein production. Plant Biotechnol J. 2010;8(5):564–87.
Schillberg S, Fischer R, Emans N. Molecular farming of recombinant antibodies in plants. Cell Mol Life Sci CMLS. 2003;60(3):433–45.
Wroblewski T, Tomczak A, Michelmore R. Optimization of Agrobacterium-mediated transient assays of gene expression in lettuce, tomato and Arabidopsis. Plant Biotechnol J. 2005;3(2):259–73.
Maliga P. Engineering the plastid genome of higher plants. Curr Opin Plant Biol. 2002;5(2):164–72.
Su J, et al. Low cost industrial production of coagulation factor IX bioencapsulated in lettuce cells for oral tolerance induction in hemophilia B. Biomaterials. 2015;70:84–93.
Pal SK, Pegram M. Targeting HER2 epitopes. In seminars in oncology. Elsevier; 2006.
Jost C, Plückthun A. Engineered proteins with desired specificity: DARPins, other alternative scaffolds and bispecific IgGs. Curr Opin Struct Biol. 2014;27:102–12.
Boersma YL, Plückthun A. DARPins and other repeat protein scaffolds: advances in engineering and applications. Curr Opin Biotechnol. 2011;22(6):849–57.
Ladjemi MZ, et al. Anti-HER2 vaccines: new prospects for breast cancer therapy. Cancer Immunol Immunother. 2010;59(9):1295–312.
Ross JS, et al. The HER-2 receptor and breast cancer: ten years of targeted anti–HER‐2 therapy and personalized medicine. Oncologist. 2009;14(4):320–68.
Goldstein R, et al. Development of the designed ankyrin repeat protein (DARPin) G3 for HER2 molecular imaging. Eur J Nucl Med Mol Imaging. 2015;42(2):288–301.
Zahnd C, et al. Selection and characterization of Her2 binding-designed ankyrin repeat proteins. J Biol Chem. 2006;281(46):35167–75.
Ehsasatvatan M, et al. The production of the first functional antibody mimetic in higher plants: the chloroplast makes the DARPin G3 for HER2 imaging in oncology. Biol Res. 2022;55(1):1–18.
Zahnd C, et al. Efficient tumor targeting with high-affinity designed ankyrin repeat proteins: effects of affinity and molecular size. Cancer Res. 2010;70(4):1595–605.
Bouchnak I, et al. Unraveling Hidden Components of the Chloroplast Envelope Proteome: Opportunities and limits of Better MS Sensitivity*[S]. Mol Cell Proteom. 2019;18(7):1285–306.
Zahnd C, et al. A designed ankyrin repeat protein evolved to picomolar affinity to Her2. J Mol Biol. 2007;369(4):1015–28.
Shipunova VO, et al. Comparative evaluation of engineered polypeptide scaffolds in HER2-targeting magnetic nanocarrier delivery. ACS Omega. 2021;6(24):16000–8.
Vorobyeva A, et al. Comparison of tumor–targeting properties of directly and indirectly radioiodinated designed ankyrin repeat protein (DARPin) G3 variants for molecular imaging of HER2. Int J Oncol. 2019;54(4):1209–20.
Shilova O, et al. Internalization and recycling of the HER2 receptor on human breast adenocarcinoma cells treated with targeted phototoxic protein DARPinminiSOG. Acta Naturae (англоязычная версия). 2015;7(3):126–32.
Daniell H, Rai V, **ao Y. Cold chain and virus-free oral polio booster vaccine made in lettuce chloroplasts confers protection against all three poliovirus serotypes. Plant Biotechnol J. 2019;17(7):1357–68.
Obembe OO, et al. Advances in plant molecular farming. Biotechnol Adv. 2011;29(2):210–22.
Scotti N, Bellucci M, Cardi T. The chloroplasts as platform for recombinant proteins production, in translation in mitochondria and other organelles. Springer; 2013. pp. 225–62.
Adem M, Beyene D, Feyissa T. Recent achievements obtained by chloroplast transformation. Plant Methods. 2017;13(1):30.
Staub JM, et al. High-yield production of a human therapeutic protein in tobacco chloroplasts. Nat Biotechnol. 2000;18(3):333–8.
Kumar S, Daniell H. Engineering the chloroplast genome for hyperexpression of human therapeutic proteins and vaccine antigens, in recombinant gene expression. Springer; 2004. pp. 365–83.
Kwon KC, et al. Oral delivery of bioencapsulated exendin-4 expressed in chloroplasts lowers blood glucose level in mice and stimulates insulin secretion in beta‐TC 6 cells. Plant Biotechnol J. 2013;11(1):77–86.
Singh R, et al. Affordable oral health care: dental biofilm disruption using chloroplast made enzymes with chewing gum delivery. Plant Biotechnol J. 2021;19(10):2113–25.
Kwon K-C, et al. Oral delivery of human biopharmaceuticals, autoantigens and vaccine antigens bioencapsulated in plant cells. Adv Drug Deliv Rev. 2013;65(6):782–99.
Lakshmi PS et al. Low cost tuberculosis vaccine antigens in capsules: expression in chloroplasts, bio-encapsulation, stability and functional evaluation in vitro. PLoS ONE. 8(1), e54708 (2013).
Kwon KC, et al. Expression and assembly of largest foreign protein in chloroplasts: oral delivery of human FVIII made in lettuce chloroplasts robustly suppresses inhibitor formation in haemophilia A mice. Plant Biotechnol J. 2018;16(6):1148–60.
Nakahira Y et al. Mass Production of Virus-Like particles using Chloroplast Genetic Engineering for highly immunogenic oral vaccine against Fish Disease. Front Plant Sci. 12 (2021).
Loibl S, Gianni L. HER2-positive breast cancer. The Lancet. 2017;389(10087):2415–29.
Wolff AC, et al. Human epidermal growth factor receptor 2 testing in breast cancer: american society of clinical Oncology/College of american pathologists clinical practice guideline focused update. Arch Pathol Lab Med. 2018;142(11):1364–82.
Zahnd C, Amstutz P, Plückthun A. Ribosome display: selecting and evolving proteins in vitro that specifically bind to a target. Nat Methods. 2007;4(3):269–79.
Garousi J, et al. Imaging using radiolabelled targeted proteins: Radioimmunodetection and beyond. EJNMMI Radiopharmacy and Chemistry. 2020;5(1):1–26.
Bragina O, et al. Phase I trial of 99mTc-(HE) 3-G3, a DARPin-based probe for imaging of HER2 expression in breast cancer. J Nucl Med. 2022;63(4):528–35.
Gomes AR, et al. An overview of heterologous expression host systems for the production of recombinant proteins. Adv Anim Veterinary. 2016;4(7):346–56.
Vieira Gomes AM, et al. Comparison of yeasts as hosts for recombinant protein production. Microorganisms. 2018;6(2):38.
Rosano GL, Ceccarelli EA. Recombinant protein expression in Escherichia coli: advances and challenges. Front Microbiol. 2014;5:172.
Shanmugaraj B, Bulaon CJI, Phoolcharoen W. Plant molecular farming: a viable platform for recombinant biopharmaceutical production. Plants. 2020;9(7):842.
Ahmad M, et al. Protein expression in Pichia pastoris: recent achievements and perspectives for heterologous protein production. Appl Microbiol Biotechnol. 2014;98(12):5301–17.
Boehm R. Bioproduction of therapeutic proteins in the 21st century and the role of plants and plant cells as production platforms. Ann N Y Acad Sci. 2007;1102(1):121–34.
Streatfield SJ, Howard JA. Plant production systems for vaccines. Expert Rev Vaccines. 2003;2(6):763–75.
Daniell H, et al. Plant-made vaccine antigens and biopharmaceuticals. Trends Plant Sci. 2009;14(12):669–79.
Arlen PA, et al. Field production and functional evaluation of chloroplast-derived interferon‐α2b. Plant Biotechnol J. 2007;5(4):511–25.
Zoubenko OV, et al. Efficient targeting of foreign genes into the tobacco plastid genome. Nucleic Acids Res. 1994;22(19):3819–24.
Ehsasatvatan M et al. Physical and Biologically Effective Parameters in Develo** Transplastomic Tobacco Plants by Particle Bombardment Method using PDS-1000/He. Genetic Engineering and Biosafety Journal. 10(2) (2022). https://dorl.net/dor/20.1001.1.25885073.1400.10.2.10.2.
Ehsasatvatan M, Baghban Kohnehrouz B. Homoplasmic Stability and cytoplasmic inheritence of DARPin G3 Scaffold protein in Generative and vegetative propagation of Transplastoic Tobacco plants. Plant Genetic Researches. 2023;9(2):1–14.
Acknowledgements
This work was supported by the Department of Plant Breeding and Biotechnology, Faculty of Agriculture, University of Tabriz, Tabriz, Iran. We thank Iranian Research Institute for Scientific Information and Documentation (IRANDOC) for providing instrumental facility and Immunology Research Center of Tabriz medical sciences university for technical supports.
Funding
Not applicable.
Author information
Authors and Affiliations
Contributions
M.E., designed the experiments, performed the experiments, analyzed the data, and wrote the manuscript. B. B. K., conceived, designed, organized, and supervised the study, provided specialized scientific and technical support, and revised the paper. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Ehsasatvatan, M., Kohnehrouz, B.B. The lyophilized chloroplasts store synthetic DARPin G3 as bioactive encapsulated organelles. J Biol Eng 17, 63 (2023). https://doi.org/10.1186/s13036-023-00383-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13036-023-00383-3